
Abstract
Light absorption can trigger biologically relevant protein conformational changes. The light-induced structural rearrangement at the level of a photoexcited chromophore is known to occur in the femtosecond timescale and is expected to propagate through the protein as a quake-like intramolecular motion. Here we report direct experimental evidence of such ‘proteinquake’ observed in myoglobin through femtosecond X-ray solution scattering measurements performed at the Linac Coherent Light Source X-ray free-electron laser. An ultrafast increase of myoglobin radius of gyration occurs within 1 picosecond and is followed by a delayed protein expansion. As the system approaches equilibrium it undergoes damped oscillations with a ~3.6-picosecond time period. Our results unambiguously show how initially localized chemical changes can propagate at the level of the global protein conformation in the picosecond timescale.
Similar content being viewed by others

Introduction
Chemical reactions often involve the motion of both electrons and nuclei of the participating molecules. In the case of proteins, the molecular machines of living organisms, the study of the dynamics of molecular motions is particularly fascinating since localized ultrafast chemical events, such as bond breaking, may trigger biologically relevant concerted motions of thousands of atoms usually referred to as protein conformational changes1. Many studies have shown that proteins are dynamic objects with a hierarchy of various intramolecular motions spanning a wide range of time and length scales1,2. In this regard, photosensitive proteins, which can be excited with ultrashort light pulses, serve as excellent model systems since they allow studying dynamics in a wide time window extending from a few tens of femtoseconds to seconds or more3. Many of such studies have used myoglobin (Mb), a relatively small protein (~18 kDa) that has been named the hydrogen atom of biology4 and has played a central role for our understanding of protein dynamics5. Mb is a monomeric protein consisting of 153 amino acids folded into eight α-helices connected by short loops. Small ligands such as molecular oxygen (O2) and carbon monoxide (CO) are able to bind reversibly to Mb and the bond between the ligand and the haem molecule contained in the active site of the protein can be photolyzed with high efficiency within 50 fs6. The events following photolysis have been extensively studied with a variety of techniques such as time-resolved optical absorption7,8,9,10, circular dichroism11, resonance Raman12,13,14, photoacoustic calorimetry15 and transient grating spectroscopy16,17. The results of these investigations indicate that an initial ultrafast rearrangement of the haem molecule, which ensues a so-called doming of the haem structure18 and an out-of-haem-plane motion of the central iron ion19, triggers a series of structural changes that extend from the amino acids close to the haem through the entire polypeptide chain and solvent hydration layer20. The initial ultrafast protein response to the breaking of the bond between the ligand and the protein has been described as a quake-like motion of Mb, since the propagation of the strain released upon photoexcitation through the protein is similar to the propagation of acoustic waves during an earthquake21. Such ‘proteinquake’ model has been used to describe the structural dynamics of other haem22 and non-haem proteins23,24,25. The analysis of the elastic response of the protein to the active site rearrangement is complicated by the simultaneous dissipation of the excess energy that is deposited by the photolysis pulse on the haem chromophore. Transient resonance Raman experiments19,26 and molecular dynamics simulations27 have demonstrated that most of haem cooling occurs within a few ps mostly through direct haem-solvent energy transfer. Dissipation of residual excess kinetic energy occurs through the polypeptide chain at a longer timescale (few tens of picoseconds) as demonstrated by transient grating spectroscopy28. Moreover, analogous experiments performed on deoxyMb16 have suggested the non-thermal origin of the quake-like response observed after photolysis of carbonmonoxy myoglobin (MbCO). A first experimental evidence of a proteinquake based on the use of a direct structural probe has been recently reported by Arnlund et al.29 in the case of multiphoton excitation of a bacterial photoreaction centre. These authors have used time-resolved X-ray solution scattering, an experimental technique able to track the structural dynamics of proteins in solution30, to show that the backbone carbon atoms of the protein helices increase their distance from the interior of the photoreaction centre on a picosecond timescale. However, under the extensive protein multiphoton absorption conditions employed, protein motions occurred in the presence of a very large excess energy deposited in the system, and the observed protein response may not reflect physiologically relevant protein functioning conditions. In the case of Mb, the experimental investigations performed so far were based either on spectroscopic measurements or did not have sufficient time resolution to measure the timescale of the proteinquake. Indeed, time-resolved X-ray solution scattering recently performed on Mb have demonstrated31, 32, 33 that, although subtle protein structural changes have been observed in the nanoseconds-to-microseconds timescale, the most significant global structural rearrangements, corresponding to a relative motion of helices33 (as revealed by changes in the wide-angle X-ray scattering (WAXS) signal) and an estimated 22 Å3 volume change31 (as probed with small-angle X-ray scattering (SAXS)), occur within the time resolution of 100 ps available in those experiments, and in agreement with previous transient grating spectroscopy results17.
