
Abstract
Nanoparticle–polymer composites, or polymer–nanoparticle composites (PNCs), exhibit unusual mechanical and dynamical features when the particle size approaches the random coil dimensions of the host polymer. Here, we harness favourable enthalpic interactions between particle-tethered and free, host polymer chains to create model PNCs, in which spherical nanoparticles are uniformly dispersed in high molecular weight entangled polymers. Investigation of the mechanical properties of these model PNCs reveals that the nanoparticles have profound effects on the host polymer motions on all timescales. On short timescales, nanoparticles slow-down local dynamics of the host polymer segments and lower the glass transition temperature. On intermediate timescales, where polymer chain motion is typically constrained by entanglements with surrounding molecules, nanoparticles provide additional constraints, which lead to an early onset of entangled polymer dynamics. Finally, on long timescales, nanoparticles produce an apparent speeding up of relaxation of their polymer host.
Similar content being viewed by others

Introduction
Addition of rigid nanoscale fillers to polymer melts is a well-known technique for augmenting various properties of polymeric materials, such as mechanical strength1,2,3, thermal stability3,4, barrier properties5,6, dimensional stability7 and wear resistance8. Remarkably, these particle-induced property enhancements are detected even at low volume fractions, where continuum analysis suggests minimal effect of the particles. As remarkable are the observations for polymer–nanoparticle composites (PNCs) in which the particle diameter (D) approaches the random coil radius (Rg) of their host polymer. In these systems, even at low particle loadings NPs cause unusual viscosity reduction of the PNC relative to the particle-free host. This is in stark contrast to expectations based on the Einstein–Batchelor9,10 law for spherical particle suspensions, wherein addition of particles to a fluid always increases the viscosity of the fluid. In their seminal 1975 study, Malinskii and co-workers11,12 reported that addition of small amounts of unfunctionalized particulate fillers to high molar mass polymers produce an unexpected decrease in viscosity, followed by an increase at higher filler contents. These findings have since been extended to a variety of PNCs on the basis of unfunctionalized as well as polymer-functionalized NPs, including polystyrene13, magnetite14 and fullerene14,15 nanoparticles in polystyrene hosts, silsesquioxane–polymer composites16,17,18, and tethered silica–polymer composites19. The most recent studies have also established empirical NP and polymer size requirements to achieve the viscosity reductions, which have been investigated in detail by simulations20 and theory21,22; however, how and why NPs violate the Einstein–Batchelor viscosity law in polymers remains a puzzle.
It is understood that achieving good dispersion of particles in their host polymer is difficult, but a requirement for meaningful studies of PNCs. Malinskii and co-workers11,12 used a rapid quenching procedure, wherein particle/polymer solutions in a volatile co-solvent were quickly frozen, followed by lyophillization to remove the solvent. This approach is designed to trap the particles in the configurations they adopt in dilute solution, but is also known to produce large density variations and aging phenomena in the PNCs. More recent studies have employed steric stabilization with small molecules and polymers to weaken the characteristic, strong attractive surface forces between NPs; however, phase separation of even these hairy NPs has been reported to be the normal case in PNCs formulated using high molecular weight polymers23,24,25. Notwithstanding these challenges, multiple physical processes have been argued to contribute to the observed viscosity reductions. It has been proposed that NPs enhance the available free volume for polymer chains11,12 or act akin to molecular plasticizers and speed-up relaxation of the entangled host polymer15,17,19. Other lines of work suggest that NPs diffuse faster in high molar mass, entangled polymers and thereby lower the lifetime of entanglements in a manner analogous to constraint release14,26 produced by a lower molecular weight polymer in a polydisperse melt. The recent molecular dynamics study by Kalathi et al.20 and theoretical studies by Ganesan et al.21 and Wang et al.22 suggest polymer slip at a NP polymer interface as an additional factor that may explain how particles lower the polymer viscosity.
In this article we report on the phase behaviour and dynamics of model PNCs and highlight multiple new and subtle effects that shed light on the role NPs play on relaxation behaviours of entangled polymers. PNCs studied here are created by dispersing poly(ethylene glycol) (PEG)-grafted SiO2 nanoparticles in high molecular weight (Mw) poly(methyl methacrylate) (PMMA) melts. By taking advantage of the slightly negative Flory–Huggins interaction parameter χ between tethered PEG and host PMMA molecules, we first show by means of small-angle X-ray scattering (SAXS) that it is possible to create PNCs with uniformly distributed NPs in entangled polymer hosts. Utilizing the structural simplicity of the materials, we study their relaxation dynamics over timescales spanning the complete range from fast monomer/segmental-scale motions to slow terminal relaxation. In particular, we find that NPs slow down polymer dynamics on all timescales, even at low particle concentrations. We also report that for well-entangled polymers, NPs play an active role in enforcing entanglement constraints on short timescales; their diffusion on longer timescales facilitates polymer relaxation and produces an apparent decrease in the terminal viscosity of the host polymer.
Results
Synthesis of model PNCs
Silica (SiO2) nanoparticles with average diameter Davg=10.8±0.3 nm (Supplementary Fig. 1) are densely grafted (Σ∼1.5–2 chains nm−2), with linear PEG of Mw=450 and 2,000 g mol−1 using previously reported procedures27. Composites of these hairy NPs in linear PMMA and PEG of different molecular weights are created using chloroform as a co-solvent. Removal of the co-solvent followed by thermal annealing yields a library of PNCs with varying particle core fraction φ, host polymer chemistries and tethered and host polymer molar weights.
Structural characterization of PNCs using SAXS
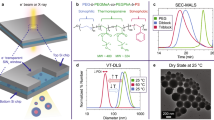
Particle dispersion in these PNCs is investigated through SAXS measurements performed at Sector 12-ID-B of the Advanced Photon Source. In Fig. 1a, we report the scaled scattering intensity C*I(q) for PNCs comprising SiO2 particles tethered with PEG (Mw=2 kg mol−1) suspended in high (Mw=280 kg mol−1) atactic PMMA with polydispersity index (PDI)= ) with φ∼2%. Here C is a scaling factor used to displace the curves vertically for better representation. Absolute values of I(q) for different samples are reported in Supplementary Fig. 2a. A long plateau in I(q) in the low wave vector (q) region for the SiO2–PEG2K/PMMA280K, a signature of well-dispersed particles in PNCs, is apparent in the figure. In contrast, for PNCs based on SiO2–PEG450 particles in either of the PEG (Mw=50 kg mol−1; PDI=1.4 and 203 kg mol−1; PDI=1.14) or PMMA (Mw=280 kg mol−1; PDI=1.7) hosts with φ∼2% loadings, I(q) increases as an approximate power-law function, I(q)∼q−α in the low q range.
