
Abstract
Oranges are an important nutritional source for human health and have immense economic value. Here we present a comprehensive analysis of the draft genome of sweet orange (Citrus sinensis). The assembled sequence covers 87.3% of the estimated orange genome, which is relatively compact, as 20% is composed of repetitive elements. We predicted 29,445 protein-coding genes, half of which are in the heterozygous state. With additional sequencing of two more citrus species and comparative analyses of seven citrus genomes, we present evidence to suggest that sweet orange originated from a backcross hybrid between pummelo and mandarin. Focused analysis on genes involved in vitamin C metabolism showed that GalUR, encoding the rate-limiting enzyme of the galacturonate pathway, is significantly upregulated in orange fruit, and the recent expansion of this gene family may provide a genomic basis. This draft genome represents a valuable resource for understanding and improving many important citrus traits in the future.
Similar content being viewed by others


Main
Citrus is a large genus that includes several major cultivated species, including C. sinensis (sweet orange), Citrus reticulata (tangerine and mandarin), Citrus limon (lemon), Citrus grandis (pummelo) and Citrus paradisi (grapefruit). In 2009, the global citrus acreage was 9 million hectares and citrus production was 122.3 million tons (FAO statistics, see URLs), which is the top ranked among all the fruit crops. Among the 10.9 million tons (valued at $9.3 billion) of citrus products traded in 2009, sweet orange accounted for approximately 60% of citrus production for both fresh fruit and processed juice consumption (FAO statistics, see URLs). Moreover, citrus fruits and juice are the prime human source of vitamin C, an important component of human nutrition.
Citrus fruits also have some unique botanical features, such as nucellar embryony (nucellus cells can develop into apomictic embryos that are genetically identical to mother plant). Consequently, somatic embryos grow much more vigorously than the zygotic embryos in seeds such that seedlings are essentially clones of the maternal parent. Such citrus-unique characteristics have hindered the study of citrus genetics and breeding improvement1,2. Complete genome sequences would provide valuable genetic resources for improving citrus crops.
Citrus is believed to be native to southeast Asia3,4,5, and cultivation of fruit crops occurred at least 4,000 years ago3,6. The genetic origin of the sweet orange is not clear, although there are some speculations that sweet orange might be derived from interspecific hybridization of some primitive citrus species7,8. Citrus is also in the order Sapindales, a sister order to the Brassicales in the Malvidae, making it valuable for comparative genomics studies with the model plant Arabidopsis.
We aimed to sequence the genome of Valencia sweet orange (C. sinensis cv. Valencia), one of the most important sweet orange varieties cultivated worldwide and grown primarily for orange juice production. Normal sweet oranges are diploids, with nine pairs of chromosomes and an estimated genome size of ∼367 Mb9. To reduce the complexity of the sequenced genome, we obtained a double-haploid (dihaploid) line derived from the anther culture of Valencia sweet orange10. We first generated whole-genome shotgun paired-end–tag sequence reads from the dihaploid genomic DNA and built a de novo assembly as the citrus reference genome; we then produced shotgun sequencing reads from the parental diploid DNA and mapped the sequences to the haploid reference genome to obtain the complete genome information for Valencia sweet orange. In addition, we conducted comprehensive transcriptome sequencing analyses for four representative tissues using shotgun RNA sequencing (RNA-Seq) to capture all transcribed sequences and paired-end–tag RNA sequencing (RNA-PET) to demarcate the 5′ and 3′ ends of all transcripts. On the basis of the DNA and RNA sequencing data, we characterized the orange genome for its gene content, heterozygosity and evolutionary features. The genome and transcriptome analyses presented in this study yield new insights into the origin of sweet orange and the genomic basis of vitamin C metabolism and provide a rich resource of genetic information for citrus breeding and genetic improvement.
Results
Genome sequencing and assembly
From the dihaploid DNA of sweet orange (Supplementary Fig. 1), we generated 785 million high quality paired-end–tag sequencing reads (2 × 100 bp) from various DNA fragment sizes (∼300 bp, 2 kb, 10 kb and 20 kb) using the Illumina GAII sequencer, representing 214-fold base-pair coverage and 4,889-fold physical coverage of the estimated citrus genome (Supplementary Table 1). The sequence reads were assembled by SOAPdenovo11 and Opera12, resulting in 16,890 assembled sequence contigs (N50, 49.89 kb) and 4,811 scaffolds (N50, 1.69 Mb) (Table 1). The total contig sequence length (320.5 Mb) covers 87.3% of the estimated sweet orange genome, and more than 80% of the genome assembly is in 135 scaffolds that are larger than 713 kb and up to 8.2 Mb. To evaluate the accuracy of the citrus genome assembly, we assessed the quality of the scaffolds by BAC clone analysis, alignment to the established citrus genetic linkage map and cytogenetic analysis.
A BAC clone library of sweet orange has previously been constructed and characterized13, and the estimated size range of BAC clone inserts was 120 kb. We picked individual clones for BAC-end capillary sequencing. Of the 735 pairs of BAC-end sequence reads in which both ends of a pair were mapped within the same contig, the average distance was 125,875 ± 26,788 bp (s.d.). This is consistent with the previously estimated length of BAC clones13, suggesting the correctness of the contig assembly. To further evaluate the accuracy of the assembly on a local scale, we analyzed seven complete BAC sequences of sweet orange that are available in GenBank and also anchored in the sweet orange genetic map. The alignments between these BAC sequences and the citrus genome assembly showed an overall identity of 97.6% (Supplementary Fig. 2 and Supplementary Table 2). Together, the BAC clone sequencing analyses validated that the assembly is of high quality.
