
Abstract
The midge, Belgica antarctica, is the only insect endemic to Antarctica, and thus it offers a powerful model for probing responses to extreme temperatures, freeze tolerance, dehydration, osmotic stress, ultraviolet radiation and other forms of environmental stress. Here we present the first genome assembly of an extremophile, the first dipteran in the family Chironomidae, and the first Antarctic eukaryote to be sequenced. At 99 megabases, B. antarctica has the smallest insect genome sequenced thus far. Although it has a similar number of genes as other Diptera, the midge genome has very low repeat density and a reduction in intron length. Environmental extremes appear to constrain genome architecture, not gene content. The few transposable elements present are mainly ancient, inactive retroelements. An abundance of genes associated with development, regulation of metabolism and responses to external stimuli may reflect adaptations for surviving in this harsh environment.
Similar content being viewed by others

Introduction
Loss of the land bridge between South America and the Antarctic Peninsula isolated the southernmost continent ~33 million years ago1, yielding a cold, desert environment inhospitable to most forms of terrestrial life. Although the surrounding ocean nurtures an abundance of marine life, and offshore islands offer summer breeding grounds for birds and seals, few animals are found year-round in Antarctica’s terrestrial habitat. Insects, the dominant life form on most continents, are represented by a single endemic Antarctic species, a wingless midge, Belgica antarctica (Diptera: Chironomidae)2,3, a species first noted by a naturalist aboard the S.Y. Belgica, a Belgian exploratory ship that plied the waters off the Antarctica Peninsula at the end of the 19th century4. In its patchy island habitat along the Antarctic Peninsula, B. antarctica is subjected to a range of environmental onslaughts including temperature extremes, periodic desiccation, exposure to both fresh water ice melt and high-salinity sea water, intense ultraviolet exposure, high nitrogen generated from penguin rookeries and elephant seal wallows and high winds5,6,7. The adults, like those of many other species living on wind-swept islands, are apterous (wingless). The larvae (Fig. 1a), encased in ice for most of the year, require 2 years to complete their development and then pupate and emerge as adults (Fig. 1b) at the beginning of their third austral summer. The apterous adults crawl over surfaces of rocks and other substrates, mate, lay eggs and die within 7–10 days after emergence.

Fourth (final) instar larvae of the Antarctic midge B. antarctica (a) and an adult male (b). This is the southernmost insect and the only insect species endemic to Antarctica. Larval length is 6–7 mm, and the adult male is ~3 mm. Photo by Richard E. Lee Jr.
Unusual adaptations, including winglessness, freeze tolerance, severe desiccation tolerance and constitutive expression of heat shock proteins8, allow this fly to survive in the inhospitable climate of rocky outcrops along the Antarctic Peninsula. The genome of this fly can be expected to offer insights into genomic processes and genome evolution essential to its survival. At the molecular level, select genes that have been examined in B. antarctica include those encoding heat shock proteins8, the antioxidant enzymes catalase and superoxide dismutase7, a collection of genes that responded to changes in hydration state9,10 and an aquaporin11. It is clear from these molecular studies that this species displays some unusual patterns of gene expression. For example, unlike most organisms, the messenger RNAs encoding heat shock proteins, catalase and superoxide dismutase are expressed at high levels all the time, not just in response to a sudden stress. Interestingly, there are also Antarctic fish species that have constitutive expression of heat shock protein 70 (refs 12, 13). The novelty of these responses suggests there are unique genomic adaptations to cope with extreme environments.
The genome we present for the extremophile B. antarctica is the first for a dipteran in the family Chironomidae; it consists of 99 megabase pairs assembled using over 100-fold depth coverage of the genome. It is the smallest insect genome discovered thus far. This unusually small genome has low repeat content and general lack of transposable elements (TEs), which are mainly limited to retroelements. The gene content is similar to other Diptera; however, intron length and repeat elements are greatly reduced. Genes that are abundant compared with the related dipteran Anopheles gambiae are associated with development, regulation of metabolism and responses to external stimuli. The genome provides a foundation for studying extremophile biology and insect genome evolution.