) with φ∼2%. Here C is a scaling factor used to displace the curves vertically for better representation. Absolute values of I(q) for different samples are reported in Supplementary Fig. 2a. A long plateau in I(q) in the low wave vector (q) region for the SiO2–PEG2K/PMMA280K, a signature of well-dispersed particles in PNCs, is apparent in the figure. In contrast, for PNCs based on SiO2–PEG450 particles in either of the PEG (Mw=50 kg mol−1; PDI=1.4 and 203 kg mol−1; PDI=1.14) or PMMA (Mw=280 kg mol−1; PDI=1.7) hosts with φ∼2% loadings, I(q) increases as an approximate power-law function, I(q)∼q−α in the low q range.

Scaled intensity C*I(q) versus wave vector q profile for a. SiO2–PEG450/PEG50k, SiO2–PEG450/PEG203k, SiO2–PEG450/PMMA280k and SiO2–PEG2k/PMMA280k at φ=2%. The insets (i), (ii) and (iii) are transmission electron micrographs of the SiO2–PEG450/PEG50k, SiO2–PEG450/PEG203k and SiO2–PEG2k/PMMA280k materials, respectively. (b) SiO2–PEG2k/PMMA55k\165k\280k at φ=0.5% (circles), 1% (diamonds), 2% (upright triangles), 5% (inverted triangles) and 10% (squares). I(q) curves are displaced vertically for clarity of presentation. Scale bars in the TEM images represent the length of 200 nm. C is a scaling factor used to displace I(q) curves vertically in both the plots, for clarity of presentation. Errors in I(q) are the s.d.’s of the counts on the two-dimensional detector pixels with same q value, with the I(q) being the mean of all those values. The error bars shown in the figure are consistently smaller than the symbols.
The strong dependence of I(q) on q at low wave vectors is a known characteristic of phase-separated or aggregated structures24 whose dimensions are beyond the length scales probed by the SAXS measurements. For the phase-separated PNCs, the power-law exponent α is between 3 and 4, indicating that NPs form compact aggregates and scatter similar to surface fractals28,29. From the transmission electron micrographs shown in the insets in Fig. 1a for (i) SiO2–PEG450/PEG50k, (ii) SiO2–PEG450/PEG203k and (iii) SiO2–PEG2k/PMMA280k materials at φ=2% and in the inset of Fig. 1b for SiO2–PEG2k/PMMA280k at φ=5% NPs appear to exist as irregularly shaped islands in the former two cases and as a homogeneous dispersion in the latter ones. It is interesting to note that even in the phase-separated domains, NPs appear not to be aggregated, and a transmission electron microscopy (TEM) measurement that inadvertently sampled only one of these domains would conclude that the NPs are well dispersed, but with interparticle distances substantially lower than expected for a φ=2% suspension. Previous reports23,30 have shown how the degree of polymerization (P) of the host polymer to that of the tethered polymer (N) determines the degree of interpenetration and mixing between grafted layers and polymer melts. In particular, Srivastava et al.23 reported that PNCs with highly grafted NPs and characterized by P/N≥5 typically exist as phase-separated materials. For SiO2–PEG450/PEG203k, P/N≈450, it is therefore hardly a surprise that these NPs are phase-separated. In contrast, SiO2–PEG2k/PMMA280k (P/N≈140) shows no evidence of particle phase separation, but SiO2–PEG450/PMMA280k (P/N≈620) does. Blends of linear PMMA and PEG are widely known to be miscible because of favourable enthalpic monomeric interactions (χ<0; ref. 31). Although there is still no consensus regarding the exact value of χ for PMMA–PEG mixtures32,33,34, the attractive enthalpic interactions between the particle-tethered and host polymer can explain the better dispersion of SiO2–PEG2k in PMMA hosts. Moreover, the enthalpic contribution to the overall PNC free energy is proportional to the number of overlapping segments  between the tethered PEG and host PMMA chains35,which would be lower for PEG450 and perhaps explains why SiO2–PEG450/PMMA280k PNCs are phase-separated. In Fig. 1b we further report C*I(q) for a large number of SiO2–PEG2k/PMMA PNCs with varying core volume fractions and P/N values ranging from 140 to 27. Absolute values of I(q) for different samples are shown in Supplementary Fig. 2b–d. All of these systems manifest stable I(q) plateaus at low q, indicating that NPs are well dispersed. We also measured the second Virial coefficient B2 for our PNCs (B2,PNC) and for hard spheres (B2,HS), using a modified Zimm analysis, limq→01/S(q)=1+2B2C (ref. 36), where S(q)=I(q)/I(q=0) is the structure factor obtained from SAXS measurements. The results are shown in Supplementary Table 1. It is evident that B2,PNC is always positive and consistently larger than B2,HS, implying that the tethered PEG chains are highly stretched out on the particle surface, consistent with expectations for polymer-grafted particles in a good solvent36,37. Our observations imply that the stability of the particles originates from the strong steric stabilization provided by the extended brush of tethered PEG chains, interacting enthalpically with the host PMMA chains. This finding agrees with previous reports, which demonstrate enhanced phase stability of polymer-grafted nanoparticles in host polymers that interact favourably with the grafted chains38.
between the tethered PEG and host PMMA chains35,which would be lower for PEG450 and perhaps explains why SiO2–PEG450/PMMA280k PNCs are phase-separated. In Fig. 1b we further report C*I(q) for a large number of SiO2–PEG2k/PMMA PNCs with varying core volume fractions and P/N values ranging from 140 to 27. Absolute values of I(q) for different samples are shown in Supplementary Fig. 2b–d. All of these systems manifest stable I(q) plateaus at low q, indicating that NPs are well dispersed. We also measured the second Virial coefficient B2 for our PNCs (B2,PNC) and for hard spheres (B2,HS), using a modified Zimm analysis, limq→01/S(q)=1+2B2C (ref. 36), where S(q)=I(q)/I(q=0) is the structure factor obtained from SAXS measurements. The results are shown in Supplementary Table 1. It is evident that B2,PNC is always positive and consistently larger than B2,HS, implying that the tethered PEG chains are highly stretched out on the particle surface, consistent with expectations for polymer-grafted particles in a good solvent36,37. Our observations imply that the stability of the particles originates from the strong steric stabilization provided by the extended brush of tethered PEG chains, interacting enthalpically with the host PMMA chains. This finding agrees with previous reports, which demonstrate enhanced phase stability of polymer-grafted nanoparticles in host polymers that interact favourably with the grafted chains38.