Here, we use the femtosecond X-ray pulses produced by the Linear Coherent Light Source (LCLS) X-ray free-electron laser (X-FEL) to visualize the structural response of Mb after photolysis. We show how the perturbation at the active site level propagates to the global protein structure with a timescale set by the acoustic speed of sound, thus confirming the proteinquake hypothesis21. The time evolution of both the radius of gyration and the volume of Mb reveals an oscillatory collective motion of the protein atoms that is damped in few ps, thus highlighting the relevance of underdamped low-frequency vibrations in proteins. Our data illustrate how ultrafast studies are potentially able to capture the intrinsic ballistic-like nature of protein motion that is generally hidden in ensemble measurements at longer timescales.
Results
Time-resolved X-ray scattering difference patterns
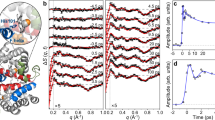
To decipher the exact timescale of the ultrafast structural dynamics in Mb, we have performed femtosecond time-resolved X-ray solution scattering experiments on Mb at the LCLS X-FEL (Fig. 1, see Methods). Laser-induced time-resolved difference scattering curves obtained by photoexciting a solution of horse MbCO with 250 fs pulses at 538 nm are shown in Fig. 2.

The sample (a 2.35-mM MbCO solution) was circulated in a closed loop through a fused-silica capillary (0.3 mm diameter) positioned at the overlap between the X-ray pulses (9 keV, 30 fs) produced by the LCLS (~1010 photons per pulse at 9 keV are transmitted by a silicon monochromator) and the photolysis pulses (538 nm, 250 fs) produced by the optical laser system (~0.3 mJ mm−2). By monitoring the pattern of the X-rays scattered by the sample at different time delays between the optical and X-ray pulses, it was possible to track the structural changes occurring in the sample after photolysis.

A significant change in both intensity and shape is evident in the SAXS region (a); continuous black lines are fittings in terms of Guinier analysis (see Methods). Data in the WAXS region (b, magnified by a factor 20) show a difference signal that develops within 1 ps and then changes in shape, while increasing in amplitude, with a timescale of the order of a few picoseconds. Data have been vertically offset for clarity.
The probe X-ray pulses (~30 fs long) hit the sample at several time delays after photolysis, spanning a time range up to 100 ps with a time resolution of ~500 fs (see Methods). Changes in the X-ray scattering signal have been simultaneously monitored both in the SAXS and WAXS regions. The overall shape of the difference signals is similar to what has been observed in the past for Mb with a much lower (100 ps) time resolution30,31,32,33, with the 100 ps difference pattern measured in the present experiment (see Fig. 2) being essentially identical to what has been reported in the literature31,32. The signal changes in shape and grows in intensity mostly within 10 ps, and a clear difference signal is observed both in the SAXS and WAXS regions already at 0.8 ps from photolysis.
