
Abstract
The catalytic hydrogenation of nitroarenes is an environmentally benign technology for the production of anilines, which are key intermediates for manufacturing agrochemicals, pharmaceuticals and dyes. Most of the precious metal catalysts, however, suffer from low chemoselectivity when one or more reducible groups are present in a nitroarene molecule. Herein we report FeOx-supported platinum single-atom and pseudo-single-atom structures as highly active, chemoselective and reusable catalysts for hydrogenation of a variety of substituted nitroarenes. For hydrogenation of 3-nitrostyrene, the catalyst yields a TOF of ~1,500 h−1, 20-fold higher than the best result reported in literature, and a selectivity to 3-aminostyrene close to 99%, the best ever achieved over platinum group metals. The superior performance can be attributed to the presence of positively charged platinum centres and the absence of Pt–Pt metallic bonding, both of which favour the preferential adsorption of nitro groups.
Similar content being viewed by others

Introduction
Functionalized anilines are industrially important and valuable intermediates for a variety of agrochemicals, pharmaceuticals, dyes and pigments1. Currently, the commercial production of functionalized anilines mainly uses non-catalytic reduction of the corresponding nitroarenes using stoichiometric reducing agents such as sodium hydrosulfite, iron, tin or zinc in ammonium hydroxide2. Such processes, however, have serious environmental issues. In this context, there is a great need to search for an environmentally benign and highly efficient method to produce functionalized anilines. Catalytic hydrogenation with supported metal catalysts is the preferred choice3. Nevertheless, when one or more reducible groups (for example, C=C, C≡C, C=O, C≡N and so on) are present in a nitroarene molecule, the chemoselective hydrogenation of the nitro group has been a grand challenge. In some cases, through modification of transition metal with well-chosen additives, for example, Pt/C-H3PO2-VO(acac)2 and Pt-Pb/CaCO3-FeCl2-nBu4NCl, chemoselective reduction of nitroarenes was obtained at the expense of activity4. Moreover, the addition of soluble metal salts still causes environmental problems. Therefore, the development of true heterogeneous catalysts without the use of any organic/inorganic additives is highly desirable.
IB-group metal catalysts, especially supported Au and Ag nanoparticle catalysts, have been reported to show excellent chemoselectivity for the hydrogenation of a variety of substituted nitroarenes5,6,7,8,9. However, the intrinsically poor capability of IB-group metals for H2 activation renders them at least one order of magnitude less active than the Pt-group metals. To enhance the activity of IB-group metals, delicate control of metal-support interactions10,11,12,13,14 or alloying with a small amount of Pt-group metals15,16 was tested. However, the achieved chemoselectivity of these catalysts was not satisfactory. Very recently, noble metal-free catalysts based on cobalt– or iron–phenanthroline complexes have been developed for the chemoselective hydrogenation of nitroarens, with excellent chemoselectivities for numerous structurally diverse substrates17,18, but similar to the IB-group metals they require relatively demanding reaction conditions, for example, long reaction time (more than 12 h), high temperature (120 °C) and high H2 pressure (50 bar).
The chemoselectivity to the hydrogenation of nitro groups arises from the preferential adsorption of nitro groups on the specific sites of metal nanoparticles or the support13,14. Although the hydrogenation of nitro groups over metal nanoparticles appears to be size insensitive, the hydrogenation of C=C and C≡C groups is indeed highly sensitive to the particle size, with the reaction rate usually increasing with increasing metal particle size19. Accordingly, by reducing the particle size of Pt-group metals and by tuning the metal–support interactions, Corma et al.20 achieved high chemoselectivity (90%–95%) over Pt/TiO2, Pt/C, Ru/TiO2 and even Ni/TiO2 catalysts while maintaining a high activity.
The lowest limit of downsizing metal particles is to disperse the metal as exclusively isolated single atoms, which we previously denoted as ‘single-atom catalyst (SAC)’21. The SAC has exhibited superior activity in CO oxidation22,23, water–gas shift reaction24,25,26, methane conversion27 and other important reactions28,29,30. On account of the uniform geometry of single atoms and their good capability for H2 dissociation31, it is expected that the SAC may have superior performance in selective hydrogenation reactions.
