
Abstract
Mitochondrial diseases are systemic, prevalent and often fatal; yet treatments remain scarce. Identifying molecular intervention points that can be therapeutically targeted remains a major challenge, which we confronted via a screening assay we developed. Using yeast models of mitochondrial ATP synthase disorders, we screened a drug repurposing library, and applied genomic and biochemical techniques to identify pathways of interest. Here we demonstrate that modulating the sorting of nuclear-encoded proteins into mitochondria, mediated by the TIM23 complex, proves therapeutic in both yeast and patient-derived cells exhibiting ATP synthase deficiency. Targeting TIM23-dependent protein sorting improves an array of phenotypes associated with ATP synthase disorders, including biogenesis and activity of the oxidative phosphorylation machinery. Our study establishes mitochondrial protein sorting as an intervention point for ATP synthase disorders, and because of the central role of this pathway in mitochondrial biogenesis, it holds broad value for the treatment of mitochondrial diseases.
Similar content being viewed by others

Introduction
Mitochondrial dysfunction has been implicated in a host of diseases with a wide range of symptoms and molecular phenotypes, which present significant challenges to diagnosis, let alone treatment1,2,3,4. Many of these disorders affect mitochondrial ATP synthase, which produces most of the cellular ATP required in humans through the process of oxidative phosphorylation (OXPHOS). Defects in the structure or assembly of this crucial enzyme result in severe diseases that manifest primarily in children, often shortly after birth. Various ATP synthase disorders resulting from defects in the structure or assembly of this enzyme complex have been described, including neuropathy, ataxia and retinitis pigmentosa (NARP), a fatal encephalopathy known as Leigh syndrome, and hypertrophic cardiomyopathy5,6,7. Many of the molecular consequences of these disorders, ranging from deficient ATP production to widespread effects on mitochondrial biogenesis, structure and metabolism have been studied using yeast models, due to the extensive conservation of mitochondrial functions and the ability to manipulate the yeast mitochondrial and nuclear genomes7.
One relevant model of ATP synthase disorders is a strain lacking Fmc1 (fmc1Δ), a factor required for the assembly of the F1 sector of ATP synthase at high temperatures8. Loss of Fmc1 directly and severely impairs ATP synthase assembly, while secondarily impairing the respiratory chain biogenesis and activity as well as mitochondrial membrane potential9. These secondary consequences on the respiratory chain are frequently observed in the context of reduced ATP synthase activity10,11,12,13, and are believed to reflect a regulatory mechanism that maintains a balanced production of ATP synthase and respiratory chain complexes7,12,14. Because of these deficiencies, fmc1Δ yeast display impaired growth on non-fermentable carbon sources like glycerol. Their drastic reduction in fully assembled ATP synthase along with their impaired mitochondrial respiration result in 90% less mitochondrial ATP synthesis8,9. Since these phenotypes are also found in multiple patients15,16, we have leveraged this model system to develop a stringent, high-throughput screening assay for the identification of compounds that suppress ATP synthase deficiency14.
In this study, we apply our screening assay to search for pathways that can be therapeutically targeted for the treatment of ATP synthase disorders. By following up one of the hits using chemical–genomic analysis, we identify the major mitochondrial protein sorting pathway mediated by the inner membrane presequence translocase (TIM23 complex) as a potential intervention point for these disorders. We show that a specific modulation of this pathway, which we recapitulate using a genetic modification, is sufficient to rescue several phenotypes associated with ATP synthase deficiency. Notably, modulating this pathway is also effective in cells derived from ATP synthase-disorder patients, demonstrating the conservation of these therapeutic effects. Our study thus provides the first indication that mitochondrial protein sorting is a promising therapeutic target for the treatment of ATP synthase disorders.
Results and discussion
Sodium pyrithione improves growth of ATP synthase-deficient cells
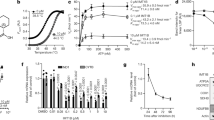
From a drug repurposing library17, our screening assay identified sodium pyrithione (NaPT), a small molecule currently used as an antiseptic. NaPT markedly improved the respiratory growth of fmc1Δ yeast in a dose-dependent manner (Fig. 1a). Notably, this compound was also effective in a cybrid (cytoplasmic hybrid) cell line derived from NARP patients carrying the atp6-T8993G mutation, a mutation also implicated in maternally inherited Leigh’s syndrome7,18. This mutation severely perturbs OXPHOS in both patient tissues and cells derived from them, resulting in extremely poor survival of the latter in glucose-deprived medium18,19,20. Treatment with NaPT improved the survival of atp6-T8993G cybrids, in a dose-dependent manner, up to 1.8-fold, but did not affect control cybrids (Fig. 1b; Supplementary Fig. 1a; Supplementary Data Set 1). These data suggest that NaPT rescues ATP synthase deficiency via a pathway that is conserved between yeast and humans.

(a) NaPT rescues respiratory growth of the fmc1Δ mutant, a yeast model for ATP synthase disorders, in a dose-dependent manner. fmc1Δ cells were spread onto solid rich glycerol medium, indicated amounts of NaPT were spotted onto filters, and plates were incubated at 36 °C for 6 days. High concentrations of NaPT are toxic, as indicated by the regions with no growth immediately surrounding the filters. The halos of growth at dose-dependent distances from the filters correspond to the lower therapeutic concentrations. DMSO (−), compound vehicle. (b) NaPT treatment improves survival of atp6-T8993G (JCP239) cybrids derived from ATP synthase disorder patients in glucose-deprived medium in a dose-dependent manner. Data from three replicates per condition are indicated by points; height of the bar represents the mean. Statistical significance: *P<0.05, (Wilcoxon’s test) relative to the untreated sample (0). DHLA, positive control, drug currently in clinical trials for mitochondrial encephalopathies14,18. (c) The chemical–genomic profile of NaPT identifies mitochondrial protein sorting as a potential target. On treatment of the yeast genome-wide deletion collection with 8.35 μM NaPT, TIM17 and TIM23, essential genes involved in mitochondrial protein import, displayed pronounced deletion sensitivity/haploinsufficiency (calculated relative to the same strain collection treated with DMSO as a negative control; see Methods). Increasing data density is depicted as lighter shades of blue. (d) TIM17-deletion sensitivity is specific to NaPT. Frequency distribution of the TIM17+/− mutant z-scores (see Methods) from 726 published chemical–genomic profiles23 across 332 compounds compared with the score obtained for NaPT.