Analysis of the X-ray scattering signal time evolution
The radius of gyration (Rg) and volume (Vp) of proteins can be directly obtained from the SAXS signal34 (solid black lines in Fig. 2, see Methods). The ultrafast increase of Rg at nearly constant Vp (Fig. 3, top panel) and the subsequent increase in Vp (Fig. 3, bottom panel), which is delayed by ~1 ps, clearly shows that a change in the mass distribution of the protein precedes its expansion (see Supplementary Fig. 1 for a comparison of analyses based on different data sets). Considering the overall size of Mb (Rg~17 Å), this implies that the strain released upon photolysis roughly propagates at ~20 Å ps−1, in agreement with the expected speed of sound in proteins35. Analogous timescales are observed in the WAXS region, where a clear ultrafast difference signal (negative peak at ~0.75 Å−1) appears within 1 ps from photoexcitation and it further changes in shape with a negative peak at ~0.3 Å−1 developing in few ps. The simultaneous evolution of both SAXS and WAXS signals shows that the motion of secondary structure elements is responsible for the Rg and Vp changes. In light of our data, it is clear that the global conformational change reflected in the WAXS difference signal, already attributed to the relative motion of protein helices30,31,32,33 and in analogy to what has been found with time-resolved WAXS measurements on other photosensitive proteins21,36,37, is essentially completed in 10 ps. As already demonstrated in previous studies7,8,9,10,31,32,38, further more subtle structural changes, which complete the conformational relaxation towards the unbound Mb equilibrium state, occur at longer timescales (extending from a few nanoseconds to a few microseconds).

The analysis of time-resolved SAXS data (blue symbols) reveals that Rg increases by ~1 Å within 1 ps and exhibits damped oscillations around its equilibrium value (a; the red curve represents a damped oscillation with a 3.6 ps time period and 6 ps decay time). The increase in the protein volume (b) is delayed with respect to the Rg increase: Vp increases by 220 Å3 within 2 ps. The observation of a delayed volume expansion implies that an initial ultrafast redistribution of protein mass from the active site towards the outside solvent (as measured by the Rg increase) occurs without significant volume change within 1 ps. After few tens of ps, Vp relaxes to a value slightly higher with respect to that before photoexcitation (ΔVp~20 Å3 at 100 ps). Error bars are from the fitting procedure used to obtain ΔRg and ΔVp, and correspond to one s.d. The black vertical lines are guides to the eye and they are spaced by 0.7 ps.
Discussion
The results here presented demonstrate that Mb undergoes significant global structural changes in the ps timescale after photolysis of the bond between the protein and the CO ligand. At difference with the recent report by Arnlund et al.29, by monitoring simultaneously both the SAXS and WAXS region, we have been able to extract directly the time evolution of relevant structural parameters. Our data clearly show that, even in biophysically relevant photoexcitation conditions (we estimate that, in our experimental conditions, ~1.6 photons are absorbed per chromophore within the duration of a photolysis pulse), an ultrafast proteinquake is observed.
Such an ultrafast, ballistic-like response of the protein might seem to contrast with the exponential-like time dependencies that typically characterize protein relaxations at longer timescales. Indeed, kinetic experiments on protein ensembles typically probe conformational relaxations that are dominated by thermally activated steps and do not reflect the actual relaxation of individual proteins, but rather the average timescale of energy barriers crossing. In this respect, ultrafast experiments using extremely short photoexcitation pulses have the advantage of unveiling the intrinsic nature of protein elementary motions before thermally activated processes start to play a role (Fig. 4). Similar evidence of such elementary stepwise protein motions have been recently obtained with ultrafast infrared spectroscopy39 and single-molecule fluorescence resonance energy transfer experiments40. We speculate that individual ballistic-like motions (e.g., one for each bond formation/breaking), rather than gradual structural transitions from one protein state to the other, could be a general feature of protein functional motions, although more experiments will be needed to confirm such a hypothesis.

We postulate that elementary motions at the level of a single protein have a quake-like ballistic nature. (a) Time evolution of an observable in an idealized single-molecule experiment (red curve). Experiments on large protein ensembles (blue curve) are able to detect such quake-like transitions only at very short times (step I), when the initial synchronization established by the photoexcitation pulse is still preserved. At longer times (step II), the observed time evolution is dominated by thermally activated transitions from one protein state to the other, which result in the ‘exponential-like’ kinetics typically observed in most time-resolved experiments. (b) Simplified free-energy diagram illustrating the difference between a downhill transition along the energy landscape (step I) and a thermally activated process; in the latter case, different protein molecules will undergo the same structural transition but with a statistically distributed onset.