In this study we report that FeOx-supported Pt single-atom and pseudo-single-atom structures are extremely active, chemoselective and recyclable for the hydrogenation of a variety of functionalized nitroarenes. More importantly, characterizations by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-ray absorption fine structure spectroscopy (XAFS) reveal that the pseudo-single-atom structure is composed of a few to tens of atoms, which are loosely and randomly associated with each other but do not form strong Pt–Pt metallic bonding, resembling the structure and function of isolated single atoms. For the chemoselective hydrogenation of 3-nitrostyrene, the turnover frequency (TOF) of the SAC and pseudo-SACs reaches as high as ~1,500 h−1, 20-fold higher than that of the best catalyst reported in literature (Pt/TiO2)15. Furthermore, the selectivity toward 3-aminostyrene is close to 99%, again the best result ever achieved over Pt-group metals.
Results
HAADF-STEM characterization
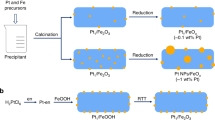
The series of Pt/FeOx catalysts were prepared by co-precipitation at 50 °C with H2PtCl6 and Fe(NO3)3 as the metal precursors and Na2CO3 as the precipitation agent22. The resultant precipitate was filtered, washed thoroughly with deionized water, dried at 60 °C overnight, calcined in air at 400 °C and reduced with 10% H2/He at 200 °C or 250 °C for 30 min. By varying the amount of Pt loading and changing the reduction temperature, the dispersion of Pt metal was finely tuned with their morphologies varying from isolated single atoms, loosely associated ensembles, subnanometre clusters to three-dimensional (3D) nanoparticles. Figure 1 presents the representative HAADF-STEM images of the freshly reduced catalysts. When the Pt loading was 0.08 wt% with a reduction temperature of 200 °C (0.08%Pt/FeOx-R200), all the Pt species existed exclusively as isolated single atoms; neither subnanometre clusters nor nanoparticles were detected (Fig. 1a; for more images, see Supplementary Fig. 1), which is in accordance with our previous observations22. On the other hand, when the 0.08 wt% Pt/FeOx catalyst was reduced at a slightly higher temperature, for example, 250 °C or above, clustering of single atoms occurred and very small clusters composed of loose and random ensembles of several to tens of atoms, with dimensions <0.8 nm, were formed (Fig. 1b and Supplementary Fig. 2). Based on the brightness variations of these clusters in HAADF images and by comparing their intensities with those of the single atoms, we concluded that most of these subnanometre clusters possessed a two-dimensional (2D) raft-like morphology, suggesting the occurrence of strong metal–support interactions32. However, with a higher Pt loading (0.31–4.30 wt%) and a reduction temperature at 250 °C, 3D nanoparticles with sizes of 1–2 nm were found to co-exist with the 2D rafts and single atoms (Fig. 1c–f and Supplementary Figs 3–5).

(a) 0.08%Pt/FeOx-R200. Scale bar, 1 nm. (b) 0.08%Pt/FeOx-R250. Scale bar, 2 nm. (c) 0.31%Pt/FeOx-R250. Scale bar, 2 nm. (d) 0.75%Pt/FeOx-R250. Scale bar, 2 nm. (e) 4.30%Pt/FeOx-R250. Scale bar, 10 nm. (f) Histogram of size distributions. The circles, squares and triangles in the figures represent the single atoms, 2D rafts and 3D clusters/particles, respectively.
XAFS characterization
The single-atom and 2D raft structures of Pt were further characterized with extended XAFS (EXAFS) spectroscopy. As shown in Table 1 and Supplementary Fig. 6, for the 0.08%Pt/FeOx-R200 catalyst there were a Pt-O contribution at 2.01 Å with a coordination number of 3.9 and a Pt-Fe contribution at 3.05 Å with a coordination number of 3.3. The much longer distance of Pt-Fe coordination (denoted as Pt-Felong) than that in Pt-Fe alloy (2.54 Å) suggests that Pt does not directly coordinate with Fe; instead, according to our previous model it coordinates with Fe via bridging oxygen22. The relatively higher coordination number of Pt–O (about 4) also supports this conclusion. Of most importance, there was no direct Pt–Pt coordination in this catalyst, suggesting that the Pt existed exclusively as isolated single atoms, in good agreement with the HAADF-STEM result as well as our previous observations of similar catalysts22. On the other hand, for the 0.08%Pt/FeOx-R250 catalyst, in addition to the Pt–O and Pt–Felong contributions, there were also Pt–Fe and Pt–Ptlong contributions at 2.55 and 2.99 Å, respectively, with