Chemical-genomic profiling of NaPT-treated cells
To identify the pathway(s) through which NaPT could be rescuing ATP synthase deficiencies, we carried out systematic chemical–genomic profiling using the yeast genome-wide deletion collection (Fig. 1c; Supplementary Data Set 2). In this approach, pronounced sensitivity of heterozygous deletion mutants (that is, haploinsufficiency) to inhibitory concentrations of a chemical indicates the genes and pathways involved in the chemical’s mechanism of action21,22,23. The 10 deletion mutants most sensitive to inhibitory concentrations of NaPT implicated two essential mitochondrial pathways: protein sorting and iron-sulfur cluster biogenesis. The latter is consistent with a previous report that the zinc salt of PT inhibits yeast growth through copper influx and inactivation of iron-sulfur cluster proteins24. The mitochondrial protein sorting pathway was indicated by the genes TIM17 and TIM23, which encode core components of the highly conserved presequence translocase of the inner mitochondrial membrane (TIM23 complex)25,26. The sensitivities of the TIM17+/− and TIM23+/− mutants to NaPT were far greater than their sensitivity to hundreds of previously profiled compounds23 (Fig. 1d; Supplementary Fig. 1b), indicating that the chemical-genetic interaction between NaPT and the TIM23 machinery is specific.
The TIM23 complex mediates the sorting of nuclear-encoded mitochondrial proteins with amino terminal presequences as targeting signals to either the inner mitochondrial membrane or the matrix, which includes many subunits of ATP synthase and respiratory chain complexes27,28,29. Notably, a recent study has demonstrated that the import of respiratory chain proteins into mitochondria via TIM23 can be directly coupled to their assembly into active complexes30. As outlined above, the activity of these respiratory chain complexes is often perturbed in ATP synthase disorders12,13,15,16. Taken together, these observations suggested that the sorting of presequence-carrying proteins into mitochondria could be an attractive therapeutic target for ATP synthase deficiencies.
NaPT modulates mitochondrial protein sorting via TIM23
The expression levels of subunits of the TIM23 complex and the presequence-translocase-associated import motor, which is essential for matrix translocation27,28,29, were not affected by NaPT treatment in wild-type or fmc1Δ cells (Supplementary Fig. 1c). Therefore, to directly test the effect of NaPT on both mitochondrial protein import activities mediated by TIM23, we carried out in vitro import assays using established model substrates targeted to the inner membrane (cytochrome b2-DHFR) or the matrix (cytochrome b2Δ-DHFR, lacking the membrane-sorting signal)31,32 (Fig. 2a,b; Supplementary Fig. 6; Supplementary Data Set 3). In the presence of NaPT, import of matrix-targeted cytochrome b2Δ-DHFR was significantly inhibited (33% slower, P<5.5 × 10−13, Fig. 2a). In contrast, import of inner membrane-targeted cytochrome b2-DHFR was enhanced (46% faster, P<1.3 × 10−8; Fig. 2b). When importing a saturating amount of cytochrome b2Δ-DHFR (using recombinantly expressed purified protein versus the in vitro-synthesized substrate above, see Methods), the impairment of matrix import by NaPT and its dose dependence were striking (Fig. 2c; Supplementary Fig. 6). We verified that NaPT does not disrupt general properties required for mitochondrial protein import, including membrane integrity and inner membrane potential (Δψ) (Supplementary Fig. 2a,b). Moreover, other major mitochondrial protein import pathways, including the assembly of β-barrel proteins into the outer membrane by the SAM (TOB) complex and insertion of proteins with internal import signals into the inner membrane by the TIM22 complex27,28, were not affected by NaPT (Supplementary Fig. 2c,d). We conclude that NaPT does not interfere with the general import competence of mitochondria, but selectively and differentially modulates import of presequence proteins via the TIM23 pathway. These findings are in line with recent studies indicating that the downregulation of mitochondrial protein import via TIM23 can be employed as a stress-responsive mechanism to maintain protein homeostasis in mitochondria33,34.

(a) Import of in vitro-synthesized, radiolabelled cytochrome b2Δ-DHFR, a model protein targeted to the mitochondrial matrix, into isolated yeast mitochondria (p=precursor; i=intermediate) in the presence or absence of 100 μM NaPT. The lower panel displays quantifications of the bands detected by autoradiography in three independent experiments (imported, protease-resistant i-form). The signal obtained in the presence DMSO after the longest incubation time was set to 100%. Error bars represent the s.e.m. Δψ, mitochondrial inner membrane potential. (b) Import of in vitro synthesized, radiolabelled cytochrome b2-DHFR, targeted to the inner membrane, into isolated mitochondria (m=mature). The lower panel displays quantifications of the bands (imported, protease-resistant m-form) as in a. (c) Importing saturating amounts of recombinantly expressed and purified cytochrome b2Δ-DHFR causes a pronounced, concentration-dependent inhibition by NaPT. Import reactions were analysed by immunoblotting using an antibody against DHFR.
To our knowledge, this is the first report of a compound with highly specific effects on TIM23-mediated protein sorting, a central pathway of mitochondrial biogenesis. Nevertheless, we also found that some effects of NaPT on protein sorting in vitro were influenced by the presence of copper (Supplementary Figs. 3,7c–e), which is consistent with previous reports24 and could complicate both the drug’s in vivo effects35 and its viability as a drug development candidate. We therefore sought an independent approach to confirm the role of TIM23-mediated mitochondrial protein sorting in suppressing ATP synthase deficiencies.
Increased Tim21 levels rescue ATP synthase-deficient cells
To study the therapeutic potential of modulating mitochondrial protein sorting in isolation, we applied a genetic modification that recapitulates the effects of NaPT on the TIM23 pathway. Overexpression of Tim21, a regulatory subunit of the TIM23 complex, shifts the activity state of TIM23 from matrix translocation towards inner membrane insertion31,32, which we confirmed in fmc1Δ mitochondria (Fig. 3a,b; wild-type import results, confirmation of Tim21 overexpression and steady-state levels of other mitochondrial proteins in Supplementary Figs 4a–c and 6d,e). We found that Tim21 overexpression caused a substantial restoration of respiratory growth of the fmc1Δ mutant, while wild-type yeast was unaffected (Fig. 3c; Supplementary Fig. 4d). Likewise in human, overexpression of TIMM21 led to a significant increase in the survival of atp6-T8993G NARP patient-derived cybrids (2.4-fold, P<0.05, Fig. 3d; Supplementary Data Set 1; confirmation of TIMM21 overexpression in Supplementary Fig. 1d), while wild-type cybrids were unaffected (Supplementary Fig. 1e). These observations support TIM23’s involvement in mutant rescue and attest to an evolutionarily conserved therapeutic effect of modulating mitochondrial protein sorting on ATP synthase dysfunction.