Another important result of our study is that the Mb structure oscillates with a period of ~3.6 ps as evidenced by the time dependence of Rg and Vp. Interestingly, a collective vibrational mode of Mb with a comparable time period (~4 ps) has been suggested to be strongly coupled to the conformation of haem and of the proximal histidine and is activated by the strain release induced by photolysis41. Figure 5 illustrates how the collective mode involves large protein regions from the active site to the protein surface. Such coupling with the mode at ~4 ps was already suggested on the basis of transient grating investigations17, although oscillations of the Mb structure could not be detected in those experiments.

Cartoon representation of the Mb three-dimensional structure; the segments of the polypeptide chain contributing to the collective mode having a ~4 ps period (which is activated by ligand release) are highlighted in red (see Methods for details). This vibrational mode involves the haem, the helix to which the haem is covalently bound (F), and parts of the A, C, E, G and H helices. Note that the regions involved participate to define the haem pocket and extend to the protein surface.
The observation of oscillations in Mb structural parameters (Rg and Vp) demonstrates that the elastic response of Mb is not dominated, at least in the ultrafast timescale, by damping mechanisms. Although evidences of underdamped protein collective motions have been recently obtained with THz spectroscopy42, this is a surprising result since theoretical arguments would suggest that low-frequency (<50 cm−1) protein oscillations are overdamped in a liquid environment. Indeed, a simple model approximating Mb as a homogeneous elastic sphere oscillating in water predicts a damping time of ~1.5 ps for an oscillation period close to that observed in our experiment (3.6 ps, see Methods). More sophisticated computational analysis based on atomic resolution protein models predict low-frequency protein collective motions to be overdamped43. At difference, our data show that Mb can also undergo underdamped collective vibrations at ~10 cm−1 after the bond between the protein and the ligand is broken.
The results here presented demonstrate that the combination of sub-picosecond time resolution available at X-FEL sources and the direct structural sensitivity of X-ray solution scattering techniques can be advantageously used to investigate the intrinsic nature of protein elementary motions.
From the point of view of biology, the present study may be of general relevance for understanding the structural basis of biological reactions that involve ultrafast chemical processes, as it is the case of electron transfer in photosynthesis. It is likely that the initial primary charge separation events, which are known to be photoinduced and occur in the picosecond timescale, are modulated by global protein motions analogous to those reported for Mb in the present paper.
Methods
Sample preparation and data acquisition
The experiment was performed at the X-ray Pump and probe (XPP) endstation of the LCLS X-FEL (SLAC National Accelerator Laboratory). The sample was a solution of horse MbCO in 0.1 M phosphate buffer at pH 7.4 with a protein concentration of 2.35 mM (~40 mg ml−1) and a threefold molar excess of sodium dithionite. The vial containing the 20 ml of CO-saturated Mb solution was connected to a liquid flowing system driven by an HPLC pump able to circulate the solution (3 ml min−1 flow speed) in a closed loop through a 0.3 mm fused-silica capillary (10 μm wall thickness) positioned at the intersection of the optical and X-ray laser beams. Photolysis of the bond between Mb and CO was achieved with circularly polarized pulses (~250 fs full-width at half-maximum; 25 μJ per pulse; 538 nm) focused to a 300 μm full-width at half-maximum circular spot (energy density ~0.3 mJ mm−2 at the sample), in nearly collinear (~1°) geometry. The absorbance of the sample at the pump wavelength was ~1.0 optical density. Given the size, energy of the photolysis beam, the concentration and molar extinction coefficient of the haem, ~1.6 photons are expected to be absorbed by each haem molecule at each photolysis pulse. A monochromatic X-ray beam at 9 keV was produced by a Si(111) double-crystal monochromator and focused to 100 μm by beryllium refractive lenses. Time-resolved X-ray scattering images were acquired with a two-dimensional charge-coupled device detector (Rayonix SX165, 2,048 × 2,048 pixels) at 16.8 cm from the sample. Each image is the result of the accumulation of 360 X-ray shots. Investigated time delays ranged between −3 and 100 ps (with negative time delays corresponding to a probe pulse arriving before the photolysis one). In particular, we measured every 0.5 ps between −3 and 10 ps; every 5 ps between 10 and 50 ps; every 50 ps at longer time delays. Reference images at a time delay of −100 ps were also recorded every 7 other images. The scattering pattern at each time delay is the result of an average over 50 repetitions. The repetition rate of the experiment was 120 Hz and the estimated time resolution was ~500 fs.