a coordination number of ~1. In comparison with the 0.08% Pt/FeOx-R200 catalyst, the Pt–O coordination number in the 0.08%Pt/FeOx-R250 catalyst decreased by 50% as a consequence of elevating the reduction temperature; concurrently, the Pt–Fe metallic bonding was formed, suggesting that with the assistance of Pt, a very small portion of the Fe3+ in the FeOx nanocrystallites was reduced to Fe0. Consistent with this result, the X-ray diffraction pattern of the 0.08%Pt/FeOx-R250 catalyst showed only Fe3O4 phase, while that of the 0.08%Pt/FeOx-R200 catalyst presented both Fe3O4 and Fe2O3 phases (Supplementary Fig. 7), indicating that reduction at 250 °C removed more lattice oxygen from the FeOx support. The coordination number of 1.0 for the Pt–Fe bond may suggest that on the more severe reduction treatment at 250 °C, Pt atoms moved to reside at oxygen vacancies in the FeOx support. Similar to the 0.08%Pt/FeOx-R200 catalyst, there was no Pt–Pt contribution at 2.76 Å (metallic bonding in Pt foil) in the 0.08%Pt/FeOx-R250 catalyst, while the occurrence of Pt–Ptlong coordination at a distance of 2.99 Å may suggest the presence of the loosely clustered Pt single atoms as observed in the HAADF-STEM images. Therefore, the structure and behaviour of the 0.08%Pt/FeOx-R250 catalyst, in essence, resembles those of the 0.08%Pt/FeOx-R200 catalyst and therefore can be considered as a pseudo-SAC. The underlying reason for the formation of pseudo-single-atom structure is not known yet. One speculation is that the residue sodium impurity in the catalyst (from the sodium carbonate used as the precipitating agent, see Supplementary Table 1) may have played a role and, furthermore, sodium cations have been reported to act as a key component to stabilize the single atoms of Pt33.
With further increase of Pt loading to 0.75, 2.73 and 4.30 wt% while retaining the reduction temperature of 250 °C, the Pt–O coordination decreased while the Pt–Pt metallic bonding started to occur and then steadily increased. The Pt–Pt coordination number, however, was relatively low (0.5–2.3) for the above three catalysts, indicative of the formation of very small Pt clusters or particles. This conclusion corroborates the HAADF-STEM observations that the sizes of the majority of the Pt clusters/particles were below 1 nm, even for the 4.30%Pt/FeOx-R250 catalyst. On the other hand, the distance of Pt–Pt coordination decreased from 2.73 to 2.62 Å when the Pt loading increased from 0.75–4.30 wt%. It has been reported that the metal–metal bond distance usually contracts when the metal particles are very small34. However, as the percentage of isolated single atoms and pseudo-single-atom ensembles decreases with increasing Pt loading and the fact that the Pt–Pt distance in the pseudo-single-atom ensembles is much longer, the observed decrease of the Pt–Pt distance with increasing Pt loading is understandable. Thus, the EXAFS data qualitatively reflect the percentage of single-atom or pseudo-single-atom structures in the supported Pt catalysts with different Pt loadings.
Figure 2 shows the normalized X-ray absorption near-edge structure (XANES) spectra. The white-line intensity, reflecting the oxidation state of the Pt species, increased steadily with a decrease of the Pt loading, suggesting that the Pt became more positively charged with lower Pt loading. In other words, lower Pt loading results in higher percentage of single-atom and pseudo-single-atom structures in the catalyst, and more electron transfers from Pt to the FeOx support22. Moreover, with the same Pt loading of 0.08 wt%, the 250 °C reduction treatment resulted in lower white-line intensity than that of the catalyst with a 200 °C reduction treatment. The changing pattern for the white-line intensity is consistent with that of the Pt–O coordination number, providing further evidence that the electronic property of Pt can be effectively tuned by forming unique nanoscale or atomic scale Pt structures.

The increased white-line intensity with decreasing the Pt loading indicates that single-atom and pseudo-single-atom structures of Pt are more positively charged than others.
By combining HAADF-STEM and XAFS techniques, the single-atom, pseudo-single-atom, as well as subnanometre and nanometre structures of Pt have been identified unequivocally. Subsequently, these catalysts with different Pt structures were tested for chemoselective hydrogenation of nitroarenes.