(a,b) Tim21 overexpression induces NaPT-like modulation of TIM23-mediated mitochondrial protein import. Assays as in Fig. 2a,b were performed with mitochondria isolated from fmc1Δ cells carrying Tim21 overexpression (Tim21↑) versus empty plasmids. Data are representative of three independent experiments. (c) Tim21 overexpression suppresses the respiratory growth defect of fmc1Δ yeast. Strains were grown in liquid rich glycerol media (YPG) at 36 °C until stationary phase; their maximum growth rate (in generations per day, see Methods) is displayed in a boxplot (WT (wild type), n=3; fmc1Δ, n=5). (d) Overexpression of TIMM21, the human ortholog of yeast TIM21, improves cybrid survival. NARP atp6-T8993G cybrids were transduced with lentiviral particles carrying TIMM21 and GFP or RFP alone as a negative control. After 6 days, surviving cells were counted using flow cytometry. Individual replicates are displayed as points; height of the bar represents the mean; * indicates statistical significance (P<0.05, Wilcoxon’s test).
Tim21 promotes OXPHOS biogenesis in a yeast disease model
Another evolutionary parallel between yeast and human Tim21 is their interaction with OXPHOS complexes: the former physically couples respiratory chain supercomplexes to the TIM23 machinery36, and the latter facilitates the assembly of nuclear-encoded subunits into cytochrome c oxidase (complex IV)30. Notably, perturbations in complex IV biogenesis have been observed in ATP synthase disorders10,12. Moreover, we observed that Tim21 overexpression reduced the accumulation of matrix-localized aggregates composed of unassembled ATP synthase subunits in fmc1Δ mitochondria (Supplementary Fig. 4e). Altogether, these findings led us to investigate whether Tim21 could be facilitating the assembly of imported proteins into mature OXPHOS complexes in yeast. We indeed discovered pronounced improvements in OXPHOS complex assembly specific to our disease model (Fig. 4a,b; import controls in Supplementary Figs 5g and 8f,g). Our results demonstrate that Tim21 can promote the biogenesis of deficient OXPHOS complexes, and indicate that the connection between mitochondrial protein import and complex assembly could be an important target for alleviating mitochondrial dysfunction.

(a,b) Tim21 overexpression accelerates the incorporation of subunits Qcr8 and Su h into cytochrome bc1-containing respiratory chain supercomplexes and ATP synthase, respectively, in fmc1Δ mitochondria. Radiolabelled, in vitro-synthesized proteins were imported into isolated mitochondria and their assembly into their respective complexes (indicated with arrows) was followed using BN-PAGE and autoradiography. Δψ, mitochondrial inner membrane potential; III2/IV2, III2/IV1, III2: respiratory chain supercomplexes formed by complex III (cytochrome bc1) and IV (cytochrome c oxidase, COX); V1 and V2, monomeric and dimeric F1Fo-ATP synthase (complex V), respectively. (c) Genetic modulation of mitochondrial protein sorting improves the mitochondrial inner membrane potential (Δψ) of fmc1Δ yeast. The Δψ of indicated mitochondria was monitored using a DiSC3(5) fluorescence quenching assay (see Methods). The difference in fluorescence before and after addition of valinomycin indicates the magnitude of Δψ of the analysed mitochondria. (d) Genetic modulation of mitochondrial protein sorting improves the bioenergetic capacity of fmc1Δ yeast. The following parameters were measured in isolated mitochondria: maximal/uncoupled respiratory rate (upper left), maximal/uncoupled cytochrome c oxidase (COX) activity with ascorbate/TMPD as a substrate (upper right), oligomycin-sensitive ATP hydrolysis using a colorimetric ATPase assay (lower left), and oligomycin-sensitive ATP synthesis using a luciferase assay (lower right). Bar height corresponds to median value of individual measurements displayed as points (both technical and biological replicates). Significance according to a t-test is represented as **P<0.01 or *P<0.05.
Tim21 enhances ATP synthesis in disease model mitochondria
Finally, we investigated the impact of modulating TIM23-dependent mitochondrial protein sorting on the defective bioenergetic properties of fmc1Δ mitochondria that are also found in other mutants with deficient ATP synthase8,9,10,11,12. An improvement in respiratory chain activity in response to Tim21 overexpression was revealed by marked increases in both mitochondrial inner membrane potential (Δψ) and mitochondrial respiration (maximal uncoupled rates in whole cells and isolated mitochondria; Fig. 4c,d; Supplementary Fig. 5i; Supplementary Data Set 4). Consistent with the improvement in OXPHOS complex assembly, the specific activities of cytochrome c oxidase and ATP synthase were also significantly enhanced (Fig. 4d; latter as oligomycin-sensitive ATP hydrolysis, corroborated by in-gel ATPase activity in Supplementary Fig. 5f; Supplementary Data Set 4). These activity increases were reflected in the elevated steady-state levels of OXPHOS complexes (Supplementary Figs 5a–e and 8a–e). Most importantly, mitochondrial ATP synthesis was increased by greater than twofold (Fig. 4d). None of these bioenergetic properties were affected by Tim21 overexpression in wild-type yeast (Supplementary Fig. 5j; Supplementary Data Set 4). Taken together, our findings demonstrate that modulation of mitochondrial protein sorting via the TIM23 pathway alleviates not only a primary ATP synthase deficiency, but also an array of critical downstream phenotypes found in these disorders10,12,15,16.
