
Abstract
Fast synchronous neurotransmitter release is triggered by calcium that activates synaptotagmin-1 (syt-1), resulting in fusion of synaptic vesicles with the presynaptic membrane. Syt-1 possesses two Ca2+-binding C2 domains that tether membranes via interactions with anionic phospholipids. It is capable of crosslinking membranes and has recently been speculated to trigger fusion by decreasing the gap between them. As quantitative information on membrane gaps is key to understanding general cellular mechanisms, including the role of syt-1, we developed a fluorescence-lifetime based inter-membrane distance ruler using membrane-anchored DNAs of various lengths as calibration standards. Wild-type and mutant data provide evidence that full-length syt-1 indeed regulates membrane gaps: without Ca2+, syt-1 maintains membranes at distances of ~7–8 nm. Activation with 100 μM Ca2+ decreases the distance to ~5 nm by binding the C2 domains to opposing membranes, respectively. These values reveal that activated syt-1 adjusts membrane distances to the level that promotes SNARE complex assembly.
Similar content being viewed by others

Introduction
Neurotransmission is accomplished by exocytosis following the fusion of synaptic vesicles with the presynaptic plasma membrane1,2,3. The neurotransmitters stored within the vesicles are released into the synaptic cleft at a millisecond timescale on Ca2+ influx4, with syt-1, residing on the synaptic vesicles and bearing two Ca2+-binding C2 domains (C2A and C2B), being the indispensable Ca2+ sensor5,6 (Fig. 1). The C2A and C2B domains, after coordinating to multiple Ca2+ ions7,8,9, bind to membranes containing negatively charged phospholipids, which complete the Ca2+-coordination sites8. Moreover, a patch of four lysine residues (KKKK, 324–327) located on the side of C2B interacts specifically with the poly-anionic phosphatidylinositol(4,5)-bisphosphate (PIP2) in a Ca2+-independent manner10. However, despite extensive research using either the full-length syt-1 (including the transmembrane domain, TMD) or the truncated soluble C2AB fragment11,12,13, the questions how syt-1 functions at a molecular level as a Ca2+-trigger and how it interacts with the SNARE proteins (soluble N-ethylmaleimide-sensitive factor attachment protein receptor), the membrane fusion machinery1,2, remained unsettled.

Red dots indicate Ca2+-binding sites of the two Ca2+-binding domains C2A and C2B, whereas the dark blue dot represents the poly-lysine patch (KKKK) of the C2B domain. Plus and minus signs indicate clusters of positive and negative charges on the linker. Bright blue dots represent neurotransmitter molecules. PIP2, phosphatidylinositol(4,5)-bisphosphate; TMD, transmembrane domain.
Before the formation of fully zippered SNARE complexes, which exert forces on the membranes and lead consequently to fusion, the synaptic vesicles are first primed to the presynaptic plasma membrane and await the Ca2+ activation of syt-1. The models describing the molecular activation of the fusion reaction by synaptotagmin fall into two groups (summarized in ref. 14). In the first scenario, the SNARE complexes are arrested at a partially zippered state during the priming process. Syt-1 may either serve as a fusion clamp that is released on Ca2+ triggering15, or alternatively, it may displace the inhibitory protein complexin from the SNARE complexes16. In the second scenario, the SNAREs do not assemble before arrival of the Ca2+ signal, although syt-1 may already be in contact with the plasma membrane via the KKKK–PIP2 electrostatic interaction10. Following Ca2+ triggering, the SNAREs rapidly progress through zippering and fusion17.
In line with the latter scenario, recently, it has been proposed that syt-1 triggering is based on Ca2+-dependent regulation of the gap between the vesicle and presynaptic membranes. In the absence of Ca2+, syt-1 connects membranes via (i) its own transmembrane domain and (ii) binding to PIP2 via the KKKK polybasic stretch. Assuming that the 61-residue linker18 between C2A and the transmembrane domain is unstructured and stretched, a maximal distance of ~28 nm is feasible. On the basis of non-quantitative Förster resonance energy transfer (FRET) between labelled lipids in the participating membranes, it was suggested that addition of Ca2+ might reduce the distance to a range of ~2–7.5 nm (ref. 19). Such distance shortening might operate upstream of the SNARE complex formation, which requires a membrane distance below 8 nm (ref. 20). However, experimental support for this model was still elusive, primarily because of the lack of experimental evidence for distance variations at the appropriate length scale.