Activity test
The catalytic activity test began with the chemoselective hydrogenation of 3-nitrostyrene, the most demanding reaction among a variety of substituted nitroarenes, because the C=C group in this molecule is highly reactive towards reduction over typical Pt catalysts6. Before the activity tests, all the catalysts were in situ pretreated in toluene at 40 °C, 1 MPa H2 for 1 h to remove the surface contamination by oxygen during sample handling. The reaction was conducted under a mild condition (T=40 °C, P=3 bar) and the results are summarized in Table 2. Over the SAC 0.08%Pt/FeOx-R200, the conversion of 3-nitrostyrene attained 95.6% within 49 min (entry 1), which was only 1/17 of the time required to achieve similar conversion level over the Pt/TiO2 catalyst under identical conditions (entry 8). More importantly, the selectivity to 3-aminostyrene was as high as 98.4% at 95.6% conversion. The only byproduct was 3-ethylaniline and no phenylhydroxylamine was detected.
As expected, the pseudo-SAC, 0.08%Pt/FeOx-R250, yielded almost the same activity and selectivity as the 0.08%Pt/FeOx-R200 SAC did, implying that both catalysts possessed the same or similar active Pt centres. By assuming that all the single/pseudo-single Pt atoms were accessible to the reactants, we estimated the TOF of both catalysts (at <20% conversion). The results showed that both catalysts gave a TOF of ~1,500 h−1, 20-fold higher than that of the best reported catalyst Pt/TiO2 (ref. 15). Moreover, when the reaction was carried out at 80 °C the TOF was as high as 11,064 h−1, while the selectivity to 3-aminostyrene remained above 91% (entry 3, Table 2). In contrast, the Pt/TiO2 was reported to give a dramatically decreased selectivity (69.7%) at 85 °C15. For the chemoselective hydrogenation of nitroarene, to the best of our knowledge, our SACs and pseudo-SACs are the most active and selective catalysts ever reported in literature, far outperforming all the reported catalysts, including the IB-group metals5,6,7,8,9,10,11,12,13,14,15 and the modified Pt-group metals20.
The kinetic studies demonstrated that no harmful intermediates such as hydroxylamine and azoxy were produced during the reaction; the only byproduct was 3-ethylaniline (Supplementary Fig. 8). Control test with the bare FeOx support (either Fe2O3 or Fe3O4) under identical conditions did not show any activity at all, highlighting the critical role of the isolated Pt single atoms and pseudo-single atoms for the hydrogenation of nitroarenes.
In contrast to the 0.08%Pt/FeOx SACs and pseudo-SACs, when the Pt loading was between 0.31 and 4.30 wt%, and the reduction treatment was performed at 250 °C, the resultant catalysts exhibited declined activity and selectivity with the increase of the Pt loading (entries 4–7). For example, the 4.30%Pt/FeOx-R250 catalyst gave a TOF of 762 h−1 based on the total amount of Pt and a selectivity of 92.7%. The decreased activity and selectivity should be associated with the presence of 3D particles bigger than 1 nm in diameter where Pt–Pt metallic bonding formed, which reduced the accessibility of Pt atoms on one hand and accelerated the rate of the side reaction C=C hydrogenation on the other hand. It can be found that the change in the activity and selectivity with Pt loading is consistent with that in the white-line intensity of the XANES spectra (Fig. 2), suggesting that the positively charged Pt single and pseudo-single atoms should be the most active sites for the chemoselective reduction.
Interestingly, the selectivity exceeding 90% achieved over the high-loading Pt catalysts (entries 6, 7) was unexpected and in remarkable contrast to the case of Pt/TiO2 or Pt/C where a high selectivity (>90%) was only achieved with a low loading of Pt (0.2 wt%)20. Although the underlying reason is yet to be investigated, the FeOx support may play an important role. As shown in Table 2, even if at a low loading of Pt (0.08 wt%), neither the Pt/SiO2 nor the Pt/Al2O3 could give rise to an acceptable chemo-selectivity (entries 9–10) in spite of the very small Pt particle sizes in those catalysts (Supplementary Fig. 9). According to previous studies11, the nitro group in the substrate could be preferentially adsorbed on the support surface when a basic or reducible support was used. The strong interaction between the Pt single and pseudo-single atoms with the surfaces of the FeOx nanocrystallites plays an important role in determining the selectivity.
To further understand the degree of the metal–support interaction, we conducted temperature-programmed reduction with H2 (H2-TPR). With a decrease of Pt loading, the first peak of H2-TPR shifted to higher temperatures (Supplementary Fig. 10), which is consistent with the XANES result that the Pt species became more positively charged with the reduction of Pt loading. Calculation of the H2 consumption (Supplementary Table 2) revealed that the first peak was not only due to the reduction of Pt(IV) but also of Fe(III) via the spillover of H2. The difference between the total H2 consumption at the first peak and the H2 consumed for Pt(IV) can be considered as an indicator of the extent of H2 spillover; the more extensive H2 spillover, the stronger the metal–support interaction. To provide a quantitative evaluation of the metal–support interaction, herein we define a parameter, the ratio of the moles of Fe3+ reduced to Fe2+, to the moles of Pt4+ to Pt0. As shown in Supplementary Table 2, the ratio increases with a decrease of Pt loading or reduction of Pt cluster size and maximizes at 0.08 wt% Pt loading, indicating that the interaction between single or pseudo-single atoms of Pt with the FeOx support is the strongest among various Pt structures. Such strong interaction favours the chemoselective reduction of nitroarenes.