Conclusions and outlook
Our study reveals mitochondrial protein sorting as a promising new intervention point for the treatment of ATP synthase disorders. This constitutes the first demonstration that this fundamental process central to mitochondrial biogenesis and functionality can be fine-tuned via pharmacological or genetic intervention to suppress disease-associated phenotypes. While a more general inhibition of such a major pathway would likely be detrimental, the slight modulations applied here are beneficial in the context of mitochondrial dysfunction, just as in a recent study that improved survival of a mouse model of an ATP synthase disorder by inhibiting the mTOR signalling pathway37. That we were led to this precise, unexpected mode of action by a large-scale phenotypic screen and a genome-wide mutant profile attests to the power of system approaches for uncovering candidate intervention points. Our findings indicate that the therapeutic benefits of modulating protein sorting for ATP synthase deficiencies are conserved between yeast and humans. Other studies have indicated that moderately downregulating this pathway can help to maintain mitochondrial proteostasis under stressful conditions33,34,38, which along with future work may help to define how exactly the modulations we describe here exert their therapeutic effects. The numerous regulatory factors and mechanisms involved in mitochondrial protein sorting now present new avenues to explore therapeutic strategies for ATP synthase disorders. Due to the fundamental role of this pathway, the therapeutic potential of mitochondrial protein sorting could extend to a variety of diseases involving mitochondrial dysfunction.
Methods
Data analysis
All statistical analyses in this study were performed using R; the specific methods and packages are detailed in the appropriate sections.
Chemical screening assay
This assay was performed as previously described14: 240 μl of exponentially growing fmc1Δ yeast, adjusted to an OD600 of 0.2, was spread homogeneously using sterile glass beads (~3 mm diameter) on a square Petri dish (12 × 12 cm) containing rich solid yeast-peptone-glycerol (YPG) medium (1% (w/v) BD Bacto yeast extract, 2% (w/v) BD Bacto peptone, 3% (v/v) glycerol). Sterile filters were placed on the agar surface, and 2.5 μl of each compound from the Prestwick chemical library dissolved in DMSO was applied to the filters, with DMSO separately applied as a negative control. The plates were incubated at 37 °C for 6 days and scanned using a Snap Scan1212 (Agfa).
Yeast strain generation and in vivo assays
Growth assays with the yeast ATP synthase disorder model strain fmc1Δ (MC6) on rich glycerol media with NaPT (Fig. 1a) were carried out as previously described14. The strain background MC1 was used as wild type (MATa ade2–1 his3–11,15 trp1–1 leu2–3,112 ura3–1 (Δi ER OR); fmc1Δ/MC6 is the same genotype plus Δfmc1::HIS3)8. For Tim21 overexpression, we used the plasmid pYEP352-TIM21, a multicopy expression vector with an insert of the TIM21 gene and its endogenous promoter and terminator regions31. Strains overexpressing Tim21 (and control strains carrying the empty vector pYEP352) were constructed by standard yeast plasmid transformation39, except that the heat shock step was omitted due to the thermosensitivity of the strains and instead, samples were left shaking gently (200 r.p.m.) overnight at room temperature (22 °C). Cells were plated on synthetic glucose media lacking uracil for selection using the URA3 marker; the resulting transformants were then restreaked to obtain single colonies. For growth tests (Fig. 3c and Supplementary Fig. 4), strains were grown overnight at 30 °C in SD-Ura (1.7 g l−1 yeast nitrogen base without amino acids (BD Difco), 20 g l−1 glucose (Sigma), 1.92 g l−1 Yeast Synthetic Dropout Medium Supplement without Uracil (Sigma)). The next day, cells were either: (a) directly transferred to rich glycerol medium (YPG, 5 μl preculture in 100 μl medium) in a 96-well plate (NunclonΔ Surface) and grown at 36 °C in a TECAN GENios multiwell plate reader until saturation (48 h; liquid growth tests, Fig. 3c); or (b) diluted to OD600~0.5 in SD-Ura and grown for 5 hours to reach exponential phase (spot dilutions, Supplementary Fig. 4). The cultures were subsequently diluted to OD600=0.08 and then fivefold serially diluted in SGly-Ura (synthetic glycerol media lacking uracil as above, but with 3% glycerol (v/v) instead of glucose). Five microliters of each cell suspension were spotted on SGly-Ura agar plates that were then incubated for 6–7 days at 35 °C and scanned with a Canon CanoScan LiDE 600F. Growth rates (Fig. 3c) were estimated as the maximum slope of smooth fits of the raw data (local polynomial fit of order 2, bandwidth of 3 h) using the R/Bioconductor cellGrowth package.
Chemical–genomic profiling
The homozygous and heterozygous yeast deletion pools were grown in YPD+25 mM HEPES (pH adjusted to 6.8) supplemented with 1% DMSO (compound vehicle, negative control) or 8.35 μM of NaPT for 5 and 20 generations, respectively, as previously described21. Genomic DNA extraction, PCR amplification of molecular tags and Genflex Tag16k array (Affymetrix) hybridization, washing and scanning were performed as previously described21.
Analysis of chemical–genomic data
Each probe on the Genflex Tag16k array (Affymetrix), known as the Tag4 array, is represented by five replicate features40. Signal intensities were first log2-transformed. Next, each tag was summarized by the median intensity across all (mostly five) matching probes on the array. We then adjusted for overall signal intensity and PCR amplification of up and down tags as follows: For each pool, some tags typically do not yield a signal above background levels, either because the strain has vanished from the pool, it does not grow or it has acquired too many mutations and therefore does not hybridize to the array. Only tag intensities above 8 (a cutoff set from visual inspection of the intensity distributions as separating a low- from a high-intensity subpopulation) were considered above background. The intensity distributions of the up- and down-tag populations in each microarray were shifted by a separate constant so that all intensity distributions above background have equal medians. Differential strain enrichment between DMSO and NaPT treatment was determined using limma’s moderated t-test41 treating tag as an additive covariate to control for difference in hybridization efficiency between up and down tags. P values were corrected for multiple testing with the Benjamini–Hochberg method42, all using the R/Bioconductor package limma. The fold changes calculated can be found in Supplementary Data Set 2.
Comparison to previously generated deletion sensitivity profiles
The z-score for every homozygous or heterozygous deletion strain was computed following the same procedure as in Hillenmeyer et al.23, that is, z=(μctrl–μexp)/σctrl, where μctrl and μexp are the means of the normalized log2 intensities in the DMSO controls and the NaPT (8.35 μM) experiments, respectively, and σctrl is the sample standard deviation of log2 intensities in the controls. The z-scores for the data from this study can be found in Supplementary Data Set 2. These z-scores were then directly compared with those for 726 experiments comprising 332 compounds from profiles in ref. 23.