Effect of nanoparticles on the dynamics of host polymer chains
The phase stability of SiO2–PEG2k nanoparticles in entangled PMMA melts allows us to use these materials as structure-less, model PNCs for studying how NPs affect their host polymer dynamics. In Fig. 2a, we compare the complex viscosity of PMMA melts and SiO2–PEG2k/PMMA55k\165k\280k PNCs with φ=2%. It is apparent that NPs lower the polymer viscosity in all PNCs studied, particularly at low rates, which is opposite to large increases in viscosity, observed in PNCs with phase-separated particles39. It also defies the Einstein–Batchelor prediction9,10 for the viscosity increase in suspensions of spherical particles. Similar behaviour is observed up to φ∼5% (inset to Fig. 2a); however, the polymer viscosity is seen to increase with particle loading at higher φ values. Our results are evidently consistent with earlier reports that addition of NPs to high molar mass polymers at low concentrations lowers the viscosity13,14,15,16,17,18,19. In this case, however, NPs lower the host polymer’s viscosity irrespective of the number of entanglements per chain Z≈Mw/Me, where Me is the entanglement molecular weight, which varies from ∼5 (PMMA55k) to 28 (PMMA280k); Me,PMMA, is 10 kg mol−1 (ref. 40).

(a) Plot of complex viscosity (η*) versus shifted frequency (at ω) for neat PMMA and for PNCs SiO2–PEG2k/PMMA55k, SiO2–PEG2k/PMMA165k and SiO2–PEG2k/PMMA280k with φ =2% at T =190 °C. Similar behaviour was observed in steady shear measurements (Supplementary Fig. 4). Inset shows the relative viscosity (ηr=ηPNC/ηneat) for SiO2–PEG2k/PMMA55k (triangles) from steady shear measurements at  ∼0.02 s−1 at T=180 °C, for SiO2–PEG2k/PMMA165k (circles) at
∼0.02 s−1 at T=180 °C, for SiO2–PEG2k/PMMA165k (circles) at  ∼0.02 s−1 at T=190 °C and for SiO2–PEG2k/PMMA280k (diamonds) at
∼0.02 s−1 at T=190 °C and for SiO2–PEG2k/PMMA280k (diamonds) at  ∼0.001 s−1 at T=170 °C. The dashed line is the predicted relative viscosity from the Einstein–Batchelor equation9,10. Error bars smaller than symbol size are not shown. (b,c) Tg values obtained using DSC for SiO2–PEG2k/PMMA55k and SiO2–PEG2k/PMMA280k, respectively. Closed symbols correspond to PNCs and open symbols correspond to corresponding particle-free neat blends. Dashed lines represent the data computed using the mixing rule predicted by the Fox equation
∼0.001 s−1 at T=170 °C. The dashed line is the predicted relative viscosity from the Einstein–Batchelor equation9,10. Error bars smaller than symbol size are not shown. (b,c) Tg values obtained using DSC for SiO2–PEG2k/PMMA55k and SiO2–PEG2k/PMMA280k, respectively. Closed symbols correspond to PNCs and open symbols correspond to corresponding particle-free neat blends. Dashed lines represent the data computed using the mixing rule predicted by the Fox equation  . DSC measurements are performed at different scan rates (2, 5, 10, 15 and 20 K min−1) and obtained Tg values showed no dependence on scan rates (Supplementary Figs 5 and 6). Results for SiO2–PEG2k/PMMA165k are shown in Supplementary Fig. 7.
. DSC measurements are performed at different scan rates (2, 5, 10, 15 and 20 K min−1) and obtained Tg values showed no dependence on scan rates (Supplementary Figs 5 and 6). Results for SiO2–PEG2k/PMMA165k are shown in Supplementary Fig. 7.
The glass transition temperature (Tg) for the SiO2–PEG2k/PMMA composites and corresponding particle-free PMMA/PEG blends are measured through differential scanning calorimetry (DSC; Supplementary Figs 5 and 6). As shown in Fig. 2b,c, a decreasing trend of Tg with decreasing PMMA volume fraction (φPMMA) for both the SiO2–PEG2k/PMMA PNCs and the PMMA/PEG blends is observed. Decreasing Tg with decreasing glassy polymer content in a homogeneous mixture is the expected response to a plasticizer, which increases free volume of the polymer. The dashed lines are the values predicted by the Fox relation41 for Tg of plasticized polymers.
It is clear that free PEG has an appreciably larger plasticizing effect than expected from the Fox relation. Such negative deviations in Tg of PEG/PMMA mixtures, relative to the Fox relation, have been reported previously and argued to be a consequence of the segmental-scale attraction of PEG and PMMA42. At the same time, a more pronounced decrease observed for free PEG chains relative to tethered chains can be understood both in terms of the lower number of chain ends43 and lower mobility near SiO2 particle surface44 for the latter.
The effect of nanoparticles on the dynamics of PMMA was studied in detail using frequency-dependent oscillatory shear measurements of pure PMMA and SiO2–PEG2k/PMMA PNCs at multiple discrete temperatures, T>Tg. The time–temperature superposition procedure was used to create dynamic maps or master curves shifted (Supplementary Fig. 8) at a fixed temperature distance with respect to the measured Tg values for respective material. This shifting procedure eliminates the trivial plasticization effect caused by the increase in free volume resulting from addition of nanoparticles. As illustrated in Fig. 3a,b, these measurements provide complete information on how the NPs affect the host polymer dynamics on timescales that span the entire range from segmental-scale motions to terminal/reptation relaxation. It is also evident from both figures that with successive addition of NPs, the frequency-dependent dynamic moduli for SiO2–PEG2k/PMMA55k and SiO2–PEG2k/PMMA280k shift to the left, that is, towards lower frequency.