In addition to the support effect, the nature of the catalytically active metal significantly affects the selectivity of the reaction. As shown in Supplementary Table 3, the Ir1/FeOx SAC with an iridium loading of 0.2 wt% (see also STEM images in Supplementary Fig. 11) gave a conversion of 98.4% with a selectivity to 3-aminostyrene of 98.7% within 240 min, demonstrating the power of single atoms in directing the chemoselective hydrogenation. On the other hand, 0.17 wt% Pd/FeOx catalysed the complete hydrogenation of both –NO2 and –C=C groups, resulting in a poor selectivity to 3-aminostyrene (16.0%) in spite of its high activity. The 0.17 wt% Rh/FeOx catalyst yielded only a moderate selectivity (66.1%), whereas the 0.17 wt% Au/FeOx was totally inactive under such a mild reaction condition. These extensive experimental data clearly demonstrate that with the same type of FeOx nanocrystallites as support material, different metals, even if highly dispersed, behave differently. The appropriate tuning of the metal–support interaction is essential to developing high-performance SACs. For the specific chemoselective hydrogenation of nitroarenes, Pt SACs and pseudo-SACs, among various FeOx-supported noble metals, gave the best activity and selectivity.
To unveil the underlying factors that provide the superior selectivity in the chemoselective hydrogenation of nitroarenes, we performed control experiments on both the SAC 0.08%Pt/FeOx-R200 and the pseudo-SAC 0.08%Pt/FeOx-R250, with styrene and nitrobenzene as the substrates, respectively. As shown in Supplementary Table 4, the TOFs for the hydrogenation of styrene and nitrobenzene over the 0.08%Pt/FeOx-R200 catalyst were 1,453 and 2,414 h−1, respectively, demonstrating that the SAC was intrinsically more active for the nitrobenzene hydrogenation. In contrast, for the pseudo-SAC, the TOFs for the two reactions were comparable (2,377 h−1 versus 2,175 h−1). Clearly, in spite of the absence of strong Pt–Pt metallic bond in the pseudo-SAC, it was still significantly active for C=C hydrogenation due to the clustering of single atoms of Pt (formation of Pt–Ptlong) or the formation of Pt–Fe metallic bond. However, with the presence of both functional groups, that is, when a mixture (1:1) of styrene and nitrobenzene was introduced, the TOF of the pseudo-SAC for styrene hydrogenation dramatically dropped from 2,377 to 139 h−1, while the TOF for the hydrogenation of nitrobenzene was slightly enhanced from 2,175 to 2,538 h−1. A very similar case also occurred for the SAC. If the nitro group preferentially adsorbed on the catalyst surface, resulting significantly suppressed adsorption and the subsequent hydrogenation of the C=C bond, then the above experimental observation can be explained. Similar result was also reported on the Pt/TiO2 catalyst35.
In comparison with homogeneous catalysts, the most outstanding advantage of the heterogeneous catalyst is its capability for easy separation and recycling. The employment of the magnetic FeOx support allows for easy separation of the catalyst from the reaction medium, as shown in Supplementary Fig. 12. The recovered Pt pseudo-SAC, 0.08%Pt/FeOx-R250, can be reused for at least five times without any loss of the selectivity, although the activity has a slight decay. The HAADF-STEM and XANES characterizations of the recycled catalyst (Supplementary Figs 13–14) revealed that both the pseudo-single-atom structure and the oxidation state of Pt remained unchanged, demonstrating the excellent stability of the catalyst.
Substrate tolerance is another important criterion to evaluate the application potential of such catalysts. Our pseudo-SAC showed good tolerance to a broad spectrum of substituted nitroarenes containing –C=C, -C=O, –C≡N and –X functional groups. In most cases, the selectivity to functionalized anilines was >97% at the nitroarene conversion level of >90% (Table 3).