Results
The genome of B. antarctica is the smallest yet reported for an insect. The estimate of total genome size based on flow cytometry is 1C=99.25±0.4 megabase pairs (Mbp) for the female and 1C=98.4±0.1 Mbp for the male (Supplementary Methods; Supplementary Fig. 1; Supplementary Table 1). On the basis of the raw sequence reads, we estimate the size of the B. antarctica genome to be >89.5 Mbp but <105 Mbp (Supplementary Fig. 2). Previous cytological preparations of polytene chromosomes from salivary glands indicate B. antarctica has three linkage groups (2n=6)14. We used a single larva of B. antarctica of unknown sex from Cormorant Island, near Palmer Station, Antarctica for the reference genome, using Illumina sequencing technology and Velvet de novo15 for assembly. Several assemblers were compared (Supplementary Table 2). Paired-end reads from RNA-seq data10 were used to improve the assembly by scaffolding contigs, resulting in 5,064 scaffolds. One Pacific Biosciences RSII SMRTbell library was generated to scaffold the assembly, which added minimal scaffolding owing to the limited amount of DNA in a single individual. The size of the assembled genome was 89.6 Mbp, including ambiguous bases; this represents over 90% of the total genome (Table 1). The assembly consists of 5,003 contigs >300 bp (Supplementary Fig. 3), with an N50 contig length of 98.2 kilobases (kb) and an average coverage of × 177 (Supplementary Fig. 4). A total of 83.89 Mbp (93.7% of the assembled genome) was contained in 1,256 contigs >10 kb. The longest contig assembled was over 622 kb. These multiple lines of evidence as well as the identification of nearly all (97%) core eukaryotic genes suggest a high-quality assembled genome (see also Supplementary Methods).
The genome of B. antarctica is smaller than even the tiny genomes reported for the body louse (104.7 Mbp) and Strepsiptera (108 Mbp)16,17. Previously published genome size estimates for three chironomid species, as well as new flow cytometry estimates for three additional members of the family Chironomidae (1C=108–118 Mbp), further suggest that the B. antarctica genome is small even for a chironomid (Supplementary Methods; Supplementary Table 1). The small genome found in this Antarctic midge does not conform to the coupling frequently reported between low temperatures and large genomes18, thus suggesting that alternative evolutionary forces are driving the small size of this genome. The only other Diptera with genomes near 100 Mbp are Colboldia fuscipes (Scatopsidae) and Psycoda cinerea (Psychodidae), cosmopolitan species whose genomes sizes may be constrained by early developmental traits19.
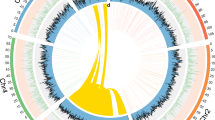
Amplification, deletion and rearrangements of repeated DNA sequences may account for intraspecific variations in genome size20. In B. antarctica the small size of the genome is a function of a paucity of repeats, including a reduction in the number of TEs and the reduced length of introns (Fig. 2). Analysis of the repeat content of the genome assembly revealed that repeat elements comprise only 0.49% of the assembled genome and 10% of the entire genome, assuming that the discrepancy between the assembled genome size and the flow cytometry estimate is due to repeat elements. Most of the repeat elements we identified were found in low-complexity sequences (Table 2; Supplementary Methods; Supplementary Data 1; Supplementary Tables 3–5). Using known TE libraries and examining raw reads, we estimate that only 0.016% of the genome failed to assemble due to TE insertions. Furthermore, no species-specific TEs were detected (Supplementary Methods). The B. antarctica genome has ~0.12% of the genome as TEs, a small proportion compared with Aedes aegypti (47%)21, Anopheles gambiae (16%)22, Culex quinquefasciatus (29%)23 and Drosophila melanogaster (20%)24,25. In contrast to the above, the body louse Pediculus humanus humanus, similar to B. antarctica, has a small genome (1C=105 Mbp) associated with a low TE proportion (1.03% of genome)17.

Each panel is ordered with respect to overall assembled genome size. The four panels represent the total amount of sequence in each annotation: genome size, intron, coding sequence (CDS) and transposable elements (TEs).
The TEs found in the B. antarctica genome were of multiple origins. The TEs represent 154 TE families from the three main TE orders (DNA elements, retroelements with long terminal repeat (LTR) and non-LTR retroelements). A total of 513 TE insertion locations were identified in the assembled genome (Table 2). Of those 513 TE insertions, 74 were nested with >1 TE insertion, while the remaining 439 clearly correspond to unique TE insertions. An additional 23 TE insertions were detected as absent from the assembly, as they were located at the flanking regions of the contigs. Most of the unique insertions are from retroelements. The reduced number of TEs in the genome was reflected in ribosomal genes. R1 and R2 non-LTR retroelements are present in nearly all arthropods and have been identified in the ribosomal DNA (rDNA) loci of nearly all arthropod lineages examined to date26. However, based on reconstruction of the rDNA region, B. antarctica lacks both R1 and R2 non-LTR retroelements. All lines of evidence suggest that the TE insertions in B. antarctica are of ancient origin.