In this study, we first developed a quantitative membrane distance assay based on accurate fluorescence lifetime detection of multiple inter-membrane FRET interactions between tethered small unilamellar liposomes using membrane-anchored double-stranded DNA (dsDNA) of defined lengths as calibration standards. This calibrated system was then used to measure membrane distances of liposomes tethered by full-length syt-1 wild-type as well as Ca2+-binding mutants in the absence and presence of physiological Ca2+ concentrations and when the content of anionic lipids required for syt-1 binding was varied. These data provide evidence that Ca2+-activated syt-1 indeed decreases membrane gaps to a level of ~5 nm, which corresponds to the inter-membrane distance spanned by a trans-SNARE complex.
Results
Membrane distance ruler
The fluorescence lifetime of donor molecules labelled on lipid headgroups (Oregon Green 488 DHPE) of the DNA- or proteoliposomes was chosen as a robust indicator of FRET, as it is insensitive to variations in the concentration of liposomes within the confocal volume of ~1 femtoliter or to variations in alignment conditions of the experimental set-up. In contrast to FRET-based distance measurements within or between interacting proteins21,22,23, distances between membranes cannot be easily extracted. In proteins, single donor and acceptor dyes are attached to defined sites and a single donor–acceptor pair is distant from the other fluorophores. In these cases, the average distance between the pair can be easily derived, providing that the Förster distance R0 for the selected donor and acceptor is known. On liposomes, contrarily, the fluorophores are distributed across the surfaces of both the inner and outer leaflets. Hence, in a single tethered cluster, each donor molecule may transfer the excitation energy to multiple acceptors, and the probability of each donor undergoing FRET is different and depends on its position relative to the acceptor liposomes containing Texas Red DHPE (Fig. 2a), which in turn leads to different lifetimes. Therefore, we measured first a calibration curve using tethered liposomes with predefined spacings so that the average donor fluorescence lifetimes can be correlated properly with membrane distances between donor and acceptor liposomes. The calibration of this membrane distance ruler was done using membrane-anchored dsDNA of various well-defined lengths as discrete spacers24. In general, dsDNA shorter than its persistence length of 50 nm, or 150 base pairs (bp), is known to have high bending rigidity25. Herein three different lengths well below the persistence length were prepared (Fig. 2), 3.3 nm (10 bp), 5.0 nm (15 bp) and 8.3 nm (25bp). The membrane anchor lipid 1,2-O-dioctadecyl-(rac)-glycerol was attached to the 5′-end of DNA oligonucleotides by solid-phase synthesis using a synthetic lipid phosphoramidite26. The complementary strands were then individually reconstituted into the donor and acceptor liposomes, respectively. In addition, 10% anionic lipids phosphatidylserine (PS) were included in both types of DNA liposomes. The negative charges on the liposome surfaces and the negatively charged DNA backbone further stabilized the perpendicular orientation of the DNA already observed in the absence of any repulsive charges on the membranes27.

The upper panels show the experimental design of the membrane distance ruler for small unilamellar liposomes, with (a) 3.3, (b) 5.0 and (c) 8.3 nm spacings. FRET decreases as the distance increases, as reflected in the longer amplitude-weighted lifetime (τamp). (d) The complementary DNA sequences for the respective lengths, where X denotes the lipid-phosphoramidite.
An excess of acceptor liposomes was used to drive the equilibrium so that each donor liposome is surrounded by an equal (maximal) number of acceptor liposomes (Fig. 2a–c), to eliminate the contribution of differences in the degree of tethering28. To determine the required ratio, we gradually increased the acceptor liposome concentration while keeping the donor liposomes constant. The donor fluorescence decay curves converged after the ratio exceeded 1:10 (Supplementary Fig. 1). Therefore, a ratio of 20 acceptor liposomes to 1 donor liposome was selected for all experiments.