In conclusion, we have developed highly active and chemoselective catalysts for hydrogenation of substituted nitroarenes by tuning the interactions of noble metal, especially Pt, single atoms and pseudo-single atoms with the metal oxide supports, especially FeOx nanocrystallites. The pseudo-single-atom ensembles consisting of several to tens of single atoms that are loosely associated with each other possess unique geometric and electronic properties resembling those of the SAC and provide excellent performance for the chemoselective hydrogenation reactions. Under mild reaction conditions (40 °C, 3 bar H2), both the SACs and pseudo-SACs gave rise to a TOF as high as ~1,500 h−1 and a selectivity to functionalized anilines >98%, with good tolerance to substrates and excellent reusability. The superior performance of these catalysts can be attributed to strong metal–support interaction, resulting in significant electron transfer from the Pt atoms or ensembles to the FeOx support. The presence of positively charged Pt centres, the absence of Pt–Pt metallic bonding and the appropriately reduced metal oxide surfaces favour the preferential adsorption of nitro groups, leading to significantly enhanced performance. The strategy and approach discussed in this paper are general and can be extended to guide the design and development of novel catalysts for a broad spectrum of green synthesis of fine chemicals. This work also demonstrates the potential of single-atom structures in bridging homogeneous and heterogeneous catalysis.
Methods
Preparation of Pt/FeOx catalysts
The Pt/FeOx catalysts were prepared by co-precipitation of an aqueous solution of chloroplatinic acid (H2PtCl6·6H2O, 7.59 × 10−2 mol l−1) and ferric nitrate (Fe(NO3)3·9H2O, 1 mol l−1), with sodium carbonate at 50 °C, with the pH value of the resulting solution controlled at about 8. The recovered solid was dried at 60 °C overnight and calcined at 400 °C for 5 h. The preparation of Pd/FeOx, Rh/FeOx and Au/FeOx catalysts was similar to that of Pt/FeOx. Ir1/FeOx catalyst was synthesized by co-precipitation of an aqueous solution of H2IrCl6 and Fe(NO3)3 with sodium hydroxide at 80 °C, with the pH value of the resulting solution controlled at about 8. The recovered solid was dried at 80 °C overnight without any further heat treatment. Pt/SiO2 and Pt/Al2O3 catalysts were prepared by incipient wetness impregnation. Pt/TiO2 was prepared according to the literature.20 Before characterization and reactivity test, the samples were reduced in 10% H2/He at 200 or 250 °C for 0.5 h.
Measurements of catalytic activities
The hydrogenation of nitroarenes was conducted in an autoclave equipped with pressure control system. Before the activity test, the catalyst was first put into the autoclave, to which 5 ml toluene was added, and then the autoclave was charged with 1 MPa hydrogen and heated at 40 °C for 1 h to allow for the reduction treatment of the catalyst (Supplementary Table 5). After that, toluene was removed and a mixture of nitroarene substrate, toluene and internal standard O-xylene, in a total volume of 5 ml, was put into the autoclave. Next, the autoclave was flushed with 10 bar hydrogen for five times. After being sealed, the autoclave was charged with H2 until 3 bar, then it was heated to 40 °C in a water bath with a magnetic stirrer to initiate the reaction. The H2 pressure decreased gradually during the reaction as a result of hydrogenation. After reaction, the product was analysed by gas chromatography–mass sprctrometry. The TOF value was separately measured by keeping the substrate conversion below 20% and the calculation of the TOF was based on the total Pt loading in the catalyst.
Characterization techniques
The loadings of Pt were measured on inductively coupled plasma spectrometer on an IRIS Intrepid II XSP instrument (Thermo Electron Corp.).
X-ray diffraction analysis was performed on a PANalytical X’ pert diffractometer equipped with a Cu Ka radiation source with a scanning angel (2θ) of 10–80°, operated at 40 kV and 40 mA.
The H2-TPR was performed on an Auto Chem II 2,920 automatic catalyst characterization system (the detection limit was 5 p.p.m. H2). For each measurement, ~50 mg of a catalyst was loaded into a U-shape quartz reactor and purged with He at 150 °C for 0.5 h, to remove adsorbed carbonates and hydrates. Next, after cooling to room temperature the flowing gas was switched to a 10 vol% H2/Ar and the catalyst was heated to 800 °C at a ramping rate of 10 °C min−1.
HAADF-STEM images were obtained on a JEOL JEM-ARM200F equipped with a CEOS probe corrector, with a guaranteed resolution of 0.08 nm. Before microscopy examination, the samples were suspended in ethanol with an ultrasonic dispersion for 5–10 min and then a drop of the resulting solution was dropped onto a holey carbon film supported by a TEM copper grid.