We then asked whether the assembly of the genome sequences matches well with the established sweet orange genetic linkage map14. We mapped the scaffolds to 768 markers with known sequences in the genetic linkage groups, and 160 scaffolds (each >10 kb) were anchored, accounting for 239 Mb of the assembled citrus genome (Supplementary Table 3). Furthermore, 83 scaffolds that accounted for the majority of the length of the anchored scaffolds (178.7 Mb, or 75% of total anchored scaffolds) were in matched orientation within the genetic map (Supplementary Note), suggesting high alignment accordance between the anchored genetic markers and the sequence scaffolds. The discordance in alignment could reflect technical accuracy, such as insufficient marker density, genotyping errors or individual variations between the sequenced genome (Valencia sweet orange) and the genetic map that was based on three crosses between three sweet orange cultivars14. Valencia orange is known to have chromosome rearrangements relative to seedy sweet oranges15. Combining the assembled genome sequences and the genetic linkage groups, we constructed the pseudomolecules for each of the nine chromosomes and ordered them on the basis of genetic length (Fig. 1).

Assembled scaffolds (blue; 239 Mb, or 75% of the assembled genome sequence) were anchored in the nine linkage groups (LG1–LG9, yellow) with the corresponding genetic markers (black bars). The pseudochromosome numbers are assigned on the basis of the estimated length of the genetic linkage groups14.
We then took some cytological markers, including two ribosomal genes (45S and 5S) and a tandem repeat sequence (107 bp), for FISH analyses to further validate the accordance of the assembled citrus genome sequences. The cytologically marked chromosomes showed tentative consistence with the genomically defined pseudochromosomes (Supplementary Table 4). For example, pseudochromosomes 1 and 8, bearing the 45S region at proximal locations, correspond to two type-B chromosomes, with one in the fragile site (Supplementary Fig. 3); pseudochromosome 6, with the mapped scaffolds containing 45S and 5S ribosomal DNA sequences at distal regions, corresponds to the type-D chromosome with a fragile site; and chromosome 7 contains a large number of copies of the 107-bp tandem repeat sequence at two distal locations, which corresponds to the type-C chromosome (Supplementary Fig. 3). Collectively, our cytological experiments provided sequence-matched evidence for chromosomes 1, 6, 7 and 8. Additional cytological evidence is needed for other chromosomes.
Genome characterization
Most plant genomes are featured with highly repetitive regions with transposable elements and unique gene-coding regions. We analyzed the citrus genome in these two aspects to reveal the basic structural and functional features of the genome.
Transposable elements are major components of plant genomes and important contributors to genome evolution. However, very few transposable elements have previously been identified in Citrus. To analyze the transposable elements of the orange genome, we first constructed a custom digital transposable element library using ab initio, homology- and structure-based transposable element prediction tools and then applied this dataset to annotate the citrus reference genome. Such analysis led to the identification of 61.7 Mb of sequence in the assembled contigs, accounting for 20.5% of the C. sinensis genome (Supplementary Table 5). This level of repetitiveness is similar to those of Arabidopsis16 and rice17, suggesting that sweet orange has a relatively compact genome.
Class I long terminal repeat retrotransposons occupy over 89% of the transposable elements, consisting of 198 Copia families and 130 Gypsy families. Similar to other compact genomes such as those of Arabidopsis18 and rice19, the intact elements in each family have less than 250 members, whereas more than 90% of long terminal repeat retrotransposon families contain only one to five full-length elements. Estimation of insertion time indicated that more than 80% of the intact elements were amplified within the last 2 million years, and 10% are younger than 50,000 years (Supplementary Table 6). In contrast, class II DNA transposons only comprised ∼11% of the transposable elements, similar to in the apple20 and papaya21 genomes. Among the class II DNA transposons, miniature inverted-repeat transposable elements (MITEs) occupied 67% of the sequence. Notably, we identified a new type of MITE, named MiM (MITE inserted in microsatellite), in the citrus genome (Supplementary Table 5).
To annotate the citrus genome for protein-coding genes, we used a comprehensive strategy that combined ab initio gene predictions, protein-based homology searches and experimental supports (ESTs, RNA-Seq and RNA-PET) (Supplementary Fig. 4). We used four ab initio gene-finding programs (Fgenesh, GeneID, Genscan and GlimmerHMM) on the repeat-masked genome sequences. In addition, we collected 917,610 proteins from the UniProt database22 and 958,121 Citrus EST reads from GenBank and generated 964.6 million RNA-Seq reads from four citrus tissues (callus, leaf, flower and fruit) (Supplementary Table 1). In total, we identified 29,445 protein-coding loci (gene models) with 44,387 transcripts (Fig. 2 and Supplementary Table 7), and 23,421 gene models were allocated in nine pseudochromosomes. The gene transcripts have an average length of 1,817 bp, a mean coding sequence size of 1,255 bp and an average of 5.8 exons per gene. The RNA-Seq and EST data suggested that 26% of the gene models (7,640 gene loci) encoded two or more transcript isoforms (Supplementary Fig. 5a). Overall, 85% of the gene models contain multiple exons (Supplementary Fig. 5b), and 99% of the predicted coding sequences and 80% of the splice junction sites were supported by the RNA-Seq data (Fig. 2b and Supplementary Table 7), showing the high accuracy of gene annotation in the orange genome. We evaluated and corrected all predicted gene models using the RNA-PET sequencing data (which is complementary to the RNA-Seq data) for demarcating the gene boundaries (Supplementary Fig. 6). We generated approximately 100 million RNA-PETs from four tissues (callus, leaf, flower and fruit) and used them for mapping, clustering and annotation of the genome (Supplementary Table 1), confirming 70% of the 5′ and 3′ boundaries of the gene transcripts (Fig. 2c and Supplementary Table 8). Notably, we identified 967 transcripts for 744 putative gene models solely on the basis of the combined RNA-Seq and RNA-PET data (Supplementary Table 9 and Supplementary Fig. 7), representing a collection of potentially new and citrus-specific genes. In addition, we identified a total of 181 ribosomal RNAs, 451 transfer RNA, 39 small nucleolar RNAs and 104 small nuclear RNAs in the citrus genome (Supplementary Note). We also identified 40 conserved microRNA (miRNA) families with 227 members in the orange genome on the basis of previous small RNA sequencing data23 (Supplementary Table 10), and most of the miRNA genes are distributed evenly across the genome, with several small clusters (Supplementary Fig. 8).