Experimental time resolution
The relative timing between X-ray and visible pulses has been monitored using the timing tool developed44 at the XPP endstation of the LCLS X-FEL45, which exploits the ultrafast free-carrier generation induced by X-rays in a Si3N4 membrane to encode the relative arrival time of X-ray and visible pulses. The timing tool data show that over the ~6 h of data collection the X-ray/laser timing has been stable to about ~150 fs and that the jitter was about 200 fs. The time resolution of the experiment is also affected by the group velocity mismatch between the X-ray and optical pulses propagating inside the 300 μm thick protein solution. By taking into account both effects and the duration of photolysis pulses (~250 fs), we estimate an overall time resolution of ~500 fs.
Calculation of scattering difference patterns
Two-dimensional scattering images recorded with a Rayonix SX165 detector were azimuthally averaged to give one-dimensional scattering patterns. The scattering angle θ was converted to momentum transfer q using the formula q=4π/λsin(θ/2), where λ=1.377 Å is the X-rays wavelength. Since the changes induced in the scattering patterns by optical photoexcitation account for less than a percent of the absolute signal, data were normalized30 at 1.4±0.1 Å−1 before calculating difference patterns with respect to the pattern measured at −100 ps. For a given time delay, each scattering difference is compared with the average difference and a simple reduced  criterion is used to discard the differences that are too far away from the average one. In the formula above, Ii refers to the scattered intensity for a given time delay and q-bin, Ii is the scattered intensity averaged over all repetitions, and σi is the error bar on the experimentally measured intensity. The differences that satisfy the above criterion are then used to produce the averages difference patterns shown in the paper. This procedure usually rejects ~15–25% of the differences.
criterion is used to discard the differences that are too far away from the average one. In the formula above, Ii refers to the scattered intensity for a given time delay and q-bin, Ii is the scattered intensity averaged over all repetitions, and σi is the error bar on the experimentally measured intensity. The differences that satisfy the above criterion are then used to produce the averages difference patterns shown in the paper. This procedure usually rejects ~15–25% of the differences.
Analysis of SAXS data
The scattered intensity of a dilute protein solution in the small-angle region can be approximated by the following expression46:

where Δρ=ρp—ρb is the electron density difference between the protein and surrounding buffer solution, Vp is the protein volume, q is the magnitude of the scattering vector and Rg is the protein radius of gyration. A conformational change of the protein may affect Vp and Rg, but not the total number of electrons it contains (ρpVp) that has thus been kept constant (~9,900 electrons) in the fits of SAXS signals. Since the experimental data are SAXS difference patterns, they have been fitted using the following expression:

with  ,
,  and ρb=0.34 electron Å−3.
and ρb=0.34 electron Å−3.
Representation of Mb activated vibrational mode
To obtain a graphical representation of the residues most affected by the Mb collective mode having a ~4 ps period, we have followed the reasoning by Seno and Go41. Starting from their normal mode analysis, the authors have evaluated the magnitude of the ligand-induced conformational change by calculating, for each residue (including the haem molecule), the mass-weighted square atomic displacement over all the atoms belonging to the residue. In particular, we used the data reported in figure 8 of ref. 41 to calculate the involvement of each residue of the Mb sequence in the ~4 ps collective normal mode. The residues that have the highest values (>10% of the maximum) of the mass-weighted square displacements are depicted in red in Fig. 5.