The fluorescence decay curves of the three DNA-tethered liposome mixtures were measured with time-correlated single photon counting29 (TCSPC) (Fig. 3a), and the emission wavelength was selected at the blue edge of donor fluorescence spectrum (525±10 nm) to minimize the crosstalk between donor and acceptor channels (Supplementary Fig. 2). The three curves were clearly distinguishable and decayed faster than the control sample without DNA, and the difference arises predominantly from the donor molecules that are facing the acceptor liposomes. The decay curves observed with the aforementioned excess ratio of 1:20 were fitted with two components, and the amplitude-weighted lifetimes (τamp, see Methods section) of the donors depending on inter-membrane distances were obtained. By measuring three independent replicates, τamp for liposomes with membrane distances of 8.3, 5.0 and 3.3 nm was determined to be 3.44±0.05, 3.06±0.13 and 2.82±0.18 ns, respectively (average±s.d.). A plot of τamp versus the membrane distance can be very well fitted by a linear approximation (Fig. 3g), which later serves as the calibration curve for membrane distance determination. As control, hybridization of complementary DNA strands on the liposomes was inhibited by adding a free single strand (without the lipid anchor, Supplementary Fig. 3). Under these conditions, the lifetime was restored to the control level observed with a sample containing donor and excess acceptor liposomes, neither with DNA. An advantage of the calibration procedure (Fig. 3g) is that any nonspecific effects, such as nonspecific liposome clustering, are intrinsically corrected for. To quantify the extent of such nonspecific effects, we compared a sample containing only pure donor liposomes (grey dashed line in Supplementary Fig. 3) directly with the donor–acceptor control without any DNA (black line in Supplementary Fig. 3). This comparison demonstrates that nonspecific effects contribute only to a minor extent to the overall fluorescence decay.

Decay curves for (a) DNA-tethered liposomes with specified number of base pairs and (b) syt-1 wild-type (WT) reconstituted into donor liposomes in the absence and presence of 100 μM Ca2+. No PIP2 indicates the sample in which 2% PIP2 was substituted with 7% PS. (c) Measurements without Ca2+ for syt-1 WT reconstituted into donor liposomes with various molar ratios of the anionic lipid PS. (d–f) Decay curves for Ab*, a*B and a*b* mutants. Control stands for the experiments in which there was no protein or DNA on either type of liposomes. The symbols represent experimental data, whereas the lines are reconvolution fits using two exponentials. (g) The plot of amplitude-weighted lifetime (τamp) versus distance. A linear regression line can be constructed with the DNA-tethered liposomes and applied to determine the distance between syt-1 tethered liposomes in the absence or presence of 100 μM Ca2+. (h) τamp and the calculated distance information for the mutants. The error bars represent s.d. values obtained from three independent repeats. Asterisks (*) designate when the τamp values are statistically different (P<0.05, two-tailed unpaired t-test).
It is known from electron microscopy (EM) studies that small unilamellar liposomes (≤50 nm), owing to their high membrane curvature, still maintain the spherical shape at contact areas when tethered by proteins (see for example, cryo-electron tomography in ref. 30). To ensure additionally that the number of membrane tethering sites between donor and acceptor liposomes are comparable in all samples, we conducted fluorescence correlation spectroscopy (FCS, see Supplementary Fig. 4 and Supplementary Methods for details). FCS data showed that the average number of acceptor liposome tethering sites per donor liposome did not vary by >15% throughout all samples measured in this work (Supplementary Table 1).
Synaptotagmin-1 controls the gap between two membranes
Next, we reconstituted syt-1 in donor liposomes containing 10% PS and measured the decay when they were bound to acceptor liposomes bearing 15% PS plus 2% PIP2 (Fig. 3b)3,31. In the absence of Ca2+ (1 mM EGTA), τamp was close to that of the 8.3 nm DNA-tethered liposomes (3.33±0.03 ns) and the membrane distance was estimated to be 7.4 nm (Fig. 3b,g). To verify that the decrease in fluorescence lifetime compared with the control sample was caused by the specific interaction of the KKKK patch with PIP2, the poly-anionic PIP2 (2%) in the acceptor liposomes was replaced by increasing the concentration of mono-anionic PS by 7 to 22% PS, which maintained the net negative charge of the acceptor liposomes32. In the absence of PIP2, the fluorescence lifetime returned to the control level, showing that tethering to the syt-1 proteoliposomes was abolished. We then repeated the incubation using PIP2-containing liposomes in the absence of Ca2+ (1 mM EGTA) to allow for the KKKK-PIP2 binding and added appropriate Ca2+ buffer afterwards to achieve a final free Ca2+ concentration of 100 μM. The resulting fluorescence decay was accelerated and corresponded to that measured with liposomes tethered with the DNA-ruler at a distance of 5.0 nm (τamp=3.05±0.10) (Fig. 3b,g). We conclude accordingly that the gap between syt-1 proteoliposomes and the surrounding acceptor liposomes decreases to ~5 nm after the addition of Ca2+.