(a) Circular diagram depicting the genomic landscape of the nine sweet orange pseudochromosomes (Chr1 to ∼Chr9 on a Mb scale). The denotation of each track is listed on the right. RPKM, reads per kilobase exon model per million mapped reads. (b) Experimental data support for gene model predictions. Left, box plot of RNA-Seq reads aligned over gene features. The top and bottom of the boxes indicate the upper and lower quartiles, respectively. The middle red bars represent the median, and the red dots denote the mean value for each feature. CDS, coding sequence. Right, Venn diagram of the predicted RNA splice junctions supported by various evidences (EST, protein and RNA-Seq data). Numbers in parentheses are the number of splice junctions in each category. (c) Demarcation of gene model boundaries by RNA-PET data. Histogram plot of RNA-PET data in association with aligned gene models in relation to the 5′ (putative transcription start site, TSS) and 3′ (putative poly(A) site, PAS) boundaries.
To better understand citrus-specific biology, we identified citrus-specific genes. We performed comparative analyses of the orange genome against 21 other plant genomes and screened them by EST and peptide sequences in the public database (Supplementary Note). Of the 29,445 protein-coding genes in the orange genome, 23,804 were grouped into 14,348 gene families, with 1,691 genes being specific to citrus (Supplementary Fig. 9). Most (96%) of the citrus-specific genes are 'hypothetical' and have unknown function. Of the 58 annotated genes, the over-represented protein domains are the zinc finger transcription factor domain and the nucleotide-binding-site and leucine-rich-repeat (NBS-LRR) domain, which contain potential disease-resistance genes (Supplementary Table 11).
Heterozygosity and hybrid origin of sweet orange
Heterozygosity is a common feature in most eukaryotic organisms and has important biological functions in crops. Although citrus trees are generally perceived to have highly heterozygous traits2, the amount of heterozygosity in the whole genome is not clear. Sequencing of the haploid and diploid genomes provided the genomic data (Supplementary Table 12) for us to comprehensively assess the heterozygosity of sweet orange. From the diploid genome sequencing data, we identified 1.06 million SNPs and 176,953 small insertions/deletions (indels). More than 80% of the SNPs and indels were anchored on the nine pseudochromosomes. The overall polymorphism density is 3.6 SNPs and 0.6 indels per kb in the genome (Supplementary Table 13). Approximately one-third of the SNPs were located in genic regions (Supplementary Table 14), and 16,843 gene loci (72%) contained at least one SNP (Supplementary Fig. 10). Considering a 0.52% error rate in detecting SNPs (Supplementary Note), we set up a stringent criterion of five or more SNPs per gene to reliably call variant alleles; using this criterion, 12,297 gene loci (53%) were considered to harbor variant alleles (Supplementary Table 15). This observation suggests a notably high heterozygosity rate in the gene loci.
To verify this observation, we genotyped 307 simple sequence repeat (SSR) markers on the dihaploid and parental diploid of sweet orange and identified 155 (50%) SSR loci in a heterozygous state (Fig. 3a and Supplementary Table 16), which is similar to the heterozygosity rate in the protein-coding gene loci found by SNP analysis. Moreover, chromosome karyotyping analysis showed that 6 of the 18 chromosomes have no clear counterpart in the parental diploid, whereas the chromosomes were easily grouped into nine pairs in the dihaploid line (Fig. 3b), further supporting the observation that the diploid genome of sweet orange is highly heterozygous.

(a) The heterozygosity rate of the sweet orange genome. Homozygous and heterozygous states of genic regions were detected using SNP data from the parental diploid genome; repeat regions were evaluated by comparison of SSR marker patterns between the diploid and dihaploid. The number in the top pie chart indicates the gene number, and the number in the bottom pie chart is the number of SSR markers. (b) Chromosome karyotyping of dihaploid (left) and parental diploid (right). The 18 chromosomes (chrs) in each nucleus were easily identified into nine matched pairs in the dihaploid line, whereas the six chromosomes in the diploid nucleus have no clear counterpart, indicating high heterozygosity in the diploid state. The cytological types of chromosomes were traditionally named as Bf, B, C, Df, D and F according to the 4′-6-diamidino-2-phenylindole (blue) and chromomycin A3 (yellow) banding regions, as well as the fragile site (circle), as previously described41 (Supplementary Fig. 3). (c) SSR marker genotyping of two pummelo cultivars (P1 and P2), sweet orange (SO) and two mandarin cultivars (M1 and M2). Of the 307 SSR markers, 105 were shared among pummelo, orange and mandarin (middle), 55 were pummelo hereditary (left) and 147 were mandarin hereditary (right). One marker showed an orange-specific pattern and was excluded in this analysis. (d) The genetic origin of the sweet orange dihaploid genome determined by high-density SNP markers of pummelo and mandarin. OP, origin pattern (pummelo origin in yellow, mandarin origin in orange and undetermined in gray); SC, assembled sequence scaffold; LG, linkage group map. Pseudochromosome 1 is shown here as an example, and the other chromosomes are shown in Supplementary Figure 11. (e) A model of the sweet orange origin. With pummelo (PP) as the female parent crossed with mandarin (MM), the interspecific hybrid was backcrossed with mandarin (MM) and produced the ancient sweet orange.
We thus investigated the speculated hybrid origin of sweet orange from pummelo (C. grandis) and mandarin (C. reticulata)7,8, which are two primitive species of Citrus. To measure the possible genetic contribution to sweet orange from pummelo and mandarin, we surveyed the genotype of sweet orange, two pummelo cultivars and two mandarin cultivars using sweet orange–derived SSR markers and identified 202 polymorphic markers, 55 of which are pummelo hereditary and 147 of which are mandarin hereditary (Fig. 3c and Supplementary Table 16). It is remarkable that the numbers of pummelo-hereditary markers and mandarin-hereditary markers (55 and 147, respectively) fit well with a 1:3 ratio (χ2 = 0.54 < χ20.95 (2–1) = 3.84), indicating an interesting genetic relationship between sweet orange, pummelo and mandarin.