Elastic sphere model
To estimate the frequency and damping time of Mb vibrational modes, we have used a model that approximates the protein as an homogeneous elastic sphere oscillating in water. Under this approximation the frequencies and damping times can be calculated following the procedure described by Talati and Jha47. In particular, the (complex) normalized frequencies (s) have to satisfy the following equation:

where Z(s) is the acoustic impedance, j the imaginary unit, vl and vt are the longitudinal and transverse speed of sound, respectively, in the protein and dP is the protein mass density. The (complex) normalized frequency s=ωR/vl, where ω is the complex angular frequency and R is the spherical protein radius. The acoustic impedance Z(s) is equal to:

where dm and vm are the density and sound speed of the medium, respectively, surrounding the sphere and km=ω/vm. Finally, the damping time τD and the complex angular frequency ω are related by the following expression: τD=−Im(ω).
The relevant physical parameters used to perform the calculations are reported in Supplementary Table 1. The solution corresponding to the first three lowest-frequency vibrational modes are characterized by oscillation periods of 2.7, 1.15 and 0.75 ps, while the corresponding damping times are 1.3, 1.7 and 1.8 ps.
Additional information
How to cite this article: Levantino, M. et al. Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat. Commun. 6:6772 doi: 10.1038/ncomms7772 (2015).
References
Frauenfelder, H., Sligar, S. G. & Wolynes, P. G. The energy landscapes and motions of proteins. Science 254, 1598–1603 (1991) .
Henzler-Wildman, K. & Kern, D. Dynamic personalities of proteins. Nature 450, 964–972 (2007) .
Martin, J. L. & Vos, M. H. Femtosecond biology. Annu. Rev. Biophys. Biomol. Struct. 21, 199–222 (1992) .
Frauenfelder, H., McMahon, B. H. & Fenimore, P. W. Myoglobin: the hydrogen atom of biology and a paradigm of complexity. Proc. Natl Acad. Sci. USA 100, 8615–8617 (2003) .
Parak, F. G. & Nienhaus, G. U. Myoglobin, a paradigm in the study of protein dynamics. Chemphyschem 3, 249–254 (2002) .
Petrich, J. W., Poyart, C. & Martin, J. L. Photophysics and reactivity of heme proteins: a femtosecond absorption study of hemoglobin, myoglobin, and protoheme. Biochemistry 27, 4049–4060 (1988) .
Lim, M., Jackson, T. A. & Anfinrud, P. A. Non exponential protein relaxation: Dynamics of conformational change in Myoglobin. Proc. Natl Acad. Sci. USA 90, 5801–5804 (1993) .
Ansari, A., Jones, C. M., Henry, E. R., Hofrichter, J. & Eaton, W. A. Conformational relaxation and ligand binding in myoglobin. Biochemistry 33, 5128–5145 (1994) .
Franzen, S. & Boxer, S. G. On the origin of heme absorption band shifts and associated protein structural relaxation in myoglobin following flash photolysis. J. Biol. Chem. 272, 9655–9660 (1997) .
Esquerra, R. M., Goldbeck, R. A., Kim-Shapiro, D. B. & Kliger, D. S. Spectroscopic evidence for nanosecond protein relaxation after photodissociation of myoglobin-CO. Biochemistry 37, 17527–17536 (1998) .
Dartigalongue, T. & Hache, F. Observation of sub-100 ps conformational changes in photolyzed carbonmonoxy-myoglobin probed by time-resolved circular dichroism. Chem. Phys. Lett. 415, 313–316 (2005) .
Findsen, E. W., Scott, T. W., Chance, M. R., Friedman, J. M. & Ondrias, M. R. Picosecond time-resolved Raman studies of photodissociated carboxymyoglobin. J. Am. Chem. Soc. 107, 3355–3357 (1985) .