Isolation of mitochondria from Saccharomyces cerevisiae
Wild-type cells (YPH499; MATa ura3-52 lys2-801_amber ade2-101_ochre trp1-Δ63 his3-Δ200 leu2-Δ1 (ref. 43)) were grown in liquid rich glycerol medium (YPG) at 30 °C. fmc1Δ and corresponding wild-type cells were precultured at 28 °C in rich lactate medium (1% (w/v) BD Bacto yeast extract, 1% (w/v) BD Bacto peptone, 2% (w/v) Lactate, 1% (w/v) KH2PO4, 200 μM adenine, adjusted to pH 5.5 with KOH) and subsequently shifted to 37° for 20–24 h before harvesting. Cells transformed with plasmid pYEP352-TIM21 or pYEP352 were precultured at 28 °C in liquid synthetic minimal medium (0.67% (w/v) yeast nitrogen base, 0.077% CSM amino acid mix minus uracil, 3% (v/v) glycerol, 0.2% (w/v) glucose) to ensure maintenance of the plasmids and then transferred to rich lactate medium at 37 °C for 20–24 h. In vivo NaPT treatment was performed using fmc1Δ and corresponding wild-type cells carrying the empty vector pYEP352. After a 24-h preculture in liquid synthetic minimal medium at 28 °C, 120 nM NaPT was added and the preculture was continued for another 24 h. Subsequently, cells were transferred to rich lactate medium containing 120 nM NaPT and grown at 37 °C for 20 h. Mitochondria were isolated by differential centrifugation44. For the bioenergetic measurements (respiration and ATP synthesis/hydrolysis assays) as well as for Supplementary Fig. 5d–f, a slightly different isolation protocol was used45.
Import of precursor proteins into isolated mitochondria
For generation of radiolabelled Qcr8 and Su h, linear DNA templates with SP6 promoters were synthesized by PCR and transcribed in vitro using the mMESSAGE mMACHINE SP6 kit (Invitrogen, USA). mRNA was purified (MEGAclear kit, Invitrogen) and translated in reticulocyte lysate (Flexi Rabbit Reticulocyte Lysate System, Promega) in the presence of [35S]methionine. [35S]cytochrome b2-DHFR, [35S]cytochrome b2Δ-DHFR, [35S]Tom40 and [35S]AAC were synthesized by coupled in vitro transcription/translation using plasmid templates and the TNT SP6 Quick coupled kit (Promega) in the presence of [35S]methionine. [35S]cytochrome b2-DHFR is a fusion protein consisting of the N-terminal 220 amino acids of the S. cerevisiae cytochrome b2 precursor (including the mitochondrial import signals) fused to mouse dihydrofolate reductase46. On translocation via the TIM23 complex, the N-terminal presequence of this precursor is cleaved by the mitochondrial processing peptidase (MPP), resulting in an intermediate (i) form. Downstream of the N-terminal presequence, [35S]cytochrome b2-DHFR harbours a stop-transfer signal that prevents complete translocation of the protein into the matrix and promotes its integration into the inner mitochondrial membrane, followed by a second processing by the inner membrane protease and release of a soluble mature (m) form into the intermembrane space. [35S]cytochrome b2Δ-DHFR is similar to [35S]cytochrome b2-DHFR but lacks the stop-transfer signal (amino acids 47–65) and is therefore fully translocated across the inner membrane into the matrix and accumulates as the intermediate (i) processing form47. Recombinantly expressed cytochrome b2Δ-DHFR carries the same deletion of the stop-transfer signal as [35S]cytochrome b2Δ-DHFR but is based on a DHFR fusion of a shorter part of cytochrome b2 (amino acids 1–167). The resulting cytochrome b2Δ-DHFR fusion protein was expressed from the plasmid pUHE 73-1 in Escherichia coli strain BMH 71-18 and purified as previously described48. Protein import reactions typically contained 50 μg of isolated mitochondria (total protein amount) diluted in 100 μl import buffer (250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 5 mM methionine, 10 mM KH2PO4, 10 mM MOPS–KOH, pH 7.2, 3% (w/v) bovine serum albumin, 2 mM NADH and 4 mM ATP). No bovine serum albumin was included in import reactions using recombinantly expressed b2Δ-DHFR. Reactions were supplemented with an ATP-regenerating system (20 mM creatine phosphate, 0.1 mg ml−1 creatine kinase) for the import and assembly of Qcr8, Su h, Tom40 and AAC. Samples were prewarmed to 25 °C, incubated with either CuCl2, NaPT or DMSO for 5–15 min where indicated, and import reactions were started by the addition of precursor proteins. Import of Tom40 was stopped by transferring the reactions to ice. Import reactions of all other precursors were terminated by the addition of AVO mix (8 μM antimycin A, 1 μM valinomycin, 20 μM oligomycin; final concentrations)49. Where indicated, non-imported precursors were removed by treatment with 50 μg ml−1 proteinase K (Roche). Mitochondria were washed with SEM buffer (250 mM sucrose, 10 mM MOPS, pH 7.2, 1 mM EDTA) and pelleted by centrifugation. Pellets were either solubilised in Laemmli buffer for analysis by SDS–PAGE or in digitonin buffer (20 mM Tris-HCl, pH 7.4, 0.1 mM EDTA, 10% (v/v) glycerol, 50 mM NaCl, 1% (w/v) digitonin, 1 mM PMSF (Roth)) for analysis by blue native electrophoresis (BN-PAGE). Radiolabelled proteins were detected by digital autoradiography and quantified using the ImageQuant(TM) software (GE Healthcare Life Sciences); quantifications for Fig. 2a,b can be found in Supplementary Data Set 3. Import of the recombinantly expressed b2Δ-DHFR (Fig. 2c) was assessed by western blotting using a polyclonal antibody directed against the DHFR moiety of the preprotein.
Statistical analysis of import dynamics
For each substrate (Fig. 2a,b), three independent import time series were obtained (Supplementary Data Set 3). Within each series, the band intensities corresponding to the imported, protease-resistant proteins were normalized by dividing by the intensity for DMSO at the final time point. Next, a linear model with intercept 0 was fitted by least square regression in the presence or absence of NaPT, separately. The ratio of the fitted slopes represents how the rate of import was affected by the presence of NaPT. To assess statistical significance, the asymptotic likelihood ratio test was applied, comparing the goodness of fit of this model with two slopes against a null model assuming a common slope to both conditions (presence or absence of NaPT).