To comprehensively verify the genome composition of sweet orange inferred from SSR markers, we extended our analysis using high-density SNP makers. For this purpose, we generated whole-genome shotgun short sequence reads for three pummelo cultivars and three mandarin cultivars, each at more than 30-fold coverage of the citrus genome (Supplementary Table 12); we then mapped the sequences to the haploid orange reference genome and identified 475,450 SNPs common in three individual pummelos and 308,837 SNPs common in three individual mandarins (Supplementary Table 17). Using a 100-kb genomic window, we calculated the SNP density preference for pummelo or mandarin and then determined the hereditary origin (Supplementary Note), which allowed us to assign 39.7 Mb of the sweet orange genome from pummelo and 118.2 Mb from mandarin, which approximately fits a ratio of 1:3 in the genomic contributions from pummelo and mandarin (Fig. 3d and Supplementary Fig. 11), consistent with the SSR analysis results.
On the basis of the high heterozygosity rate (50%), the 1:3 ratio of pummelo and mandarin genetic input and the evidence that the chloroplast of sweet orange is probably derived from pummelo7 (Supplementary Fig. 12), we hypothesized that sweet orange originated from an interspecific hybridization with pummelo as the female parent and mandarin as the male parent followed by a backcross with a male mandarin (sweet orange = (pummelo × mandarin) × mandarin) (Fig. 3e).
Genome evolution
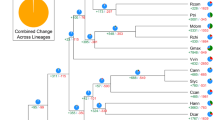
Characterization and annotation of the citrus genome provided the basic features for us to further investigate the evolutionary history of sweet oranges. Most plant species have gone through whole-genome duplication (WGD) during evolution24. The size and frequency of paralogous gene families accumulated in each genome serves as a record for such evolutionary process. Self-alignment of the citrus genome sequences on the basis of the 23,421 gene models located in the nine pseudochromosomes identified 1,296 paralogous gene groups (Fig. 4a). The mean synonymous substitution rates (Ks) in the duplicated genes (Ks = 1.27) suggested that an ancient WGD occurred in citrus (Fig. 4b and Supplementary Fig. 13). Furthermore, we analyzed intergenome collinearity between orange and cacao25, Arabidopsis16, apple20, strawberry26 and grape27 genomes and identified orange orthologous genes in syntenic blocks with respect to each of these genomes (Supplementary Table 18). A one-to-one orthologous relationship was predominant for the orange-to-cacao, orange-to-grape and orange-to-strawberry comparisons, indicating that there were no recent WGDs except the shared ancient triplication, called the γ event, which was shared by all core eudicots24. In contrast, we found an approximately one-to-two orthologous relationship for the orange-to-apple comparison and a one-to-four relationship for the orange-to-Arabidopsis comparison, supporting that an additional one and two rounds of recent WGD occurred in apple and Arabidopsis, respectively (Fig. 4c and Supplementary Fig. 14). Furthermore, on the basis of cross-species synteny block boundaries, we estimated that at least 49 events of chromosome translocations and fusions occurred in the nine citrus chromosomes evolved from chromosomes of a proposed paleohexaploid ancestor that is common to all sequenced eudicot genomes28. It is noteworthy that citrus pseudochromosome 4 had only one chromosome translation and fusion event, whereas other pseudochromosomes underwent multiple (3–12) interchromosome fusions during their evolutionary history (Supplementary Fig. 15).

(a) Schematic representation of major interchromosomal relationships within the 1,294 paralogous gene groups in the sweet orange genome. Syntenic blocks derived from the seven ancestral protochromosomes A1, A4, A7, A10, A13, A16 and A19 (ref. 28) are color coded as indicated. (b) Distribution of synonymous substitution rates (Ks) for homologous gene groups for intrachromosome and interchromosome comparisons. Gene duplication analysis in comparison to apple, Arabidopsis and cacao indicates that no recent WGDs occurred in the sweet orange genome. (c) Schematic representation of the syntenic relationship of sweet orange chromosome 9 and the corresponding chromosomes in Arabidopsis, Theobroma cacao, Malus × domestica, Fragaria vesca and Vitis vinifera. Syntenic regions derived from the seven ancestral protochromosomes are color coded as in a. Among the 59 syntenic blocks between sweet orange chromosome 9 and the Arabidopsis chromosomes, 22 (37%) show primary correspondence to four Arabidopsis blocks, 10 to three blocks, 24 to two blocks and 3 to one block; among the 79 syntenic blocks between sweet orange chromosome 9 and the apple chromosomes, 48 (61%) show primary correspondence to two Arabidopsis blocks, 20 to three blocks, 6 to four blocks and 2 to one block. Therefore, on the basis of these percentages, one-to-four and one-to-two relationships were determined to be the most probable for the orange-to-Arabidopsis and orange-to-apple comparisons, respectively. (d) Citrus phylogeny on the basis of 103 single-copy genes shared between Arabidopsis, cacao, poplar, grape, apple, strawberry, papaya and castor bean from nuclear genome data. MYA, million years ago.
This whole-genome sequence also provides comprehensive information for phylogenetic analysis. We applied the maximum likelihood method for genome-scale phylogenic analysis using 103 shared single-copy nuclear genes from orange, Arabidopsis16, cacao25, poplar29, grape27, apple20, strawberry26, papaya21 and castor bean30 (Fig. 4d and Supplementary Note). The phylogenetic tree showed that Citrus is phylogenetically closer to cacao, Arabidopsis and papaya, although it is ancient compared to them in the Malvidae, and is estimated to have diverged from Malvales 85 million years ago. Thus, our analysis provides new information on the phylogeny of citrus and other plant species on the basis of genome-scale data.
Genes specially involved in citrus fruit biology
Fruit biology is an interesting topic for citrus not only because citrus fruits are an important nutrition source for humans but also because they involve unique metabolic pathways. This citrus reference genome provides a comprehensive genomic framework of genes to help address citrus fruit biology. A clear strategy is to identify genes specifically expressed in fruit tissues. We compared the RNA-Seq data derived from callus, leaf, flower and fruit and identified 2,697 (9.2%) genes that were significantly upregulated (P < 0.01) in fruits (Supplementary Table 19). Gene Ontology analysis of upregulated genes in citrus fruits revealed that genes for oxidoreductase, oxidation reduction and vitamin binding were over-represented (Supplementary Table 20).