The XAFS spectra at Pt LIII-edge of the samples were measured at beamline 14 W of Shanghai Synchrotron Radiation Facility in China and beamline 17C1 of National Synchrotron Radiation Research Center in Taiwan. The output beam was selected by Si(111) monochromator. The energy was calibrated by the Pt foil. Before measurement, the samples were reduced under 10%H2/He at 200 or 250 °C for 0.5 h, followed by purging with pure He until cooled to room temperature. Next, the samples were evacuated and transferred to glove box without exposure to air. The sample was sealed in Kapton films in the glove box before XAFS measurement. The data were collected at room temperature under fluorescence mode by using solid state detector. Pt foil as standard compound was measured simultaneously by using the third ionization chamber so that energy calibration could be performed scan by scan. Athena software package was employed to process the EXAFS data.
Additional information
How to cite this article: Wei, H. et al. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 5:5634 doi: 10.1038/ncomms6634 (2014).
References
Ono, N. The Nitro Group in Organic Synthesis ed. Feuer H. Wiley-VCH: New York, (2001).
Blaser, H. U., Siegrist, U. & Steiner, H. Aromatic Nitro Compounds: Fine Chemicals Through Heterogeneous Catalysis eds Sheldon R. A., van Bekkum H. Wiley-VCH: Weinheim, (2001).
Downing, R., Kunkeler, P. & Van Bekkum, H. Catalytic syntheses of aromatic amines. Catal. Today 37, 121–136 (1997).
Siegrist, U., Baumeister, P., Blaser, H.-U. & Studer, M. Chemical Industries (ed. Herkes F. 207–220Marcel Dekker: New York, (1998).
Chen, Y., Wang, C., Liu, H., Qiu, J. & Bao, X. Ag/SiO2: a novel catalyst with high activity and selectivity for hydrogenation of chloronitrobenzenes. Chem. Commun. 5298–5300 (2005).
Corma, A. & Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 313, 332–334 (2006).
He, D., Shi, H., Wu, Y. & Xu, B.-Q. Synthesis of chloroanilines: selective hydrogenation of the nitro in chloronitrobenzenes over zirconia-supported gold catalyst. Green Chem. 9, 849–851 (2007).
He, L. et al. Efficient and selective room-temperature gold-catalyzed reduction of nitro compounds with CO and H2O as the hydrogen source. Angew. Chem. Int. Ed. 48, 9538–9541 (2009).
Shimizu, K. I., Miyamoto, Y. & Satsuma, A. Size- and support-dependent silver cluster catalysis for chemoselective hydrogenation of nitroaromatics. J. Catal. 270, 86–94 (2010).
Mitsudome, T. et al. Design of a silver-cerium dioxide core-shell nanocomposite catalyst for chemoselective reduction reactions. Angew. Chem. Int. Ed. 51, 136–139 (2012).
Boronat, M. et al. A molecular mechanism for the chemoselective hydrogenation of substituted nitroaromatics with nanoparticles of gold on TiO2 catalysts: a cooperative effect between gold and the support. J. Am. Chem. Soc. 129, 16230–16237 (2007).
Cárdenas-Lizana, F., Gómez-Quero, S. & Keane, M. A. Exclusive production of chloroaniline from chloronitrobenzene over Au/TiO2 and Au/Al2O3 . ChemSusChem 1, 215–221 (2008).
Liu, L., Qiao, B., Chen, Z., Zhang, J. & Deng, Y. Novel chemoselective hydrogenation of aromatic nitro compounds over ferric hydroxide supported nanocluster gold in the presence of CO and H2O. Chem. Commun. 653–655 (2009).
Shimizu, K. I. et al. Chemoselective hydrogenation of nitroaromatics by supported gold catalysts: mechanistic reasons of size-and support-dependent activity and selectivity. J. Phys. Chem. C. 113, 17803–17810 (2009).
Serna, P., Concepción, P. & Corma, A. Design of highly active and chemoselective bimetallic gold–platinum hydrogenation catalysts through kinetic and isotopic studies. J. Catal. 265, 19–25 (2009).
Wang, A., Liu, X., Mou, C. Y. & Zhang, T. Understanding the synergistic effects of gold bimetallic catalysts. J. Catal. 308, 258–271 (2013).
Westerhaus, F. A. et al. Heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes. Nat. Chem. 5, 537–543 (2013).
Jagadeesh, R. V. et al. Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines. Science 342, 1073–1076 (2013).