The distance of 7.4 nm in the absence of Ca2+ is relatively short, suggesting that the linker between the transmembrane domain and the C2A domain is not completely stretched19. Intriguingly, the linker contains a membrane-adjacent stretch of positive charges, which may interact with the more C-terminally localized stretch of negative charges33 and/or with anionic lipids in the resident membrane of the protein (a cis interaction)34. To further examine the electrostatic interactions with negative charges on the liposome membranes28, we performed a set of experiments in which the content of PS on the donor proteoliposomes was varied (Fig. 3c). When the donor proteoliposomes were neutral, that is, without PS, and only the acceptor liposomes contained anionic lipids, the measured lifetime was surprisingly short (2.82±0.09 ns) even in the absence of Ca2+ and was similar to that of the shortest set of DNA-tethered liposomes (3.3 nm). This short distance can be explained by the nonphysiological elimination of electrostatic repulsion between the two membranes, which is present in vivo between the negatively charged synaptic vesicle and presynaptic membranes3,31. In addition, the membranes may be pulled into close proximity if the positively charged lysine residues on the linker are attached in trans to the surfaces of acceptor liposomes. Figure 3c also indicates that at an intermediate level of PS (5%), there is an equilibrium between the 7.4- and 3.3-nm configurations.
Ca2+-binding mutants
As all known Ca2+ effects are mediated by the Ca2+-binding sites in the C2A and C2B domains, we repeated the distance measurements using previously characterized syt-1 mutants in which Ca2+ binding to the C2A, C2B or to both C2 domains was disrupted (a*B: D178A, D230A and D232A; Ab*: D309A, D363A and D365A; a*b*: D178A, D230A, D232A, D309A, D363A and D365A)11. In the mutant with a disrupted C2B domain (Ab*), the inter-membrane distances in the absence and presence of Ca2+ were very similar to those of the wild-type protein (Fig. 3d,g,h). In contrast, in the mutants with a disrupted C2A domain (a*B and a*b*), there was little or no distance shortening on addition of Ca2+, respectively (Fig. 3e,f,h). Summarized in Fig. 3h are the fitted τamp and the corresponding distances calculated for all mutants. These data support a model according to which the C2A domain is predominantly responsible for distance shortening due to Ca2+-mediated cis binding to its own membrane. On the contrary, the C2B domain, at least under our experimental conditions, does not appear to be capable of simultaneous cis-trans binding as previously suggested8,35. The fact that a*B still exhibits a minor decrease in distance on Ca2+ addition may rather be explained by the Ca2+-induced binding of C2B to the acceptor membrane and the insertion of its hydrophobic residues10,36.
Discussion
The calibrated fluorescence-lifetime-based membrane distance ruler with membrane-anchored DNAs of well-defined lengths allowed for direct quantitative determination of membrane gaps controlled by wild-type and mutants of the full-length neurotransmission trigger syt-1. Initially, in the absence of Ca2+, the poly-lysine patch (KKKK) located on the C2B domain is targeted to PIP2 clusters on the presynaptic plasma membrane (Fig. 4a,b)37,38. Our data demonstrate that at this stage full-length syt-1 maintains the two membrane bilayers at a distance of ~7–8 nm (Fig. 4b), which is much shorter than a maximum length of ~28 nm if the linker would be unstructured and fully stretched. The ~7–8 nm distance corresponds to that when the SNARE motifs start to assemble and form the coiled coil four-helix bundle (measured with surface forces apparatus)20, implying that syt-1 holds the two membranes at this distance so that the fusion machinery is ready for fast initiation17. Recently, such a pre-triggered state was also captured in cryo-EM images in ref. 30, which revealed that a large number of small liposomes incorporating syt-1 and synaptobrevin (the vesicle-SNARE protein) remained docked to giant liposomes including syntaxin and SNAP-25 (the target-SNAREs) at distances on the order of ~10 nm, even after extended incubation. The fact that removing synaptobrevin or adding its soluble counterpart to inhibit the full-length SNARE assembly did not alter the overall tethering suggests the major role of syt-1 in establishing a first contact to the target membrane, upstream of SNARE nucleation (Fig. 4b)19,39.