Master curves (dynamic moduli versus shifted frequency) obtained from TTS of oscillatory shear rheology measurements for: (a) SiO2–PEG2k/PMMA55k and (b) SiO2–PEG2k/PMMA280k. Master curves for different φ values have been shifted vertically for clarity of presentation, shift factor for which are mentioned in the figure legend. Frquency sweep measurements at temperatures T>Tg are shifted with respect to the material’s respective Tg’s, to create each master curve. Typical shift factors employed in obtaining the individual master curves are shown in Supplementary Fig. 10. Error bars are smaller than symbol sizes and are not shown.
The shift can be quantified in terms of the three fundamental timescales for entangled polymers45—the segmental relaxation time τo, entanglement relaxation time τe and the terminal relaxation or reptation time τt. In Fig. 4a–c we report the respective timescales45 obtained for all three PNCs (see Supplementary Fig. 8 for detailed analysis). It is apparent that addition of SiO2–PEG2k NPs to PMMA slows down polymer relaxation on all timescales. The terminal Newtonian viscosity ηo of an entangled polymer can be estimated as ηo≈Geτt where Ge is the shear modulus at the rubbery plateau. The procedure reported in ref. 46 was used to determine Ge (Supplementary Fig. 9). As shown in Fig. 4d, it can be seen that Ge increases continuously on nanoparticle addition; however, the changes are more modest than those seen forτt. In Fig. 4e we plot the relative viscosity ηr=ηPNC/ηneat for these PNCs. Comparison of the results with those in the inset of Fig. 2a shows that once the plasticization effect is taken into account by comparing viscosities at a fixed temperature distance relative to Tg of the PNC, the conventional suspension result—namely that addition of particles always increases the suspension viscosity—is qualitatively recovered. More careful inspection of Fig. 4e indicates that the observed increase in ηr at modest amounts of NPs is far larger than what is normally achieved with hydrodynamic effects alone. To make this point more concrete, we computed ηr based on a modified Einstein dilute suspension expression47  to account for the contribution of tethered PEG-chain height (Δ) in nanoparticles’ effective volume. At first, we evaluated ηr assuming tethered PEG chains to form a collapsed brush of height 2Rg (ref. 48; ∼1.7 nm for PEG2k), as shown by the dashed line. However, it is known from previous measurements44 that because of the high grafting density of tethered chains, this height can be substantially larger. Hence, we also evaluated ηr assuming the polymer brush to be comprising fully stretched out PEG chains with Δ∼N*b (∼16 nm), as shown in solid line, here N is the number of kuhn monomers in entire chain length and b is the length of a polymer chain segment. It is apparent that in both extreme limits, we are unable to reproduce the magnitude of the experimentally observed enhancements in viscosity.
to account for the contribution of tethered PEG-chain height (Δ) in nanoparticles’ effective volume. At first, we evaluated ηr assuming tethered PEG chains to form a collapsed brush of height 2Rg (ref. 48; ∼1.7 nm for PEG2k), as shown by the dashed line. However, it is known from previous measurements44 that because of the high grafting density of tethered chains, this height can be substantially larger. Hence, we also evaluated ηr assuming the polymer brush to be comprising fully stretched out PEG chains with Δ∼N*b (∼16 nm), as shown in solid line, here N is the number of kuhn monomers in entire chain length and b is the length of a polymer chain segment. It is apparent that in both extreme limits, we are unable to reproduce the magnitude of the experimentally observed enhancements in viscosity.

Effect of NP content in SiO2–PEG/PMMA PNCs on (a), (b), (c) the three relaxation times (τo,τe,τt) for: SiO2–PEG2k/PMMA55k; SiO2–PEG2k/PMMA165k; and SiO2–PEG2k/PMMA280k respectively. The relaxation times are obtained from the G’ and G” crossover (Supplementary Fig. 8) at a fixed temperature difference from Tg. (d) Plateau modulus (Ge) obtained using Van Gurp analysis (Supplementary Fig. 9), as a function of particle concentration. (e) Relative viscosity ηr=ηPNC/ηneat, where η∼Geτt. The lines are predictions based on the modified Einstein (ME) expression  (ref. 46), which accounts for a polymer brush of height; ME 1: Δ=2Rg (dashed line) or ME 2: Δ=fully stretched length (solid line). (f) Modified relative viscosity
(ref. 46), which accounts for a polymer brush of height; ME 1: Δ=2Rg (dashed line) or ME 2: Δ=fully stretched length (solid line). (f) Modified relative viscosity  .
.
In entangled polymer melts, relaxation is understood to proceed in a hierarchical manner beginning with fast, local segmental-scale processes on timescale τo, which affect dynamics on longer timescales: τe=(ae/b)4τo and τt=3Z3τe, where ae is the entanglement tube diameter, b the length of a chain segment and Z=M/Me is the average number of entanglements each chain makes with its neighbours. The large increases in ηr with increasing φ therefore to some extent reflect the strong increase in τo apparent in Fig. 4a–c. This effect can be taken into account by defining a modified relative viscosity 
 , in which τt is normalized using the measured τo values.
, in which τt is normalized using the measured τo values.
From Fig. 4f, we understand that, while  increases progressively with increasing particle content for PNCs on the basis of the 55k PMMA host, for materials based on the 165k and 280k PMMA host polymers,
increases progressively with increasing particle content for PNCs on the basis of the 55k PMMA host, for materials based on the 165k and 280k PMMA host polymers,  decreases at low φ, but increases at high φ. This effect of NPs on
decreases at low φ, but increases at high φ. This effect of NPs on  is in qualitative accord with earlier reports, but would obviously only be visible in experiments in which the NPs do not also slow down segmental-scale dynamics. In addition, in the present case, the apparent reduction in viscosity is isolated from any NP plasticization or other NP effects on segmental-scale dynamics, implying that neither of these two arguments advanced in the early literature can be the source of the behaviour.
is in qualitative accord with earlier reports, but would obviously only be visible in experiments in which the NPs do not also slow down segmental-scale dynamics. In addition, in the present case, the apparent reduction in viscosity is isolated from any NP plasticization or other NP effects on segmental-scale dynamics, implying that neither of these two arguments advanced in the early literature can be the source of the behaviour.