Approximately 19.4% (just under 19 Mbp) of the genome is protein coding in B. antarctica and contains 97% of the core eukaryotic genes (Supplementary Methods; Supplementary Tables 6 and 7). A large proportion of the genome is coding in comparison with Ae. aegypti (22 Mbp, 1.6% of the genome), An. gambiae (20.7 Mbp, 7.6%), C. quinquefasciatus (24.9 Mbp, 4.3%) and D. melanogaster (22.8 Mbp, 13.6%) (Fig. 2). A total of 13,517 protein-coding genes were annotated, underscoring that loss of gene function is not driving the small genome of B. antarctica. On the basis of a cox1 sequence data, our sample clusters with other samples collected at the same location (Supplementary Fig. 5). Of the 13,517 gene models, 12,914 gene models were supported by at least one RNA-seq read, and 9,011 models were supported by at least 100 RNA-seq reads. Among the annotated genes, 8,575 genes have unique alignments to entries in the SwissProt database, and 10,557 genes have matches in the non-redundant database. Genes are clustered in regions of relatively high GC content (GC content of coding regions is 47%, compared with a 37% GC content for the non-coding, Supplementary Table 8).
We compared the B. antarctica genome with that of four other dipteran species, three mosquitoes, Ae. aegypti, An. gambiae, C. quinquefasciatus and D. melanogaster, the insect with the most completely annotated genome. Overall, B. antarctica has an intermediate genome GC content but a lower coding GC% than any of the other four Diptera (Supplementary Table 8). Analysis of codon usage suggests that B. antarctica is not unusual compared with the other four dipteran species (Supplementary Fig. 6). Potential clusters of orthologous genes for comparative analyses were determined using annotations from the four dipteran species (Fig. 3). In an orthoMCL27 comparison between the five species, 4,910 genes were unique to B. antarctica, and 3,582 one-to-one orthologous genes between all five species were identified. Given the lack of TEs in the B. antarctica genome, we interrogated the PIWI-interacting RNA (piRNA) pathway genes (Supplementary Methods). The piRNA pathway serves to control transposon activity28. We identify several key players in the piRNA pathway that are absent from the B. antarctica genome (Supplementary Table 9).

Venn diagram of orthologous gene clusters among B. antarctica, An. gambiae, Ae. aegypti, C. quinquefasciatus and D. melanogaster. The numbers in each area indicate the number of orthologous gene clusters in each category, and the numbers in parentheses indicate the total number of genes in each area. The Venn diagram was generated at http://bioinformatics.psb.ugent.be/webtools/Venn/.
Intron size distribution was compared with protein-coding length distribution, calculated for the one-to-one orthologs as well as all genes for each of the five dipteran species (Supplementary Tables 10–12; Supplementary Fig. 7). The comparison showed that reduction in intron length also contributed to the reduced size of this genome.
While the number of genes is consistent with other Diptera, the relative proportion in different ontologies varies between B. antarctica and An. gambiae. Gene ontology (GO) terms were assigned to the gene models for B. antarctica and the published genes of An. gambiae using Blast2GO29; this yielded 8,856 and 8,653 genes, respectively, with at least one GO term. A comparison of the gene sets using Fisher’s exact test revealed 162 GO terms positively enriched in B. antarctica and 20 terms negatively enriched (Supplementary Data 2). Many of the positively enriched terms fall into two broad categories, ‘development’ (38 terms) and ‘regulation of biological processes’ (50 terms).
The effective population size of B. antarctica has been decreasing over the past 10,000 years. Mapping reads onto the assembled genome allowed us to identify 195,860 of the 88,780,579 non-repeat-masked base pairs in the assembled genome as putative heterozygotes; this is an ~0.2% heterozygosity rate, which suggests an average of one heterozygous position for every 450 bp in this single individual (in contrast D. melanogaster has an order of magnitude more heterozygosity at ~2%). The result is similar (~185,000 single-nucleotide polymorphisms (SNPs) in 83.89 Mbp) when the analysis is limited to contigs greater than 10 kb. Using the D. melanogaster single-nucleotide mutation rate of 8.4 × 10−9 per site per generation (ref. 30), we estimate that the time-averaged effective population size of B. antarctica is ~60,000 diploid individuals. We used a pairwise sequentially Markovian coalescent analysis31 to infer population changes from a single individual to make inferences about population change over time (Fig. 4). Assuming that the mutation rate estimated for B. antarctica is correct, the analysis suggests that the population reached a population size that was maximum just prior to the last glacial maximum, suggesting that the midge populations declined markedly at the glacial maximum but survived in refugia during the period of extensive glaciation. The use of alternate mutation rates would, of course, shift the estimates either higher or lower (Supplementary Figs 8 and 9). Our work is consistent with current hypotheses on Antarctic arthropod dispersal, indicating that most endemic species established well before the last glacial maximum and survived in isolated refugia during glacial periods32. Moreover, low levels of genetic diversity suggest a small effective population size, implying that strong selective pressure drove the fixation of adaptive alleles underlying these unique features of the midge genome.