Fruit ripening is an important biological feature and has tremendous economical value. Genes involved in ripening have been well studied in tomato31. Of the 16 tomato ripening genes, we found that only 3 (ETR3 (also known as NR), MADS-RIN and BP0353 (also known as PHYF)) are differentially expressed in orange fruits, whereas the majority (13) are not (Supplementary Table 21), possibly reflecting the existence of two distinct ripening mechanisms for the climacteric (tomato) and nonclimacteric (orange) fruits. Notably, MADS-RIN, a transcription factor previously shown to be necessary for the normal development and ripening of both tomato32 (climacteric) and strawberry33 (nonclimacteric), is highly upregulated in orange fruits (Supplementary Table 21), providing further support for the notion that MADS-RIN may function as a key-ripening regulator with broad impacts in both climacteric and nonclimacteric fruit ripening.
We were specifically interested in the biosynthesis of vitamin C (L-ascorbic acid (AsA), a strong antioxidant agent), as orange fruits are known to be rich in this vitamin. We analyzed the gene transcription of key enzymes involved in four known biosynthesis branch pathways upstream of AsA34,35 (Supplementary Fig. 16) and found that many of these genes are upregulated specifically in fruit. Remarkably, all three genes for the enzymes (PG, PME and GalUR) involved in the galacturonate pathway branch were substantially upregulated (Fig. 5a), of which the gene encoding D-galacturonic acid reductase (GalUR) is the rate-limiting step in this pathway36. Coincidentally, we identified 18 GalUR paralogous genes in the citrus genome (Supplementary Table 22), which is the largest number among the plant genomes (17 in apple and grapevine, 15 in strawberry, 13 in papaya, 12 in rice, maize and cocoa, 7 in Arabidopsis, 6 in Brachypodium and none in Chlamydomonas) (Supplementary Table 23). Further evolutionary analysis using the four closely related plant species within the Malvidae clade (orange, cacao, papaya and Arabidopsis) revealed two clusters (one with four genes and the other with seven genes) of recent GalUR gene expansion in the citrus genome, probably by tandem duplication (Fig. 5b). Notably, these two clusters also showed an expansion mode in papaya, another tree crop with high vitamin C content in its fruit. Noticeably, orange GalUR genes within these two clusters showed diverse expression patterns across different developmental stages. One of them (GalUR-12) was significantly upregulated in fruit (P = 5 × 10−7), whereas the other three members in the same cluster did not show a similar expression pattern (Fig. 5a,b), indicating diversification of gene transcription regulation in AsA biosynthesis. GalUR-12 shares high sequence identity (E value < 10−106) and shows a close phylogenetic relationship (Supplementary Fig. 17) with the strawberry GalUR gene, which has been further tested for specific vitamin C production36. Collectively, our data indicate that the galacturonate pathway is highly upregulated in orange fruit and GalUR may be the most important contributor to AsA accumulation in orange fruit.

(a) Heat map of the normalized RNA-Seq data for genes involved in AsA metabolism. Genes with a minimum expression level (RNA-Seq RPKM > 1 (ref. 42)) are shown. (b) Phylogenetic analysis of the GalUR gene family in C. sinensis, Arabidopsis thaliana, Carica papaya and T. cacao (top); two recent expansions are shaded with two oval circles (one cluster with GalUR-6, GalUR-11, GalUR-12 and GalUR-13 (orange oval) and the other cluster with GalUR-1, GalUR-2, GalUR-3, GalUR-4, GalUR-8, GalUR-10 and GalUR-14 (gray oval)). Note that GalUR-12 is a recent member of the family and is highly upregulated in orange fruits. The genomic organization of the 18 GalUR genes (black arrows) in the sweet orange genome is shown (bottom). Sc, scaffold.
Discussion
We sequenced the sweet orange genome using a two-step approach: first, we sequenced and assembled a dihaploid genome, and second, we mapped the parental diploid genomic DNA sequence reads to the haploid reference genome to complete the construction of the heterozygous genome map. The availability of the sweet orange genome sequence provides a valuable genomic resource for citrus genetics and breeding improvement. The sequence of sweet orange, as a member of the Rutaceae family and the Sapindales, also provides a valuable reference for plant genome evolutionary analyses.
Our genome analysis indicated that there was only an ancient (γ) triplication event and no recent WGD events in the orange genome, which partially explains the small size of the citrus genome. Phylogenetic analysis indicated that citrus is rather ancient, evolutionarily close to cacao and estimated to have diverged 85 million years ago from Malvales. An infrequent reproductive cycle in some taxa because of apomixis, male and/or female sterility, long juvenility and vegetative propagation may contribute to the restriction of genome expansion and evolution.
Heterozygosity is of great interest in crop plants. Hybridization causing a high degree of heterozygosity could create hybrid vigor known as heterosis37. The remarkably high degree of heterozygosity in the genome of cultivar Valencia sweet orange (C. sinensis cv. Valencia), as evidenced in our genomic and cytological analyses, hinted that sweet orange is an interspecific hybrid between pummelo and mandarin. On the basis of the collective evidence, we reconstructed the scenario regarding the ancient primary events of the origin of sweet orange: female pummelo crossed with male mandarin to create the initial interspecific hybrid that was further crossed again with male mandarin to produce sweet orange. This event might have happened at least 2,300 years ago, or much earlier, as sweet orange was recorded in Chinese literature as long ago as 314 BC3,38. Although additional genetic changes might have occurred afterward, it is still remarkable that this ancient hybrid genotype seems to be preserved in today's sweet orange, probably because of its strict nature of asexual reproduction (apomixis through nucellar embryo) and manmade selection and propagation by grafting. This scenario explains why most of today's sweet orange cultivars are genetically one biotype and highly heterozygous, with diversification occurring mostly through somatic mutations7,39,40.
Characterization of the citrus genome for protein-coding genes and comparative genomic analysis with other plant genomes, as well as transcriptome analyses, have yielded several insights into citrus-specific genes, particularly those that are involved in fruit biology and metabolic pathways. Our analysis suggests that the rate-limiting enzyme GalUR36 in the galacturonate pathway could be an important contributor to the high vitamin C accumulation in citrus fruit, and the recent expansion of the GalUR gene family in the citrus genome may suggest a genomic basis for regulating vitamin C production.