More insights about the environment in which polymer chains relax in PNCs can be obtained by analysing the effect of NPs on entanglement dynamics of their host polymer. The dimensionless group α≡(τe/τ0)1/4=ae/b allows this effect to be isolated. In Fig. 5a we report α for PNCs relative to the respective particle-free, neat PMMA (αneat). For PMMA55k and PMMA165k, α is virtually unchanged up to φ≈2%, where after it falls sharply below the value of the neat PMMA melt indicating that the host polymer feels an effectively smaller tube diameter in the PNC. For PMMA280k, the most entangled polymer studied, α is always lower than αneat (although moderately at low φ) meaning that the PMMA chains in PNC are in a more constricted tube than in pure melts at all φ. Through the cartoon in Fig. 5c we intend to help one visualize the role NPs might play in constraining their host polymer chains in tighter tubes. We also estimated the interparticle distance dp–p in a well-dispersed PNC on the bass of spherical packing using the formula  and calculated surface-to-surface distance ds–s=dp–p−(2Δ+D). To obtain an estimate of the order of magnitude for ds–s, we again assumed the brush height as Δ≈2Rg. In Fig. 5b we compare the calculated ds–s obtained at various φ with the equilibrium tube diameter ao(=bNe), where Ne is the number of Kuhn monomers in Me. For φ<5%, as ds–s>ao, particles are sufficiently far apart and hence they are unable to confine the host polymer. However, at φ=5%, ds−s<ao, suggesting a tube-like particle-induced confinement effect on length scales of ds–s is possible, which is consistent with our observations. For the case of a fully extended brush of tethered chains, we find that ds–s>ao only for φ=0.5%, where after the surface-to-surface distance between particles is lower than the equilibrium tube diameter. Our results therefore show that even at low concentrations NPs in a PNC may provide additional, tube-like constraints on the motions of their entangled polymeric hosts. This conclusion is consistent with recent predictions from computer simulations49,50 and theory51. It also explains the earlier onset of in-tube/reptation motion of the host polymer chains observed in PNCs.
and calculated surface-to-surface distance ds–s=dp–p−(2Δ+D). To obtain an estimate of the order of magnitude for ds–s, we again assumed the brush height as Δ≈2Rg. In Fig. 5b we compare the calculated ds–s obtained at various φ with the equilibrium tube diameter ao(=bNe), where Ne is the number of Kuhn monomers in Me. For φ<5%, as ds–s>ao, particles are sufficiently far apart and hence they are unable to confine the host polymer. However, at φ=5%, ds−s<ao, suggesting a tube-like particle-induced confinement effect on length scales of ds–s is possible, which is consistent with our observations. For the case of a fully extended brush of tethered chains, we find that ds–s>ao only for φ=0.5%, where after the surface-to-surface distance between particles is lower than the equilibrium tube diameter. Our results therefore show that even at low concentrations NPs in a PNC may provide additional, tube-like constraints on the motions of their entangled polymeric hosts. This conclusion is consistent with recent predictions from computer simulations49,50 and theory51. It also explains the earlier onset of in-tube/reptation motion of the host polymer chains observed in PNCs.

(a) The effect of NP volume fraction on α≡(τe/τ0)1/4=ae/b for PNCs with respect to the particle-free melt. (b) Surface-to-surface distance (ds–s=dp–p−(2Δ+D)), for Δ=2Rg and Δ∼Nb, as a function of particle content. The dashed horizontal line corresponds to the tube diamater (ao) of the host polymer. (c) Schematic of the mechanism through which hairy NPs exert entanglement-like constraints on polymer chains in a PNC. (d) β≡(τt/τe)1/3=31/3Z versus NP volume fraction for PNCs with respect to particle-free polymer melts.
From the corresponding results for β≡(τt/τe)1/3=31/3Z reported in Fig. 5d it is observed that for the two more entangled polymers there is a clear decrease in the effective number of entanglements β at low φ followed by a rise at high φ, whereas for PMMA55k the least entangled polymer β monotonically increases with φ. That Ge that remains essentially unchanged over the same range of φ suggests that the observed reduction in Z for more entangled PMMA is not due to increased Me (= , where ρ is polymer density and R is the universal gas constant), but rather reflects a reduction in the number of effective entanglements experienced by the host chains at the time of tube escape.
, where ρ is polymer density and R is the universal gas constant), but rather reflects a reduction in the number of effective entanglements experienced by the host chains at the time of tube escape.
This ‘constraint release’ phenomenon has previously been theorized by Mackay and co-workers14 as responsible for the observed lowering of an entangled polymer’s viscosity by NPs. In that case it was argued to originate from the fast diffusion of NPs in an entangled polymer host. Our results offer a complementary, although perhaps more straightforward explanation—because the NPs participate explicitly in the tube constraints the host polymer experiences on short timescales, their diffusion on longer timescales can reduce entanglements and perforate the tube in a manner analogous to what has been reported in bi-disperse polymer blends. In PNCs where the particle size (D∼10 nm) is only slightly larger than the host polymer entanglement mesh size (ao∼ 7 nm in our case), particle motion and host-chain entanglement dynamics are expected to be coupled52 with the particle’s escape time falling somewhere between τe and τt (ref. 50). The dissimilar effect of NPs on the terminal relaxation of weakly and well-entangled PMMA can therefore be understood in terms of competition between the rate of particle diffusion, which destroys the tube, and reptation relaxation of the polymer chains in their tube. In particular, we conjecture that for the more entangled PMMA hosts nanoparticle diffusion occurs on a timescale that causes tube reconstruction before the host is able to escape by reptation, which releases entanglements26,53. For the less entangled host, tube reconstruction by particle diffusion occurs at a rate comparable to or slower than tube escape by reptation.