Dasgupta, S., Spiro, T. G., Johnson, C. K., Dalickas, G. A. & Hochstrasser, R. M. Picosecond resonance Raman evidence for unrelaxed heme in the (carbonmonoxy)myoglobin photoproduct. Biochemistry 24, 5295–5297 (1985) .
Sato, A., Gao, Y., Kitagawa, T. & Mizutani, Y. Primary protein response after ligand photodissociation in carbonmonoxy myoglobin. Proc. Natl Acad. Sci. USA 104, 9627–9632 (2007) .
Norris, C. L. & Peters, K. S. A photoacoustic calorimetry study of horse carboxymyoglobin on the 10-nanosecond time scale. Biophys. J. 65, 1660–1665 (1990) .
Genberg, L., Richard, L., McLendon, G. & Miller, R. J. Direct observation of global motion in hemoglobin and myoglobin on picosecond time scales. Science 251, 1051–1054 (1991) .
Deak, J., Chiu, H. L., Lewis, C. M. & Miller, R. J. Ultrafast phase grating studies of heme proteins: observation of the low frequency modes directing functionally important protein motions. J. Phys. Chem. B 102, 6621–6634 (1998) .
Franzen, S., Bohn, B., Poyart, C. & Martin, J. L. Evidence of sub-picosecond heme doming in hemoglobin and myoglobin: a time-resolved resonance Raman comparison of carbonmonoxy and deoxy species. Biochemistry 34, 1224–1247 (1995) .
Kitagawa, T., Haruta, N. & Mizutani, Y. Time-resolved resonance Raman study on ultrafast structural relaxation and vibrational cooling of photodissociated carbonmonoxy myoglobin. Biopolymers 67, 207–213 (2002) .
Dantsker, D., Samuni, U., Friedman, J. M. & Agmon, N. A hierarchy of functionally important relaxations within myoglobin based on solvent effects, mutations and kinetic model. Biochim. Biophys. Acta 1749, 234–251 (2005) .
Ansari, A. et al. Protein states and proteinquakes. Proc. Natl Acad. Sci. USA 85, 5000–5004 (1985) .
Zang, C. et al. Ultrafast proteinquake dynamics in cytochrome c. J. Am. Chem. Soc. 131, 2846–2852 (2009) .
Xie, A. et al. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry 40, 1510–1517 (2001) .
Itoh, K. & Sasai, M. Dynamical transition and proteinquake in photoactive yellow protein. Proc. Natl Acad. Sci. USA 101, 14736–14741 (2004) .
Li, S. et al. Thermal-triggerd proteinquake leads to disassembly of DegP hexamer as an imperative activation step. Sci. Rep. 4, 4834 (2014) .
Mizutani, Y & Kitagawa, T. Direct observation of cooling of heme upon photodissociation of carbonmonoxy myoglobin. Science 278, 443–446 (1997) .
Zhang, Y., Fujisaki, H & Straub, J. E. Molecular dynamics study on the solvent dependent heme cooling following ligand photolysis in carbonmonoxy myoglobin. J. Phys. Chem. B 111, 3243–3250 (2007) .
Genberg, L., Heisel, F., McLendon, G. & Miller, R. J. D. Vibrational energy relaxation processes in heme proteins: model systems of vibrational energy dispersion in disordered systems. J. Phys. Chem. 91, 5521–5524 (1987) .
Arnlund, D. et al. Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nat. Methods 11, 923–926 (2014) .
Cammarata, M. et al. Tracking the structural dynamics of proteins in solution using time-resolved wide-angle X-ray scattering. Nat. Methods 5, 881–886 (2008) .
Cho, H. S. et al. Protein structural dynamics in solution unveiled via 100-ps time-resolved X-ray scattering. Proc. Natl Acad. Sci. USA 107, 7281–7286 (2010) .
Oang, K. Y. et al. Conformational substates of myoglobin intermediate resolved by picosecond X-ray solution scattering. J. Phys. Chem. Lett. 5, 804–808 (2014) .
Ahn, S., Kim, K. H., Kim, Y., Kim, J. & Ihee, H. Protein tertiary changes visualized by time resolved X-ray solution scattering. J. Phys. Chem. B 113, 13131–13133 (2009) .