(a) Syt-1 targets PIP2 clusters via the poly-lysine patch (blue). (b) The distance between two membranes is maintained at ~7-8 nm, as the linker is not fully stretched34. (c) On Ca2+ influx, the C2A domain binds cis, reducing the distance to 5 nm and potentially promoting the electrostatic zippering of the linker33. Both membranes contain PS.
Second, the ruler provides clear evidence that, on Ca2+ influx, the inter-membrane gap is compressed to ~5 nm (Fig. 4c). As a trans-SNARE complex spans two membranes at a distance of ≥4 nm (ref. 40), a distance reduction from ~7–8 nm to ~5 nm brings the two membranes to a level at which very likely full assembly of the SNAREs is promoted41, leading subsequently to the initiation of membrane fusion and neurotransmitter release. At first glance, the ~2.4 nm change in distance may not seem very large in terms of the absolute value. However, considering the above-mentioned dimensions of the SNAREs, such a change is quite significant and allows for efficient triggering at the millisecond timescale.
The membrane distance of 5 nm also correlates to cryo-EM observations35,42 for liposomes clustered by soluble C2AB fragments incorporating Ca2+, in which the C2A and C2B domains bind to opposing membranes43. Such an antiparallel conformation has been shown to be of the lowest energy from simulated annealing based on EPR-derived restraints using the soluble C2AB fragment44,45,46. Moreover, it corroborates new findings that alteration in the length and rigidity of the short (9-residue) linker between the C2 domains has a significant impact on evoked neurotransmitter release47.
In vivo mutation studies demonstrated that the C2B domain is indispensable for fast synchronous neurotransmitter release48,49. The fact that the distance regulation behaviour of the Ab* mutant is similar to that of the wild-type (Fig. 3g,h) illustrates that in contrast to the C2B domain—which plays an essential general role—C2A plays a more important role in the distance regulation itself: initially, the C2B domain is needed to tether the membranes together, especially via its poly-lysine patch also in the absence of Ca2+ (Fig. 4b). After C2B is already attached to the opposite membrane, an effective decrease of the inter-membrane gap can then be accomplished by binding of the C2A domain back to the host membrane and an even tighter binding of the C2B domain to the opposite membrane with Ca2+ coordination (Fig. 4c). Obviously, the role of C2B Ca2+ binding sites is less connected to the distance regulation function of syt-1 than to other important mechanistic functions, which might include direct interactions with the SNAREs15,21,23,50 or facilitating curvature of the presynaptic membrane51,52. Notably, recently an ~80% decrease in release has been demonstrated using a D229E mutation in Drosophila at the Ca2+-coordination site of C2A53, supporting the important function also of C2A in synchronous synaptic transmission.
As a final remark, the membrane distance ruler developed in this study using membrane-anchored DNAs will also be very beneficial to the investigation of distance regulatory steps in other biological processes of interest, such as fertilization, viral entry as well as protein trafficking54,55, in which the gap between two membranes needs to be quantified at the nm-scale.