Role of tethered chains on the dynamics of host polymer chains
We close by considering how tethered PEG chains might potentially affect the observations discussed here. From Fig. 6a,b we report the effect of PEG volume fraction (φPEG) on α and β for linear PEG2k/PMMA55\280k blends. By focusing on φPEG close to those of the corresponding tethered PEG loadings in the SiO2–PEG2k/PMMA PNCs, it is possible to characterize the effect of PEG on PMMA dynamics under conditions similar to those in the PNCs. The results clearly show that irrespective of the PMMA molecular weight, α increases and β decreases progressively with increasing φPEG, suggesting that free PEG chains dilate the PMMA entanglement network. That the effect is qualitatively unchanged for the weak- and well-entangled PMMA molecules, suggests that the observed effect of NPs is a characteristic of the particles, as opposed to the chains tethered to their surfaces alone.

(a) α≡(τe/τ0)1/4=ae/b and (b) β≡(τt/τe)1/3 =31/3Z as functions of PEG2k content for particle-free PEG2k/PMMA blends with respect to the respective pure PMMA materials.
Discussion
Our findings provide important new insights about how well-dispersed surface-functionalized nanoparticles influence the dynamics of polymers in nanocomposite materials. We show that favourable enthalpic interactions between the grafted and host chains can be used to overcome the tendency of nanoparticles to phase-separate in high molecular weight polymer hosts. In addition, we report for the first time that at a fixed temperature difference from Tg, NPs slow down polymer relaxation on all timescales, including the segmental relaxation time, and over a broad range of particle concentrations. By studying the effect of NPs on the Rouse time for entanglement segments, we find that even at low particle concentrations, polymer-functionalized NPs provide additional tube-like constraints on chain relaxation that leads to an earlier onset of reptation relaxation. On long timescales, particle motions appear to degrade the tube surroundings of their host chains, which accelerates tube escape by a process analogous to constraint release in mixtures of entangled polymers with widely differing molecular weights. In well-entangled materials these processes lead to an apparent lowering of the viscosity of the host polymer at low NP concentrations. However, the effect would only be directly observable in experiments where the particles do not also slow down the host polymer’s fast, segmental-scale motions.
Methods
Preparation of PNCs
Silane-functionalized polyethylene glycol chains (PEG, Mw,teth∼450 g mol−1, PDI: 1.2, supplied by Gelest Inc.) were tethered to SiO2 nanoparticles of diameter ∼10 nm (LUDOX SM-30; supplied by Sigma Aldrich) to prepare SiO2–PEG450 particles. To synthesize SiO2–PEG2k particles, silica particles were first functionalized with sulfonic acid using 3-(trihydroxysilyl)-1-propanesulfonic acid (supplied by Gelest Inc.). Later, amine-terminated PEG (Mw,teth∼2 kg mol−1, PDI: 1.3, supplied by Polymer Source Inc.) was added in stoichiometric ratios. Repeated precipitation using a solvent (ethanol/chloroform) and a nonsolvent (n-hexane) was performed to completely remove the untethered polymer chains in both the cases. Thermogravimetric analysis (TGA), carried out on the TA Instruments TGA Q500 for successive repeated centrifuge cycles, showed no changes in solid content by the fifth centrifuge cycle, indicating negligible amount of the unattached material content and suggested the grafting density of ∼1.5–2 chains nm−2. Composites of the nanoparticles were prepared by mixing appropriate amounts of nanoparticles with host polymer PEG/PMMA (supplied by Polymer Source Inc) in a common solvent (chloroform) followed by removal of the solvent by heating for 12 h in convection oven at 70 °C, followed by heating for at least 12 h under vacuum at 50 °C for SiO2–PEG/PEG suspensions and at 130 °C under vacuum for SiO2–PEG/PMMA suspensions. For control measurements, particle-free neat PEG2k/PMMA blends, corresponding to the suspensions, were also synthesized. PEG2k and PMMA were mixed in relative amounts similar to their respective suspensions again in a common solvent followed by removal of the solvent by heating for at least 12 h at 130 °C under vacuum.
Determination of the glass transition temperature
DSC measurements were carried out at temperature ramp rates of 2, 5, 10, 15 and 20 K min−1 in a nitrogen environment using a TA Instruments DSC Q2000. Tg values were obtained from the infection point in heat capacity versus temperature plot as shown in Supplementary Fig. 5.
SAXS measurements
SAXS measurements were performed at Sector 12-ID-B of the Advanced Photon Source. Measurements at 12-ID-B were carried out using Pilatus 2M detector, sound-bounce monochromator and a beam energy range of 7.9–14 keV. Measurements for SiO2–PEG/PEG composites were carried out at 70 °C and for SiO2–PEG/MMA composites at 130 °C. The scattering of NPs in the PNCs is obtained by subtracting the particle-free polymer-scattering, obtained using the same procedure. Errors in scattered intensities I(q) are the s.d.’s of the counts on the two-dimensional detector pixels with same wave vector q, with the I(q) being the mean of all those values.
Rheology measurements
Steady shear rheology measurements were performed at 190 °C using a strain-controlled ARES–LS rheometer (Rheometric Scientific) outfitted with a 4-mm diameter cone and plate fixture. Measurements were performed at shear rates ( ) in the range 10−4 s−1≤
) in the range 10−4 s−1≤ ≤10−1 s−1. Oscillatory rheology measurements were performed at different temperatures (T), again using the same Rheometrics ARES rheometer outfitted with a 3-mm diameter parallel plate fixture. Measurements were performed in the linear viscoelastic regime at frequencies (ω) in the range 10−1 s−1≤ω≤102 s−1. Thus, obtained frequency sweep curves were shifted horizontally and vertically, considering the data at Tg as reference, to obtain Time Temperature Superposition master curves. A representative master curve and typical shift factors are shown in Supplementary Figs 8 and 10, respectively. Plateau modulus (Ge) values were obtained utilizing Van Gurp–Palmen plot46 in which phase angle δ (=tan−1(G′′/G′)) is plotted against |G*|. The analysis predicts that plateau modulus is approximately equal to the minimum value of |G*| (i.e. Ge∼|G*|δ min).