BN-PAGE of isolated mitochondria
For BN-PAGE analysis50, mitochondrial extracts (starting from 40–100 μg mitochondrial proteins) solubilized with digitonin (digitonin to protein ratio of 10 g/g) were separated on NativePAGE 3–12% Bis-Tris gels (Invitrogen, USA) or on homemade blue native gradient gels (5–10%, 4–16.5% or 6–16.5% polyacrylamide). After electrophoresis, gels were either stained with Coomassie blue, incubated in a solution of 5 mM ATP, 5 mM MgCl2, 0.05% lead acetate, 50 mM glycine–NaOH, pH 8.4 to detect ATPase activity51, or transferred to nitrocellulose or PVDF membranes and analysed by western blotting52. Membranes were first incubated with peptide antibodies against Cox4, Atp4 or F1β, or polyclonal antibodies raised against cytochrome b. Subsequently, they were incubated with peroxidase-conjugated secondary antibodies at a 1:5,000 dilution (Promega or Sigma) and revealed using enhanced chemiluminescence.
Membrane potential measurement
Mitochondria isolated from wild-type (YPH499) yeast or fmc1Δ and the corresponding wild type carrying Tim21 overexpression or empty plasmids were diluted in membrane potential assay buffer (0.6 M sorbitol, 0.1% (w/v) bovine serum albumin, 10 mM MgCl2, 0.5 mM EDTA, 20 mM KPi pH 7.2; supplemented with 5 mM succinate and 5 mM malate for fmc1Δ and corresponding wild type) with or without 150 μM NaPT. The mitochondrial inner membrane potential was determined by measuring fluorescence quenching of the membrane potential-sensitive dye 3,3′-dipropylthiadicarbocyanine iodide [DiSC3(5)] using an Aminco-Bowman series 2 luminescence spectrometer (Thermo Spectronic) at room temperature with an excitation of 622 nm and an emission of 670 nm (ref. 53).
Respiration rates
For whole-cell respiratory activity measurements, cells were resuspended in YP medium at 1 OD600 nm ml−1 and oxygen consumption rates were measured with a Clark electrode. The reactions were started by adding 1% pure ethanol as the substrate. Uncoupled maximal respiratory rates were measured after adding 80 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP). For respiration measurements in isolated mitochondria, oxygen consumption rates were measured with a Clark electrode in the respiration buffer (0.65 M mannitol, 0.36 mM EGTA, 5 mM Tris-phosphate, 10 mM Tris-maleate, pH 6.8) as previously described54: mitochondria (0.15 mg ml−1) were placed in a 1.2-ml thermostatically controlled chamber at 28 °C in respiration buffer. Uncoupled maximal respiratory rates were measured in the presence of 4 mM NADH as substrate and 4 μM CCCP as uncoupler. The specific activity of cytochrome c oxidase was measured using a combination of 12.5 mM ascorbate and 1.4 mM N,N,N',N'-Tetramethyl-p-phenylenediamine dihydrochloride (TMPD) to reduce cytochrome c and in the presence of 4 μM CCCP as uncoupler. All of these measurements can be found in Supplementary Data Set 4.
ATP synthesis and hydrolysis
For ATP synthesis rate measurements, mitochondria were incubated as for oxygen consumption experiments. The reaction was started by the addition of 4 mM NADH and 1 mM ADP and stopped by addition of 3.5% perchloric acid and 12.5 mM EDTA. The samples were then neutralized to pH 6.5 by the addition of KOH and 0.3 M MOPS. ATP was quantified in a luciferin/luciferase assay (Perkin Elmer) with an LKB bioluminometer. ATP hydrolysis rates were measured at pH 8.4 with a previously described procedure55. Oligomycin-sensitive ATP synthesis and hydrolysis rates were measured by adding oligomycin (20 μg mg−1 of protein) to estimate the relative amounts of fully assembled F1F0-ATP synthase. These measurements can be found in Supplementary Data Set 4.
Statistical analysis of bioenergetics data
For measurements of respiration rates (whole-cell and isolated mitochondria), specific cytochrome c oxidase activity, and ATP synthesis and hydrolysis, statistical significance of the differences between strains carrying the Tim21 overexpression versus empty plasmid was assessed using a one-sided t-test assuming equal variance in both the groups. Two biological replicates were measured per strain. Technical replicates, which displayed lower variance than biological replicates, were summarized by their median value before testing. The individual measurements can be found in Supplementary Data Set 4.
Human cell lines and culture conditions
The cybrid survival assays with NaPT treatment in Fig. 1b and Supplementary Fig. 1e were performed as previously described14: cell lines were first cultivated in high-glucose DMEM, after which the medium was removed and survival measurements were carried out in glucose-deprived DMEM supplemented with the indicated concentrations of NaPT, dihydrolipoic acid (DHLA;+control)18 or DMSO (− control); no antibiotics were used. Survival assays started with 3.5 × 104 JCP239 eb13.13 (NARP atp6-T8993G) cells18 and 2 × 104 JCP213 (control) cells being plated in 12-well plates containing DMEM with high glucose (4.5 g l−1) for 24 h. For each treatment condition, three wells were used. NaPT solutions in DMSO were diluted 1:1,000 in the medium. DHLA at 100 μM or the same volume of DMSO were used as positive and negative controls, respectively. After 6 days of incubation with the drugs, cell proliferation was estimated by cell counting using the Scepter (Millipore). Experiments were performed at least three times per condition and the values obtained can be found in Supplementary Data Set 1. Cell counts in each condition were expressed as a proportion of cell counts in DMSO control experiments (‘Percentage of live cells (relative to untreated)’). For statistical analysis, an analysis of variance model was fitted with intercept at 100% growth, and significance of the effect for each concentration was assessed by the t-test using the R package lmtest. TIMM21 overexpression experiments were performed using JCP239 eb13.13 and JCP213 cybrid cell lines transduced with lentiviral constructs overexpressing TIMM21 (pLOC-Tim21 OHS5897-101185497) or RFP as control (pLOC-RFP-control OHS5832), both provided by Open Biosystems. HIV1-based lentiviruses were produced in HEK293FT cells according to ref. 56. Both cell lines were transduced with a multiplicity of infection of 1. At 4 days post transduction, cells carrying the different constructs were seeded separately in six-well plates (105 cells per well) containing DMEM with glucose. After 24 h of growth, three wells for each line were used to estimate the total number of cells per well using a C6 flow cytometer (Accuri). The medium was removed from remaining wells and cells were further incubated with DMEM without glucose (as described above). After 6 days of incubation, cell proliferation/survival was estimated by cell counting using a C6 flow cytometer (Accuri). Experiments were performed at least three times per condition and three different wells were counted for each line in each experiment; the values obtained can be found in Supplementary Data Set 1. Cell counts in each condition were expressed as a proportion of the cell counts following the first 24 h of growth in Fig. 3d. To confirm TIMM21 overexpression, 10 μg protein of cell lysates from JCP239 cells transduced with overexpression constructs were separated on SDS–PAGE and subjected to western blot analysis using anti-TIMM21 (anti-C18orf55) affinity-purified rabbit polyclonal antibodies (Proteintech Europe) and revealed by enhanced chemiluminescence.