Methods
Sample preparation
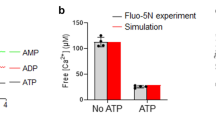
Syt-1 (Rat) wild-type and the mutants were purified as previously described28 and elaborated in Supplementary Methods. The DNA-lipid conjugates were synthesized by solid-phase synthesis using the lipid-phosphoramidite26 as the last base under otherwise standard DNA synthesis conditions56 and purified by RP-HPLC (see Supplementary Methods for details and characterization). Small unilammelar liposomes were prepared using Sephadex G-50 superfine (Sigma-Aldrich) size-exclusion columns. The size of the resulting liposomes was ~40 nm in diameter, estimated from the FCS information recorded simultaneously57 and dynamic light scattering (DLS). Liposomes were labelled either with 0.5% OG-DHPE or with 1% TR-DHPE (Invitrogen), and the DNA/protein-to-lipid ratios were kept at 1:1,000. The proteoliposomes consisted of 20% DOPE, 10% cholesterol and 10% DOPS, whereas the acceptor liposomes contained 20% DOPE, 10% cholesterol, 15% DOPS and 2% PIP2, unless otherwise specified. Ten percent DOPS was included into all DNA liposomes. The remaining lipids were adjusted with DOPC to yield 100%. All non-labelled lipids were purchased from Avanti Polar Lipids. The buffer contained 150 mM KCl, 20 mM Hepes and was adjusted to pH 7.4. Hybridization of the DNA-liposomes was achieved by incubation for 5 min at 35 °C for 10 bp and 55 °C for 15 and 25 bp, followed by slow cooling back to room temperature. Syt-1 proteoliposomes were first incubated with the acceptor liposomes for 30 min and then added to buffers containing 1 mM EGTA or a final Ca2+ concentration of 100 μM (checked with Fluo-5N, Invitrogen) for another 30 min to reach equilibrium. Final concentrations of donor liposomes were maintained at ~2 nM (one particle in the focal volume on average).
Instrumentation
Fluorescence microscopy measurements were performed on an Olympus IX71 inverted microscope with an UPlanSApo × 60/1.2 W water immersion objective. A Ti:sapphire oscillator (Chameleon, Coherent) centering at 800 nm served as the two-photon excitation source. A dichroic mirror 715DCSPXR (AHF) and a short pass filter E700SP were used to separate the excitation and emission and the dichroic mirror 590DCXR (AHF) to split the fluorescence onto the green and red avalanche photodiode detectors (SPCM-CD 2969, PerkinElmer), selected with band pass filters D525/20m and D680/30m (Chroma). Data were acquired with a router (PRT 400)-coupled TCSPC card (TimeHarp200, PicoQuant) and analysed with the SymPhoTime software version 5.3 (PicoQuant).
Amplitude-weighted lifetime (τamp)
For numerical determination of the distances, we fitted all the fluorescence decay curves with iterative reconvolution using a model function of two exponential components: I(t)=A1exp(−t/τ1)+A2exp(−t/τ2). The amplitude-weighted lifetime (τamp=a1τ1+a2τ2, where a1=A1/(A1+A2) and a2=A2/(A1+A2)), which is more sensitive to changes in the faster component, was then calculated.
Additional information
How to cite this article: Lin, C.-C. et al. Control of membrane gaps by synaptotagmin-Ca2+ measured with a novel membrane distance ruler. Nat. Commun. 5:5859 doi: 10.1038/ncomms6859 (2014).
References
Jahn, R. & Südhof, T. C. Membrane fusion and exocytosis. Annu. Rev. Biochem. 68, 863–911 (1999).
Jahn, R. & Scheller, R. H. SNAREs−engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 (2006).
Takamori, S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006).
Schneggenburger, R. & Neher, E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406, 889–893 (2000).
Chapman, E. R. How does synaptotagmin trigger neurotransmitter release? Annu. Rev. Biochem. 77, 615–641 (2008).
Brose, N., Petrenko, A. G., Südhof, T. C. & Jahn, R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256, 1021–1025 (1992).
Ubach, J., Zhang, X., Shao, X., Südhof, T. C. & Rizo, J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2‐domain? EMBO J. 17, 3921–3930 (1998).
Honigmann, A. et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat. Struct. Mol. Biol. 20, 679–686 (2013).
Fernandez, I. et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 (2001).
Bai, J., Tucker, W. C. & Chapman, E. R. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44 (2004).
Stein, A., Radhakrishnan, A., Riedel, D., Fasshauer, D. & Jahn, R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat. Struct. Mol. Biol. 14, 904–911 (2007).