≤10−1 s−1. Oscillatory rheology measurements were performed at different temperatures (T), again using the same Rheometrics ARES rheometer outfitted with a 3-mm diameter parallel plate fixture. Measurements were performed in the linear viscoelastic regime at frequencies (ω) in the range 10−1 s−1≤ω≤102 s−1. Thus, obtained frequency sweep curves were shifted horizontally and vertically, considering the data at Tg as reference, to obtain Time Temperature Superposition master curves. A representative master curve and typical shift factors are shown in Supplementary Figs 8 and 10, respectively. Plateau modulus (Ge) values were obtained utilizing Van Gurp–Palmen plot46 in which phase angle δ (=tan−1(G′′/G′)) is plotted against |G*|. The analysis predicts that plateau modulus is approximately equal to the minimum value of |G*| (i.e. Ge∼|G*|δ min).
Additional information
How to cite this article: Mangal, R. et al. Phase stability and dynamics of entangled polymer–nanoparticle composites. Nat. Commun. 6:7198 doi: 10.1038/ncomms8198 (2015).
References
Wang, H., Zeng, C., Elkovitch, M., Lee, L. J. & Koelling, K. W. Processing and properties of polymeric nano-composites. Polym. Eng. Sci. 41, 2036–2046 (2001) .
Schubert, U. Polymers reinforced by covalently bonded inorganic clusters. Chem. Mater. 13, 3487–3494 (2001) .
Giannelis, E. Polymer layered silicate nanocomposites. Adv. Mater. 8, 29–35 (1996) .
Gilman, J. Flammability and thermal stability studies of polymer layered-silicate (clay) nanocomposites. Appl. Clay Sci. 15, 31–49 (1999) .
Pavlidou, S. & Papaspyrides, C. D. A review on polymer–layered silicate nanocomposites. Prog. Polym. Sci. 33, 1119–1198 (2008) .
Kalendova, A., Merinska, D., Gerard, J. F. & Slouf, M. Polymer/clay nanocomposites and their gas barrier properties. Polym. Compos. 34, 1418–1424 (2013) .
Zeng, C., Han, X., Lee, L. J., Koelling, K. W. & Tomasko, D. L. Polymer–clay nanocomposite foams prepared using carbon dioxide. Adv. Mater. 15, 1743–1747 (2003) .
Rong, M. Z., Friedrich, K., Liu, H., Zeng, H. & Wetzel, B. Microstructure and tribological behavior of polymeric nanocomposites. Ind. Lubr. Tribol 53, 72–77 (2012) .
Batchelor, G. K. The effect of Brownian motion on the bulk stress in a suspension of spherical particles. J. Fluid Mech. 83, 97 (2006) .
Einstein, A. On the theory of Brownian movement. Ann. Phys. 19, 371–381 (1906) .
Prokopenko, V. V., Titova, O. K., Fesik, N. S., Malinski, Y. & Bakeyev, N. F. The nature of the anomalous viscosity of filled polymers as a function of concentration at low filler contents. Polym. Sci. USSR 19, 111–119 (1977) .
Guzeyev, V., Rafikov, M. & Malinskii, Y. The effect of filler dispersity of the melt viscosity of polyvinyl chloride (PVC). Polym. Sci. USSR 5, 804–806 (1975) .
Mackay, M. E. et al. Nanoscale effects leading to non-Einstein-like decrease in viscosity. Nat. Mater. 2, 762–766 (2003) .
Tuteja, A., Duxbury, P. M. & Mackay, M. E. Multifunctional Nanocomposites with Reduced Viscosity. Macromolecules 40, 9427–9434 (2007) .
Tan, H. et al. Melt viscosity behavior of C60 containing star polystyrene composites. Soft Matter 9, 6282–6290 (2013) .
Nusser, K., Schneider, G. J., Pyckhout-Hintzen, W. & Richter, D. Viscosity decrease and reinforcement in polymer-silsesquioxane composites. Macromolecules 44, 7820–7830 (2011) .
Roberts, C., Cosgrove, T., Schmidt, R. G. & Gordon, G. V. Diffusion of Poly(dimethylsiloxane) Mixtures with Silicate Nanoparticles. Macromolecules 34, 538–543 (2001) .
Schmidt, R. G. et al. A critical size ratio for viscosity reduction in poly(dimethylsiloxane)−polysilicate nanocomposites. Macromolecules 43, 10143–10151 (2010) .
Kim, D., Srivastava, S., Narayanan, S. & Archer, L. A. Polymer nanocomposites: polymer and particle dynamics. Soft Matter 8, 10813–10818 (2012) .
Kalathi, J. T., Grest, G. S. & Kumar, S. K. Universal viscosity behavior of polymer nanocomposites. Phys. Rev. Lett. 109, 198301 (2012) .
Ganesan, V., Pryamitsyn, V., Surve, M. & Narayanan, B. Noncontinuum effects in nanoparticle dynamics in polymers. J. Chem. Phys. 124, 221102 (2006) .
Wang, M. & Hill, R. J. Anomalous bulk viscosity of polymer-nanocomposite melts. Soft Matter 5, 3940–3953 (2009) .
Srivastava, S., Agarwal, P. & Archer, L. A. Tethered nanoparticle–polymer composites: phase stability and curvature. Langmuir 28, 6276–6281 (2012) .
Akcora, P. et al. Anisotropic self-assembly of spherical polymer-grafted nanoparticles. Nat. Mater. 8, 354–359 (2009) .
Chevigny, C. et al. Polymer-grafted-nanoparticles nanocomposites: dispersion, grafted chain conformation, and rheological behavior. Macromolecules 44, 122–133 (2011) .
Tuteja, A., Mackay, M. E., Narayanan, S., Asokan, S. & Wong, M. S. Breakdown of the continuum stokes-einstein relation for nanoparticle diffusion. Nano Lett. 7, 1276–1281 (2007) .
Rodriguez, R., Herrera, R., Archer, L. A. & Giannelis, E. P. Nanoscale ionic materials. Adv. Mater. 20, 4353–4358 (2008) .
Schaefer, D. W. Polymers, fractals, and ceramic materials. Science 243, 1023–1027 (1989) .
Neves, S. & Polo Fonseca, C. Determination of fractal dimension of polyaniline composites by SAXS and electrochemical techniques. Electrochem. Commun. 3, 36–43 (2001) .