Additional information
How to cite this article: Aiyar, R. S. et al. Mitochondrial protein sorting as a therapeutic target for ATP synthase disorders. Nat. Commun. 5:5585 doi: 10.1038/ncomms6585 (2014).
References
Wallace, D. C., Fan, W. & Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. Mech. Dis. 5, 297–348 (2010).
Nunnari, J. & Suomalainen, A. Mitochondria: in sickness and in health. Cell 148, 1145–1159 (2012).
Schon, E. A., DiMauro, S., Hirano, M. & Gilkerson, R. W. Therapeutic prospects for mitochondrial disease. Trends Mol. Med. 16, 268–276 (2010).
Calvo, S. E. & Mootha, V. K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44 (2010).
Houstek, J. et al. Mitochondrial diseases and genetic defects of ATP synthase. Biochim. Biophys. Acta 1757, 1400–1405 (2006).
Jonckheere, A. I., Smeitink, J. A. & Rodenburg, R. J. Mitochondrial ATP synthase: architecture, function and pathology. J. Inherit. Metab. Dis. 35, 211–225 (2012).
Kucharczyk, R. et al. Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches. Biochim. Biophys. Acta 1793, 186–199 (2009).
Lefebvre-Legendre, L. et al. Identification of a nuclear gene (FMC1) required for the assembly/stability of yeast mitochondrial F1-ATPase in heat stress conditions. J. Biol. Chem. 276, 6789–6796 (2001).
Lefebvre-Legendre, L. et al. Failure to assemble the α3β3 bubcomplex of the ATP synthase leads to accumulation of the α and β subunits within inclusion bodies and the loss of mitochondrial cristae in Saccharomyces cerevisiae. J. Biol. Chem. 280, 18386–18392 (2005).
Rak, M. et al. A yeast model of the neurogenic ataxia retinitis pigmentosa (NARP) T8993G mutation in the mitochondrial ATP synthase-6 gene. J. Biol. Chem. 282, 34039–34047 (2007).
Kucharczyk, R., Salin, B. & di Rago, J.-P. P. Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum. Mol. Genet. 18, 2889–2898 (2009).
Soto, I. C. et al. Synthesis of cytochrome c oxidase subunit 1 is translationally downregulated in the absence of functional F1F0-ATP synthase. Biochim. Biophys. Acta 1793, 1776–1786 (2009).
Trounce, I., Neill, S. & Wallace, D. C. Cytoplasmic transfer of the mtDNA nt 8993 T-->G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc. Natl Acad. Sci. USA 91, 8334–8338 (1994).
Couplan, E. et al. A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl Acad. Sci. USA 108, 11989–11994 (2011).
De Meirleir, L. et al. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 41, 120–124 (2004).
Cízková, A. et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 40, 1288–1290 (2008).
Wermuth, C. G. Selective optimization of side activities: another way for drug discovery. J. Med. Chem. 47, 1303–1314 (2004).
Mattiazzi, M. et al. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 13, 869–879 (2004).
Houstek, J., Andersson, U., Tvrdik, P., Nedergaard, J. & Cannon, B. The expression of subunit c correlates with and thus may limit the biosynthesis of the mitochondrial F0F1-ATPase in brown adipose tissue. J. Biol. Chem. 270, 7689–7694 (1995).
Geromel, V. et al. Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the neurogenic ataxia retinitis pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA. Hum. Mol. Genet. 10, 1221–1228 (2001).
Hoon, S. et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol. 4, 498–506 (2008).
Ho, C. H. H. et al. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat. Biotechnol. 27, 369–377 (2009).
Hillenmeyer, M. E. et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365 (2008).
Reeder, N. L. et al. Zinc pyrithione inhibits yeast growth through copper influx and inactivation of iron-sulfur proteins. Antimicrob. Agents Chemother. 55, 5753–5760 (2011).
Hoogenraad, N. J., Ward, L. A. & Ryan, M. T. Import and assembly of proteins into mitochondria of mammalian cells. Biochim. Biophys. Acta 1592, 97–105 (2002).
Dolezal, P., Likic, V., Tachezy, J. & Lithgow, T. Evolution of the molecular machines for protein import into mitochondria. Science 313, 314–318 (2006).
Neupert, W. & Herrmann, J. M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723–749 (2007).
Schmidt, O., Pfanner, N. & Meisinger, C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 11, 655–667 (2010).
Endo, T., Yamano, K. & Kawano, S. Structural insight into the mitochondrial protein import system. Biochim. Biophys. Acta 1808, 955–970 (2011).
Mick, D. U. et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 151, 1528–1541 (2012).
Chacinska, A. et al. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 120, 817–829 (2005).
Popov-Celeketić, D., Mapa, K., Neupert, W. & Mokranjac, D. Active remodelling of the TIM23 complex during translocation of preproteins into mitochondria. EMBO J. 27, 1469–1480 (2008).
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M. & Haynes, C. M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590 (2012).
Rainbolt, K. K., Atanassova, N., Genereux, J. C. & Wiseman, L. L. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 18, 908–919 (2013).
Schlecht, U. et al. A functional screen for copper homeostasis genes identifies a pharmacologically tractable cellular system. BMC Genomics 15, 263 (2014).
Van der Laan, M. et al. A role for Tim21 in membrane-potential-dependent preprotein sorting in mitochondria. Curr. Biol. 16, 2271–2276 (2006).
Johnson, S. C. et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528 (2013).
Pellegrino, M. W., Nargund, A. M. & Haynes, C. M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416 (2013).
Gietz, R. D. & Schiestl, R. H. Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 1–4 (2007).
Pierce, S. E. et al. A unique and universal molecular barcode array. Nat. Methods 3, 601–603 (2006).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 (2004).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995).