Wang, Z., Liu, H., Gu, Y. & Chapman, E. R. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J. Cell Biol. 195, 1159–1170 (2011).
Lee, H. K. et al. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 328, 760–763 (2010).
Jahn, R. & Fasshauer, D. Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207 (2012).
Chicka, M. C., Hui, E., Liu, H. & Chapman, E. R. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+. Nat. Struct. Mol. Biol. 15, 827–835 (2008).
Yang, X., Kaeser-Woo, Y. J., Pang, Z. P., Xu, W. & Südhof, T. C. Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory alpha helix. Neuron 68, 907–920 (2010).
Pobbati, A. V., Stein, A. & Fasshauer, D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science 313, 673–676 (2006).
Perin, M. S., Brose, N., Jahn, R. & Südhof, T. C. Domain structure of synaptotagmin (p65). J. Biol. Chem. 266, 623–629 (1991).
van den Bogaart, G. et al. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat. Struct. Mol. Biol. 18, 805–812 (2011).
Li, F. et al. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat. Struct. Mol. Biol. 14, 890–896 (2007).
Vrljic, M. et al. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat. Struct. Mol. Biol. 17, 325–331 (2010).
Brunger, A. T., Weninger, K., Bowen, M. & Chu, S. Single-molecule studies of the neuronal SNARE fusion machinery. Annu. Rev. Biochem. 78, 903–928 (2009).
Choi, U. B. et al. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 17, 318–324 (2010).
Chung, M., Koo, B. J. & Boxer, S. G. Formation and analysis of topographical domains between lipid membranes tethered by DNA hybrids of different lengths. Faraday Discuss. 161, 333–345 (2013).
Baumann, C. G., Smith, S. B., Bloomfield, V. A. & Bustamante, C. Ionic effects on the elasticity of single DNA molecules. Proc. Natl Acad. Sci. USA 94, 6185–6190 (1997).
Chan, Y. H., van Lengerich, B. & Boxer, S. G. Lipid-anchored DNA mediates vesicle fusion as observed by lipid and content mixing. Biointerphases 3, FA17–FA21 (2008).
Chung, M., Lowe, R. D., Chan, Y. H., Ganesan, P. V. & Boxer, S. G. DNA-tethered membranes formed by giant vesicle rupture. J. Struct. Biol. 168, 190–199 (2009).
Vennekate, W. et al. Cis- and trans-membrane interactions of synaptotagmin-1. Proc. Natl Acad. Sci. USA 109, 11037–11042 (2012).
Lakowicz, J. R. Principles of Fluorescence Spectroscopy 3 edn Springer Science+Business Media, LLC (2006).
Bharat, T. A. et al. SNARE and regulatory proteins induce local membrane protrusions to prime docked vesicles for fast calcium-triggered fusion. EMBO Rep. 15, 308–314 (2014).
Smith, C. U. M. Elements of Molecular Neurobiology 3 edn John Wiley & Sons, Ltd. (2002).
McLaughlin, S., Wang, J., Gambhir, A. & Murray, D. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 (2002).
Lai, Y., Lou, X., Jho, Y., Yoon, T. Y. & Shin, Y. K. The synaptotagmin 1 linker may function as an electrostatic zipper that opens for docking but closes for fusion pore opening. Biochem. J. 456, 25–33 (2013).
Lu, B., Kiessling, V., Tamm, L. K. & Cafiso, D. S. The juxtamembrane linker of full-length synaptotagmin 1 controls oligomerization and calcium-dependent membrane binding. J. Biol. Chem. 289, 22161–22171 (2014).
Araç, D. et al. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 13, 209–217 (2006).
Hui, E., Bai, J. & Chapman, E. R. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys. J. 91, 1767–1777 (2006).
van den Bogaart, G. et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 (2011).
Aoyagi, K. et al. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 280, 17346–17352 (2005).
Ellena, J. F. et al. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc. Natl Acad. Sci. USA 106, 20306–20311 (2009).
Hanson, P. I., Roth, R., Morisaki, H., Jahn, R. & Heuser, J. E. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90, 523–535 (1997).
Gao, Y. et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337, 1340–1343 (2012).