Zhao, F., Ikushima, Y. & Arai, M. Hydrogenation of nitrobenzene with supported platinum catalysts in supercritical carbon dioxide: effects of pressure, solvent, and metal particle size. J. Catal. 224, 479–483 (2004).
Corma, A., Serna, P. & Calvino, J. J. Transforming nonselective into chemoselective metal catalysts for the hydrogenation of substituted nitroaromatics. J. Am. Chem. Soc. 130, 8748–8753 (2008).
Yang, X. F. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx . Nat. Chem. 3, 634–641 (2011).
Moses-DeBusk, M. et al. CO oxidation on supported single Pt atoms: experimental and ab initio density functional studies of CO interaction with Pt atom on θ-Al2O3(010) surface. J. Am. Chem. Soc. 135, 12634–12645 (2013).
Fu, Q., Saltsburg, H. & Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 301, 935–938 (2003).
Lin, J. et al. Remarkable performance of Ir1/FeOx single-atom catalyst in water gas shift reaction. J. Am. Chem. Soc. 135, 15314–15317 (2013).
Yang, M., Allard, L. F. & Flytzani-Stephanopoulos, M. Atomically dispersed Au–(OH)x species bound on titania catalyze the low-temperature water-gas shift reaction. J. Am. Chem. Soc. 135, 3768–3771 (2013).
Guo, X. et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 344, 616–619 (2014).
Kwak, J. H., Kovarik, L. & Szanyi, J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 3, 2094–2100 (2013).
Zhang, X. et al. Catalytically active single-atom niobium in graphitic layers. Nat. Commun. 4, 1924 (2013).
Zhang, L. et al. Efficient and durable Au alloyed Pd single-atom catalyst for the ullmann reaction of aryl chlorides in water. ACS Catal. 4, 1546–1553 (2014).
Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209–1212 (2012).
Liu, J. Advanced electron microscopy of metal-support interactions in supported metal catalysts. ChemCatChem 3, 934–948 (2011).
Zhai, Y. et al. Alkali-stabilized Pt-OHx species catalyze low-temperature water-gas shift reactions. Science 329, 1633–1636 (2010).
Sanchez, S. I. et al. The emergence of nonbulk properties in supported metal clusters: negative thermal expansion and atomic disorder in Pt nanoclusters supported on gamma-Al2O3 . J. Am. Chem. Soc. 131, 7040–7054 (2009).
Serna, P., Boronat, M. & Corma, A. Tuning the behavior of Au and Pt catalysts for the chemoselective hydrogenation of nitroaromatic compounds. Top. Catal. 54, 439–446 (2011).
Acknowledgements
We are grateful to the National Science Foundation of China (21176235, 21203182, 21202163, 21303194 and 21373206), the Key Research Program of the Chinese Academy of Sciences, and the Hundred Talents Program of Dalian Institute of Chemical Physics for the financial supports. We also thank Professor Yuying Huang (14W of Shanghai Synchrotron Radiation Facility in China) for his great help in the EXAFS experiment. J.L. acknowledges the start-up fund of the College of Liberal Arts and Sciences of Arizona State University and the use of facilities in the John M. Cowley Center for High Resolution Electron Microscopy at Arizona State University.
Author information
Authors and Affiliations
Contributions
H.W. performed the catalyst preparation, characterizations and catalytic tests. X.L. and L.Z. performed measurements and data analyses of EXAFS. J.L. and S.M. conducted the STEM examinations and contributed to writing the STEM sections. B.Q., X.Y. and Y.H. helped the catalyst preparation and characterization, and discussed the result. A.W. and T.Z. conceived the idea, designed the study, analysed the data and co-wrote the paper. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-14 and Supplementary Tables 1-5 (PDF 3253 kb)
Rights and permissions
About this article

Cite this article
Wei, H., Liu, X., Wang, A. et al. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat Commun 5, 5634 (2014). https://doi.org/10.1038/ncomms6634
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6634
This article is cited by
-
Advances of graphdiyne-supported metal catalysts in thermocatalytic reactions
Nano Research (2024)
-
Atomically dispersed materials: Ideal catalysts in atomic era
Nano Research (2024)
-
Ultimate structures in catalysis: Single atoms, subnano-clusters, and electrons
Science China Materials (2023)
-
Performance Regulation of Single-Atom Catalyst by Modulating the Microenvironment of Metal Sites
Topics in Current Chemistry (2023)
-
Post-synthetic electrostatic adsorption-assisted fabrication of efficient single-atom Fe-N-C oxygen reduction catalysts for Zn-air batteries
Science China Materials (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