The availability of the orange genome and deep mining of orange-specific genes with known or unknown biochemical functions will facilitate a better understanding of many economically and agronomically important traits, including fruit color, flavor, sugar, organic acid and disease resistance in years to come. Future comparison of this sweet orange genome with the recently released high-quality clementine mandarin genome assembly draft (see URLs) should provide new insights into citrus biological and evolutionary questions.
URLs.
Food and Agriculture Organization of the United Nations (FAO statistics), http://faostat.fao.org/default.aspx; clementine mandarin genome assembly, http://www.phytozome.net/clementine.php; sweet orange genome website, http://citrus.hzau.edu.cn/orange.
Methods
Plant material and DNA sequencing.
The dihaploid line derived from another culture of C. sinensis cv. Valencia10 was used for paired-end–tag DNA sequencing using an Illumina GAII platform. Approximately 785 million 2 × 100 bp reads were generated from libraries with different fragment sizes (300 bp, 2 kb, 10 kb and 20 kb). DNA from the heterozygous diploid parent of the dihaploid line (the tree from which the anther was harvested and the dihaploid line was derived), three pummelos (C. grandis) and three mandarins (C. reticulata) was subjected to whole-genome shotgun sequencing of paired-end–tag reads from 300-bp genomic DNA fragments (Supplementary Table 12).
Genome assembly.
We first applied the software QUAKE43 to correct the reads from each library. Contig assembly, scaffolding and gap filling of sequencing data was done using the assembler SOAPdenovo11, with the three DNA-PET libraries (2 kb, 10 kb and 20 kb) being used to link contigs into scaffolds. Scaffolds and contigs were refined further with the gap-filling module in SOAPdenovo (GapCloser), which is used for bridging scaffold gaps, to produce an assembly with a scaffold N50 of 427 kb that covered 286 Mb of the citrus genome. To further improve the assembly, we used the optimal scaffolder Opera12 with DNA-PET reads (10 kb and 20 kb) and BAC ends (∼125 kb, 5,136 BAC-end sequences) in order of increasing library size to construct larger scaffolds and fill gaps using GapCloser (final genome size of 320.5 Mb and N50 of 1.69 Mb). DNA-PET reads and BAC ends were mapped respectively to the SOAPdenovo assembly with BOWTIE44 and BWA-SW45 (Supplementary Note).
Assembled scaffolds anchoring to genetic map.
The C. sinensis genetic map by Lyon et al.14 was used for anchoring the genome assembly. A total of 768 sequence-known markers in the genetic map were used to align the scaffolds by BLASTN analysis at an E value cutoff of 1 × 10−20. Hits with coverage >50% and identity >90% were considered mapped markers. The pseudomolecules were constructed on the basis of the anchoring results, and the order of chromosomal numbers was based on the linkage group length in cM.
Identification of repetitive elements in the citrus genome.
Transposable element libraries were constructed by integrating the results of different signature-based, homology-based and ab initio–based methods (Supplementary Note). Results from different methods were combined to build transposable element families as described by Wicker et al.46. At least one member for each family was selected to construct the custom transposable element libraries, which were applied to mask the citrus genome using the RepeatMasker software (http://www.repeatmasker.org/).
Gene prediction and annotation.
A hybrid approach was used, which combined de novo predictions with evidence-based data (ESTs, protein homology and RNA-Seq) analysis using the PASA and EVM47 pipeline (Supplementary Note). The predicted gene models from EVM were then updated by PASA assembly alignments. Gene functions were assigned according to the best alignment using BLASTP (E value < 10−5) to the Uniprot database (including the SWISS-PROT and TrEMBL databases). miRNAs were predicted on the basis of small RNA sequencing data23, and the criteria applied to characterize a miRNA locus is described in the Supplementary Note.
RNA-Seq analysis.
RNA-Seq reads were generated using Illumina and SOLiD platforms and aligned to the reference genome using TopHat48 (v1.2.0) and BioScope (v1.3, with default parameters), respectively (Supplementary Table 1 and Supplementary Fig. 4), to identify exons and splice junctions ab initio. The expression level of each gene in each RNA-Seq library was calculated as the RPKM42 (Supplementary Note). Biological and technical replicates were performed for gene expression analysis (Supplementary Fig. 18). For hierarchical clustering analysis, we used the clustering software Cluster 3.0 (ref. 49) to perform complete linkage hierarchical clustering on genes using uncentered Pearson's correlation as distance measure.
RNA-PET analysis.
RNA-PET analysis for full-length mRNA transcripts was performed as described in Ruan and Ruan50. Approximately 100 million RNA-PETs (Supplementary Table 1) were generated from four tissues (callus, flower, leaf and fruit) and were used for mapping, clustering and annotation of the reference genome (Supplementary Fig. 6).
Identification of potential species-specific genes.
The OrthoMCL method51 was adopted to construct gene families for the genes in 22 plant species (Supplementary Fig. 19). Proteins with no homologs in the other 21 plant genomes were used for TBLASTN searches against the Plant Unique Transcripts Assembly database (PUTs, http://www.plantgdb.org/) to exclude false-positive genes caused by incomplete genome annotation. The potential species-specific genes were rescreened by BLAST searches against the nonredundant protein database in GenBank.
SNPs and small indels analysis.
Illumina reads from heterozygous diploid orange (the diploid parent of the dihaploid line), three mandarin cultivars (C. reticulata) and three pummelo cultivars (C. grandis) were used for SNP and small indel analysis. All the sequencing reads were mapped using bwa v0.5.9 with default parameters for pair-end reads. Two variant calling tools, SAMtools52 and the Genome Analysis Toolkit (GATK53), were used (Supplementary Note). The sequence data from the dihaploid line were used as a negative control for the SNP analysis as described above to estimate the error rate of SNP detection (Supplementary Note).
Analyses for the hybrid origin of sweet orange.
The SSR and SNP markers were used for genotyping analysis to test the potential hybrid origin of sweet orange.