Sikorski, R. S. & Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 (1989).
Meisinger, C., Pfanner, N. & Truscott, K. N. Isolation of yeast mitochondria. Methods Mol. Biol. 313, 33–39 (2006).
Guérin, B., Labbe, P. & Somlo, M. Preparation of yeast mitochondria (Saccharomyces cerevisiae) with good P/O and respiratory control ratios. Methods Enzymol. 55, 149–159 (1979).
Voos, W., Gambill, B. D., Guiard, B., Pfanner, N. & Craig, E. A. Presequence and mature part of preproteins strongly influence the dependence of mitochondrial protein import on heat shock protein 70 in the matrix. J. Cell Biol. 123, 119–126 (1993).
Chacinska, A. et al. Distinct forms of mitochondrial TOM-TIM supercomplexes define signal-dependent states of preprotein sorting. Mol. Cell. Biol. 30, 307–318 (2010).
Dekker, P. J. T. et al. The Tim core complex defines the number of mitochondrial translocation contact sites and can hold arrested preproteins in the absence of matrix Hsp70–Tim44. EMBO J. 16, 5408–5419 (1997).
Stojanovski, D., Pfanner, N. & Wiedemann, N. Import of proteins into mitochondria. Methods Cell Biol. 80, 783–806 (2007).
Schägger, H. & von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231 (1991).
Grandier-Vazeille, X. & Guérin, M. Separation by blue native and colorless native polyacrylamide gel electrophoresis of the oxidative phosphorylation complexes of yeast mitochondria solubilized by different detergents: specific staining of the different complexes. Anal. Biochem. 242, 248–254 (1996).
Arselin, G., Vaillier, J., Graves, P. V. & Velours, J. ATP synthase of yeast mitochondria. isolation of the subunit h and disruption of the ATP14 gene. J. Biol. Chem. 271, 20284–20290 (1996).
Geissler, A. et al. Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b2 modulates the Δψ-dependence of translocation of the matrix-targeting sequence. Mol. Biol. Cell 11, 3977–3991 (2000).
Rigoulet, M. & Guerin, B. Phosphate transport and ATP synthesis in yeast mitochondria: effect of a new inhibitor: the tribenzylphosphate. FEBS Lett. 102, 18–22 (1979).
Somlo, M. Induction and repression of mitochondrial ATPase in yeast. Eur. J. Biochem. 5, 276–284 (1968).
Zufferey, R. et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72, 9873–9880 (1998).
Acknowledgements
We thank I. Perschil, P. Jakob, S. Clauder-Münster, N. Ezkurdia, J. Kirkpatrick and J. Krijgsveld for technical assistance; as well as N. Pfanner, B. Guiard, G. Manfredi, C. Hughes and A. Chacinska for materials and discussion. We would like to dedicate this work to the memory of Andy Robertson, who not only provided useful comments on the manuscript, but also contributed tirelessly to professional development and diversity in the biological sciences. This study was technically supported by the European Molecular Biology Laboratory’s Electron Microscopy Core Facility and the Stanford Genome Technology Center. This work was funded by grants from ANR (Agence Nationale de la Recherche), AFM (Association Française contre les Myopathies) and FRM (Fondation pour la Recherche Médicale, 2011–13) to J.-P.d.R. and M.Bl.; National Science Center of Poland (1932/B/P01/2010/39) to R.K.; National Institutes of Health (P01HG000205 and R01GM068717) and a European Research Council Advanced Investigator Grant (ERC grant agreement no. AdG-294542) to L.M.S.; Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 746 and the Excellence Initiative of the German Federal and State Governments (EXC 294 BIOSS) to M.v.d.L. and a Boehringer-Ingelheim Fonds predoctoral fellowship to M.Bo. S.S. and R.P.S. were funded by grants from the National Human Genome Research Institute (P01HG000205 and R01HG003317).
Author information
Authors and Affiliations
Contributions
R.S.A., M.v.d.L., M.Bo., J.P.d.R., M.Bl., C.V., S.D.-C., C.F., C.A. and L.M.S. designed the research; S.S. and R.P.S. generated the chemical–genomic profiles, which were analysed by J.G., R.S.A. and R.P.S.; R.S.A., M.Bo. and K.v.d.M. performed the import, assembly and biochemical assays; R.S.A., E.C., C.V., E.S.F., F.S., J.E., M.Bo. and K.v.d.M. performed the in vivo yeast work; R.S.A. and R.K. generated yeast strains; C.F. performed the electron microscopy with the help of R.S.A. and both analysed it with the help of J.G.; C.V. and F.S. assayed cybrid survival in the presence of NaPT; S.D.-C. and F.C. performed the cybrid transduction and survival assays; J.G., S.D.-C., and C.V. analysed all the cybrid data; S.D.-C., F.C. and M.Bo. performed the assays of mitochondrial bioenergetics; R.S.A., M.v.d.L., J.P.d.R., M.Bl., C.A. and L.M.S. supervised the research; and R.S.A., M.v.d.L., J.P.d.R. and L.M.S. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8, Supplementary Methods and Supplementary References (PDF 21023 kb)
Supplementary Data 1
Raw counts of surviving atp6-T8993G and control cybrids in response to NaPT treatment or TIMM21 overexpression (Figs. 1b, 3d). (XLS 50 kb)
Supplementary Data 2
Systematic chemical-genomic profiles derived from the yeast homozygous and heterozygous deletion collection treated with NaPT (Fig. 1). The z-scores as computed in Hillenmeyer et al.3, fold changes as described in Methods, and gene annotation according to the Saccharomyces Genome Database are provided, in tab-delimited format. (TXT 2935 kb)
Supplementary Data 3
Intensities of bands detected by digital autoradiography from SDSPAGE gels corresponding to radiolabeled mitochondrial proteins imported via the TIM23 complex (Fig. 2a-b), in tab-delimited format. (TXT 1 kb)
Supplementary Data 4
Measurements of mitochondrial respiration (whole-cell and isolated mitochondria), cytochrome c oxidase-specific activity, ATP synthesis, and ATP hydrolysis in fmc1Δ yeast overexpressing Tim21, in tab-delimited format. (TXT 4 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Aiyar, R., Bohnert, M., Duvezin-Caubet, S. et al. Mitochondrial protein sorting as a therapeutic target for ATP synthase disorders. Nat Commun 5, 5585 (2014). https://doi.org/10.1038/ncomms6585
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6585
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