Seven, A. B., Brewer, K. D., Shi, L., Jiang, Q. X. & Rizo, J. Prevalent mechanism of membrane bridging by synaptotagmin-1. Proc. Natl Acad. Sci. USA 110, E3243–E3252 (2013).
Connell, E. et al. Cross-linking of phospholipid membranes is a conserved property of calcium-sensitive synaptotagmins. J. Mol. Biol. 380, 42–50 (2008).
Kuo, W., Herrick, D. Z. & Cafiso, D. S. Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry 50, 2633–2641 (2011).
Herrick, D. Z. et al. Solution and membrane-bound conformations of the tandem C2A and C2B domains of synaptotagmin 1: Evidence for bilayer bridging. J. Mol. Biol. 390, 913–923 (2009).
Lai, A. L., Huang, H., Herrick, D. Z., Epp, N. & Cafiso, D. S. Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J. Mol. Biol. 405, 696–706 (2011).
Liu, H. et al. Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nat. Neurosci. 17, 670–677 (2014).
Mackler, J. M., Drummond, J. A., Loewen, C. A., Robinson, I. M. & Reist, N. E. The C2B Ca2+-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418, 340–344 (2002).
Nishiki, T. & Augustine, G. J. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J. Neurosci. 24, 8542–8550 (2004).
Bhalla, A., Chicka, M. C., Tucker, W. C. & Chapman, E. R. Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat. Struct. Mol. Biol 13, 323–330 (2006).
Martens, S., Kozlov, M. M. & McMahon, H. T. How synaptotagmin promotes membrane fusion. Science 316, 1205–1208 (2007).
Hui, E., Johnson, C. P., Yao, J., Dunning, F. M. & Chapman, E. R. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell 138, 709–721 (2009).
Striegel, A. R. et al. Calcium binding by synaptotagmin's C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J. Neurosci. 32, 1253–1260 (2012).
Martens, S. & McMahon, H. T. Mechanisms of membrane fusion: disparate players and common principles. Nat. Rev. Mol. Cell Biol. 9, 543–556 (2008).
Jahn, R., Lang, T. & Südhof, T. C. Membrane Fusion. Cell 112, 519–533 (2003).
Wachowius, F., Javadi-Zarnaghi, F. & Höbartner, C. Combinatorial mutation interference analysis reveals functional nucleotides required for DNA catalysis. Angew. Chem. Int. Ed. 49, 8504–8508 (2010).
Cypionka, A. et al. Discrimination between docking and fusion of liposomes reconstituted with neuronal SNARE-proteins using FCS. Proc. Natl Acad. Sci. USA 106, 18575–18580 (2009).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 803, to C.-C.L., A.P.-L., R.J. and P.J.W.) and in part by the Cluster of Excellence and DFG Research Center Nanoscale Microscopy and Molecular Physiology of the Brain (C.H.). We thank U. Ries for protein purification and D. Riedel for checking the quality of DNA liposomes with electron microscopy.
Author information
Authors and Affiliations
Contributions
C.-C.L., R.J. and P.J.W. wrote the paper. J.S. and C.H. synthesized the DNA lipids. A.P.-L. and R.J. provided the proteins. C.-C.L. designed the study and performed all other experiments. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figure 1-4, Supplementary Table 1, Supplementary Methods and Supplementary References (PDF 411 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lin, CC., Seikowski, J., Pérez-Lara, A. et al. Control of membrane gaps by synaptotagmin-Ca2+ measured with a novel membrane distance ruler. Nat Commun 5, 5859 (2014). https://doi.org/10.1038/ncomms6859
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6859
This article is cited by
-
Vesicle trafficking and vesicle fusion: mechanisms, biological functions, and their implications for potential disease therapy
Molecular Biomedicine (2022)
-
Conserved arginine residues in synaptotagmin 1 regulate fusion pore expansion through membrane contact
Nature Communications (2021)
-
Selected tools to visualize membrane interactions
European Biophysics Journal (2021)
-
Synaptotagmin-1 membrane binding is driven by the C2B domain and assisted cooperatively by the C2A domain
Scientific Reports (2020)
-
Different states of synaptotagmin regulate evoked versus spontaneous release
Nature Communications (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