For SSR genotyping, SSR regions were predicted using the whole-genome sequence. A total of 480 pairs of SSR primers (primers are listed in Supplementary Table 24) were selected on the basis of three primer pairs per scaffold (>10 kb). The genomic locations of these SSR loci are provided in Supplementary Figure 20. PCR amplification was conducted in a MJ-PTC-200 thermal PCR cycler (MJ Research, Waltham Mass) using the following program: 3 min of 94 °C, 32 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s and a final extension at 72 °C for 10 min. After PCR, polyacrylamide gel analysis and SSR bands were visualized with silver staining as described by Chen et al.54.
For SNP genotyping, the genome-wide patterns of the sweet orange genetic origin were investigated on the basis of the whole-genome SNP data for mandarins, pummelos and sweet orange. We used a sliding window method55 to detect the ratio (denoted as RM/P) of SNP numbers from mandarin (M) or pummelo (P) in a 100 kb window size and 10 kb sliding step. Only windows with SNP number ≥30 were used for the genetic origin analysis. If RM/P was larger than 4, we marked this region as from pummelo, and if RM/P was smaller than 0.25, we marked the region as from mandarin. Otherwise the region was regarded as undetermined.
Estimation of genome duplication.
We used two methods to detect the signature of genome duplications. The multicollinearity alignment algorithm MCscan56, with the gap size set to 15 genes and requiring at least 5 syntenic genes, was used to define a duplicated block. For each duplicate gene pair, the synonymous site divergence value (Ks) was calculated using Bioperl modules under the Jukes-Cantor corrected model (Supplementary Note).
Genomic synteny analysis.
MCscan software56 was used to identify syntenic regions between C. sinensis and two genomes (A. thaliana and T. cacao) in Malvidae, two genomes (F. vesca and M. domestica) in Fabidae and V. vinifera with the same parameters as used for the above mentioned genome duplication analysis.
Phylogenetic analysis.
Maximum parsimony–based phylogeny construction was performed using heuristic searches that consisted of tree bisection and reconnection (TBR) branch swapping in PAUP*4.0b10 soft. Maximum likelihood–based method analysis was conducted in RaxML version 7.2.8 (ref. 57) using the shared single-copy genes (Supplementary Note). DNA sequences of 103 single-copy genes were concatenated, and the GTRGAMMA substitution model was used for the phylogenetic tree construction.
Chromosome karyotype analysis.
Root tips (3–5 mm) from germinated seeds of Valencia sweet orange and suspended cells from calli of the Valencia sweet orange dihaploid line were pretreated with saturated p-dichlorobenzene solution for 2 h at 20 °C, fixed in 3:1 ethanol and acetic acid (v/v) for 24 h and stored at −4 °C in 70% ethanol solution. Roots were treated with an enzyme mixture containing 0.25% pectinase (Sigma), 0.25% pectolyase Y-23 (Yakult) and 0.5% cellulose RS (Onozuka) for 60–70 min at 37 °C. The meristem of each individual root tip was then squashed on a clean slide in a drop of 60% acetic acid and then in a drop of fixation solution with a fine needle and was then flame dried over an alcohol flame.
Chromomycin A3 (CMA) and 4′-6-diamidino-2-phenylindole (DAPI) banding pattern analysis was performed according to Guerra et al.41. The slides were double stained with 0.5 mg/ml of CMA (1 h) and 5 μg/ml of DAPI (30 min) and mounted with in antifade solution of Vectashield (Vector Laboratories, Peterborough, UK). Slides were examined under a Zeiss Scope A1 fluorescence microscope (Zeiss, Germany) equipped with a Hamamatsu digital camera. Gray-scale images were captured for each color channel. Chromosomes in at least ten metaphase cells were measured using Image-Pro Plus 5.0 software (Media Cybernetics).
Accession codes.
Genome assembly data has been deposited at DDBJ/EMBL/GenBank under the accession AJPS00000000 and the BioProject ID PRJNA86123. The version described in this paper is the first version, AJPS01000000. DNA sequence data, assembly, annotation and the browser of the sweet orange genome are available through our website at http://citrus.hzau.edu.cn/orange.
Accession codes
References
Roose, M.L. & Close, T.J. Genomics of citrus, a major fruit crop of tropical and subtropical regions. in Genomics of Tropical Crop Plants (eds. Moore, P.H. & Ming, R.) 187–201 (Springer Press, 2008).
Gmitter, F.G. et al. Citrus genomics. Tree Genet. Genomes 8, 611–626 (2012).
Webber, H.J. History and development of the Citrus industry. in The Citrus Industry (eds. Reuther, W. et al.) Chap. 1, 1–39 (University of California Press, 1967).
Scora, R.W. On the history and origin of Citrus. Bull. Torrey Bot. Club 102, 369–375 (1975).
Gmitter, F. & Hu, X. The possible role of Yunnan province, China, in the origin of contemporary Citrus species (Rutaceae). Econ. Bot. 44, 267–277 (1990).
Legge, J. The shoo king and The tribute of Yu. in The Chinese Classics, Vol. 3, Pt. 1 and Pt. 3, Bk. 1, Chap. 6, 111–112 (Trubner & Co., London, 1865).
Moore, G.A. Oranges and lemons: clues to the taxonomy of Citrus from molecular markers. Trends Genet. 17, 536–540 (2001).
Nicolosi, E. et al. Citrus phylogeny and genetic origin of important species as investigated by molecular markers. Theor. Appl. Genet. 100, 1155–1166 (2000).
Arumuganathan, K. & Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218 (1991).
Cao, H. et al. Doubled haploid callus lines of Valencia sweet orange recovered from anther culture. Plant Cell Tissue Organ Cult. 104, 415–423 (2011).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Gao, S., Sung, W.K. & Nagarajan, N. Opera: reconstructing optimal genomic scaffolds with high-throughput paired-end sequences. J. Comput. Biol. 18, 1681–1691 (2011).
Baig, M.N., Yu, A., Guo, W. & Deng, X. Construction and characterization of two Citrus BAC libraries and identification of clones containing the phytoene synthase gene. Genome 52, 484–489 (2009).
Lyon, M.P. A Genomic Genetic Map of the Common Sweet Orange and Poncirus trifoliata. PhD dissertation, University of California, Riverside, (2008).
Iwamasa, M. Reciprocal translocation in the Valencia orange. Chromosome Inform. Service 4, 9–10 (1963).