Svergun, D. I. & Koch, M. H. Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 66, 1735–1782 (2003) .
Yu, X. & Leitner, D. M. Vibrational energy transfer and heat conduction in a protein. J. Phys. Chem. B 107, 1698–1707 (2003) .
Andersson, M. et al. Structural dynamics of light-driven proton pumps. Structure 17, 1265–1275 (2009) .
Takala, H. et al. Signal amplification and transduction in phytochrome photosensors. Nature 509, 245–248 (2014) .
Tian, W. D., Sage, J. T., Srajer, V. & Champion, P. M. Relaxation dynamics of myoglobin in solution. Phys. Rev. Lett. 68, 408–411 (1992) .
Li, G., Magana, D. & Dyer, R. B. Anisotropic energy flow and allosteric ligand binding in albumin. Nat. Commun. 5, 3100 (2014) .
Diez, M. et al. Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat. Struct. Mol. Biol. 11, 135–141 (2004) .
Seno, Y. & Go, N. Deoxymyoglobin studied by the conformational normal mode analysis. II The conformational change upon oxygenation. J. Mol. Biol. 216, 111–126 (1990) .
Turton, D. A. et al. Terahertz underdamped vibrational motion governs protein-ligand binding in solution. Nat. Commun. 5, 3999 (2014) .
Kitao, A., Hirata, F. & Go, N. The effects of solvent on the conformation and the collective motions of protein: normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 158, 447–472 (1991) .
Harmand, M. et al. Achieving few-femtosecond time-sorting at hard X-ray free electron lasers. Nat. Photon. 7, 215–218 (2013) .
Emma, P. et al. First lasing and operation of Angstrom-wavelength free-electron-laser. Nat. Photon. 4, 641–647 (2010) .
Glatter, O. & Kratky, O. Small Angle X-ray Scattering Academic Press (1982) .
Talati, M. & Jha, P. K. Low-frequency acoustic phonons in nanometric CeO2 particles. Physica E 28, 171–177 (2005) .
Acknowledgements
We thank the Coherent X-ray Imaging endstation for the use of the HPLC pump. We wish to thank Tim Brandt van Driel for help during the tests with the CSPAD detector. M.Ca. acknowledges the Projets Exploratoires Premier Soutien (PEPS) project ‘SASLELX’ of the CNRS and the University of Rennes 1 for financial support. M.L. acknowledges the financial support of the University of Palermo (FFR program 2012/2013). G.S. acknowledges the PEPS project ‘SASLELX’ of the CNRS for financial support. H.I. acknowledges the support by the Institute for Basic Science (IBS) (IBS-R004-G2). Portions of this research were carried out at the Linac Coherent Light Source (LCLS) at SLAC National Accelerator Laboratory. LCLS is an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University.
Author information
Authors and Affiliations
Contributions
M.Ca. conceived the project, H.T.L. and M.Ca. led the experiment preparation. M.L. G.S., H.T.L., J.M.G., D.Z., M.Ch. and M.Ca. performed the experiment. M.L., G.S. and M.Ca. analysed the data and wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figure 1 and Supplementary Table 1 (PDF 90 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Levantino, M., Schirò, G., Lemke, H. et al. Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat Commun 6, 6772 (2015). https://doi.org/10.1038/ncomms7772
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms7772
This article is cited by
-
Carotenoid responds to excess energy dissipation in the LH2 complex from Rhodoblastus acidophilus
Photosynthesis Research (2022)
-
Filming ultrafast roaming-mediated isomerization of bismuth triiodide in solution
Nature Communications (2021)
-
Ultrafast coherent motion and helix rearrangement of homodimeric hemoglobin visualized with femtosecond X-ray solution scattering
Nature Communications (2021)
-
Tracking Membrane Protein Dynamics in Real Time
The Journal of Membrane Biology (2021)
-
Short-lived metal-centered excited state initiates iron-methionine photodissociation in ferrous cytochrome c
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