Ferreira, P. G. & Ajdari, A. Scaling law for entropic effects at interfaces between grafted layers and polymer melts. Macromolecules 31, 3994–4003 (1998) .
Zawada, J. A., Ylitalo, C. M., Fuller, G. G., Colby, R. H. & Long, T. E. Component relaxation dynamics in a miscible polymer blend: poly(ethylene oxide)/poly(methyl methacrylate). Macromolecules 25, 2896–2902 (1992) .
Hopkinson, I., Kiff, F. T., Richards, R. W., King, S. M. & Farren, T. Isotopic labelling and composition dependence of interaction parameters in polyethylene oxide/polymethyl methacrylate blends. Polymer (Guildf) 36, 3523–3531 (1995) .
Ito, H., Russell, T. & Wignall, G. Interactions in mixtures of poly (ethylene oxide) and poly (methyl methacrylate). Macromolecules 20, 2213–2220 (1987) .
Pedemonte, E., Polleri, V. & Turturro, A. Thermodynamics of poly (ethylene oxide)-poly (methyl methacrylate) blends: prediction of miscibility based on the corresponding-states theory. Polymer (Guildf) 35, 3278–3281 (1994) .
Mackay, M. E. et al. General strategies for nanoparticle dispersion. Science 311, 1740–1743 (2006) .
Dutta, N., Egorov, S. & Green, D. Quantification of nanoparticle interactions in pure solvents and a concentrated PDMS solution as a function of solvent quality. Langmuir 29, 9991–10000 (2013) .
Anderson, B. & Zukoski, C. Nanoparticle stability in polymer melts as determined by particle second virial measurement. Macromolecules 40, 5133–5140 (2007) .
Hore, M. J. A. & Composto, R. J. Using miscible polymer blends to control depletion–attraction forces between au nanorods in nanocomposite films. Macromolecules 45, 6078–6086 (2012) .
Zhang, Q. & Archer, L. A. Poly(ethylene oxide)/silica nanocomposites: structure and rheology. Langmuir 18, 10435–10442 (2002) .
Fetters, L. J., Lohse, D., Richter, D., Witten, T. A. & Zirkel, A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 27, 4639–4647 (1994) .
Couchman, P. Glass-transition temperatures of compatible polymer mixtures. Phys. Lett. A 70, 155–157 (1979) .
Kalogeras, I. M. & Brostow, W. Glass transition temperatures in binary polymer blends. J. Polym. Sci. Part B Polym. Phys. 47, 80–95 (2009) .
Fox, T. & Flory, P. The glass temperature and related properties of poly- styrene. Influence of molecular weight. J. Polym. Sci. 14, 315–319 (1954) .
Agarwal, P., Kim, S. A. & Archer, L. A. Crowded, confined, and frustrated: dynamics of molecules tethered to nanoparticles. Phys. Rev. Lett. 109, 258301 (2012) .
Rubinstein, M. & Colby, R. Polymer Physics 361–388Oxford University Press (2003) .
Gurp, M. Van & Palmen, J. Time-temperature superposition for polymeric blends. Rheol. Bull. 67, 5–8 (1998) .
Larson, R. G. The Structure and Rheology of Complex fluids 265–267Oxford University Press (1999) .
Wu, T., Efimenko, K., Vlček, P., Šubr, V. & Genzer, J. Formation and properties of anchored polymers with a gradual variation of grafting densities on flat substrates. Macromolecules 36, 2448–2453 (2003) .
Li, Y., Kröger, M. & Liu, W. K. Nanoparticle effect on the dynamics of polymer chains and their entanglement network. Phys. Rev. Lett. 109, 118001 (2012) .
Schneider, G. J., Nusser, K., Willner, L., Falus, P. & Richter, D. Dynamics of entangled chains in polymer nanocomposites. Macromolecules 44, 5857–5860 (2011) .
Yamamoto, U. & Schweizer, K. Theory of entanglements and tube confinement in rod–sphere nanocomposites. ACS Macro Lett. 2, 955–959 (2013) .
Kalathi, J. T., Yamamoto, U., Schweizer, K. S., Grest, G. S. & Kumar, S. K. nanoparticle diffusion in polymer nanocomposites. Phys. Rev. Lett. 112, 108301 (2014) .
Grabowski, C. A., Adhikary, B. & Mukhopadhyay, A. Dynamics of gold nanoparticles in a polymer melt. Appl. Phys. Lett. 94, 021903 (2009) .
Acknowledgements
This work was supported by the National Science Foundation, Award No. DMR–1006323 and by Award No. KUS–C1018–02, made by the King Abdullah University of Science and Technology (KAUST). Use of the Advanced Photon Source, operated by the Argonne National Laboratory, was supported by the US DOE under Contract No. DE–AC02–06CH11357. We acknowledge Mr Xiaobing Zuo, Assistant Physicist, Argonne National Laboratory for helpful discussions.
Author information
Authors and Affiliations
Contributions
R.M., S.S. and L.A.A. designed the study. R.M. did the experiments. R.M., S.S. and L.A.A. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
L.A.A. is the co-founder and holds a financial interest in NOHMs Technologies, a technology concern focused on commercialization of electrodes and nanoparticle-based electrolytes for secondary batteries. The remaining authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-10 and Supplementary Table 1 (PDF 448 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mangal, R., Srivastava, S. & Archer, L. Phase stability and dynamics of entangled polymer–nanoparticle composites. Nat Commun 6, 7198 (2015). https://doi.org/10.1038/ncomms8198
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8198
This article is cited by
-
A Perspective on the Dynamics Properties in Polymer Nanocomposites
Chinese Journal of Polymer Science (2023)
-
Mechano-responsive hydrogen-bonding array of thermoplastic polyurethane elastomer captures both strength and self-healing
Nature Communications (2021)
-
Molecular Motions in Polymer Matrix for Microenvironment Sensing
Chemical Research in Chinese Universities (2021)
-
Parametric influences of fiber laser micro-machining for the generation of micro-channels on PMMA
Journal of the Brazilian Society of Mechanical Sciences and Engineering (2020)
-
An unexpected N-dependence in the viscosity reduction in all-polymer nanocomposite
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