The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Goff, S.A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science 296, 92–100 (2002).
Du, J. et al. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 63, 584–598 (2010).
Gao, L., McCarthy, E.M., Ganko, E.W. & McDonald, J.F. Evolutionary history of Oryza sativa LTR retrotransposons: a preliminary survey of the rice genome sequences. BMC Genomics 5, 18 (2004).
Velasco, R. et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996 (2008).
Dimmer, E.C. et al. The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 40, D565–D570 (2012).
Xu, Q. et al. Discovery and comparative profiling of microRNAs in a sweet orange red-flesh mutant and its wild type. BMC Genomics 11, 246 (2010).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Argout, X. et al. The genome of Theobroma cacao. Nat. Genet. 43, 101–108 (2011).
Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116 (2011).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Abrouk, M. et al. Palaeogenomics of plants: synteny-based modelling of extinct ancestors. Trends Plant Sci. 15, 479–487 (2010).
Tuskan, G.A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Chan, A.P. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28, 951–956 (2010).
The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Vrebalov, J . et al. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science 296, 343–346 (2002).
Seymour, G.B. et al. A SEPALLATA gene is involved in the development and ripening of strawberry (Fragaria × ananassa Duch.) fruit, a non-climacteric tissue. J. Exp. Bot. 62, 1179–1188 (2011).
Cruz-Rus, E., Botella, M.A., Valpuesta, V. & Gomez-Jimenez, M.C. Analysis of genes involved in l-ascorbic acid biosynthesis during growth and ripening of grape berries. J. Plant Physiol. 167, 739–748 (2010).
Bulley, S.M. et al. Gene expression studies in kiwifruit and gene over-expression in Arabidopsis indicates that GDP-l-galactose guanyltransferase is a major control point of vitamin C biosynthesis. J. Exp. Bot. 60, 765–778 (2009).
Agius, F. et al. Engineering increased vitamin C levels in plants by overexpression of a D-galacturonic acid reductase. Nat. Biotechnol. 21, 177–181 (2003).
Lippman, Z.B. & Zamir, D. Heterosis: revisiting the magic. Trends Genet. 23, 60–66 (2007).
Chinese Society of Citriculture. Citrus Industry in China. (China Agriculture Press, Beijing, China, 2008).
Barrett, H.C. & Rhodes, A.M. A numberical taxonomic study of affinity relationships in cultivated Citrus and its close relatives. Syst. Bot. 1, 105–136 (1976).
Mendel, K. Bud mutations in citrus and their potential commercial value. Proc. Int. Soc. Citricult. 1, 86–89 (1981).
Guerra, M. Cytogenetics of Rutaceae. V. High chromosomal variability in Citrus species revealed by CMA/DAPI staining. Heredity 71, 234–241 (1993).
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Kelley, D.R., Schatz, M.C. & Salzberg, S.L. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 11, R116 (2010).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982 (2007).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
de Hoon, M.J., Imoto, S., Nolan, J. & Miyano, S. Open source clustering software. Bioinformatics 20, 1453–1454 (2004).
Ruan, X. & Ruan, Y. Genome wide full-length transcript analysis using 5′ and 3′ paired-end-tag next generation sequencing (RNA-PET). Methods Mol. Biol. 809, 535–562 (2012).
Li, L., Stoeckert, C.J. Jr. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Chen, C. et al. EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliata. Tree Genet. Genomes 4, 1–10 (2008).
Lai, J. et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42, 1027–1030 (2010).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463 (2005).
Acknowledgements
The sweet orange genome project was supported by the Ministry of Science and Technology of China (2011CB100600), the National Natural Science Foundation of China (30921002, 30830078 and 31071779) and the Ministry of Agriculture (2011-g21 and CARS-27). Sweet orange mapping was funded by the Citrus Research Board (5200-125) and University of California Discovery grant program (itl-bio-10155). We thank W. Xie and Z. Li for their assistance in bioinformatic analysis and cytological experiments.
Author information
Authors and Affiliations
Contributions
X.-X.D. and Y.R. conceived the project and the strategy. Q.X. coordinated the overall project. X.R. directed sequencing data generation. L.-L.C. led the genome annotation analysis. N.N., D.B. and S.G. assembled the draft genome. D.C., W.-B.J., B.-H.H., J.-W.C., Z.T., F.X., O.J.L., Y.L., X.-W.X. and H.-Y.Z. performed gene annotation, SNP analysis, transcriptome analysis and database management. A.Z., Q.X., H.K., J.C., L.W., Q.H., M.K.B., W.Z., J.-H.L., F.G., H.C., Y.-J.C., J.X., X.Y. and W.-W.G. performed repetitive elements, genome anchor, gene synteny and evolutionary analyses. C.C., H.L., X.G., Y.M. and S.X. performed cytological analyses. M.P.L. and M.L.R. developed the sweet orange linkage map. Q.X. and Y.R. wrote the manuscript with contributions from X.R., L.-L.C., D.C., A.Z., N.N., D.B., C.C., W.-B.J., F.X., H.K., M.L.R. and X.-X.D.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Tables 1–8, 11–15, 17 and 18 and Supplementary Figures 1–20 (PDF 3315 kb)
Supplementary Tables
Supplementary Tables 9,10, 16 and 19-24 (XLSX 855 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike license (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar license.
About this article
Cite this article
Xu, Q., Chen, LL., Ruan, X. et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet 45, 59–66 (2013). https://doi.org/10.1038/ng.2472
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2472
This article is cited by
-
Genome-wide identification of the GRF family in sweet orange (Citrus sinensis) and functional analysis of the CsGRF04 in response to multiple abiotic stresses
BMC Genomics (2024)
-
Evolutionary assessment of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE genes in citrus relatives with a specific focus on flowering
Molecular Horticulture (2023)
-
Transcriptome analysis of Citrus limon infected with Citrus yellow vein clearing virus
BMC Genomics (2023)
-
Comprehensive genome-wide identification and transferability of chromosome-specific highly variable microsatellite markers from citrus species
Scientific Reports (2023)
-
Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Photinia serratifolia
Scientific Reports (2023)