Pairwise sequentially Markovian coalescent (PSMC) analysis for inferred historical population sizes using variant data from the sequenced diploid individual using a mutation rate of 0.84 × 10−8 per site per generation. Populations of B. antarctica reached a maximum size coinciding with the glacial maximum at 20,000 years ago (vertical line on graph). The x axis gives time measured by pairwise sequence divergence and the y axis gives the effective population size measured by the scaled mutation rate. The green lines correspond to PSMC inferences on 100 rounds of bootstrapped sequences, while the red line corresponds to the estimate from the data.
Discussion
The small genome size of B. antarctica is achieved by a reduction in repeats, TEs and intron size, a result similar to that reported for the bladderwort plant (Utricularia gibba)33 and pufferfish (Tetraodon nigroviridis)34, whose small genomes are also attained by a great reduction in non-genic DNA. Intron size has been correlated to TE number in D. melanogaster35, suggesting that in B. antarctica small intron size may be a result of few TEs. There is strong evidence that DNA-mediated transposons (class II) are derived from horizontal gene transfer36,37; we hypothesize that horizontal gene transfer events are likely rare in the species-poor Antarctic environment, thus reducing the presence of TEs in Antarctic genomes. While gene content remains largely conserved, the absence of TEs does, however, have a major impact on certain gene classes (for example, the piRNA pathway genes). Moreover, there is evidence that the expression of Hsp90 may be a suppressor of TE movement through either direct interaction with piRNA biogenesis or transposon activation38. The constitutive expression of Hsp90 in B. antarctica larvae8 may also contribute to the lack of active TEs in the genome.
Among negatively enriched terms, ‘odourant binding’ suggests a loss of sensory perception in B. antarctica, a feature that may reflect the limited food choices available to the midge in its Antarctic environment, as well as its mating behaviour that does not involve long-distance orientation. By contrast, the mosquito relies extensively on odour reception for the complicated challenges of finding vertebrate hosts and floral nectar sources. It is worth noting that any hypotheses derived from the GO analysis are preliminary, since our comparison was restricted to a single species. The availability of closely related species with well-assembled genomes will facilitate a more in-depth analysis. Notably absent in this genome were genes encoding late-embryogenesis abundant (LEA) proteins; these are critical for surviving extreme dehydration in a close relative, the African sleeping midge, Polypedilum vanderplanki39. Dehydration is also a critically important response in larvae of B. antarctica, but in this species other genes, including constitutively expressed heat shock proteins and genes involved in regulation of autophagy, likely contribute to dehydration survival10.
Use of a single individual for sequencing imposed technological limitations due to the DNA input requirements needed for an assembly. The fresh mass of the fourth instar larvae used in the study was <1.5 mg, and it contained < 1 μg of DNA. To add Pacific Biosciences RSII (PacBio) data, we prepared the sequencing library from DNA extracted from a second individual. The PacBio data yielded only a modest improvement in assembly connectivity due to the low input of genomic DNA. This highlights the need for long-read sequencing technologies that have low input DNA requirements for assemblies of small organisms that cannot be easily reared in the laboratory or readily collected in the field.
In recent times, several additional midge species have arrived in the northern perimeter of the Antarctic Peninsula, but these species are also found in Tierra del Fuego, suggesting that they are recent immigrants from South America2. B. antarctica has a single congener, B. albipes, a species restricted to one of the sub-Antarctic islands, Isles Crozet. The B. antarctica genome sets the stage for future comparative genomics of Antarctic and Sub-Antarctic species.
As the first polar insect and first freeze-tolerant insect to be sequenced, B. antarctica offers a unique opportunity to probe the genome architecture of an extremophile. Lynch40 argues that small species-effective size should lead to genome expansion, presumably due to a reduction in efficacy of purifying selection. However, this is the opposite of what we observe. Among the genome’s conspicuous features, in fact, is its small size that is attained without reducing the number of protein-coding genes. By stripping the genome of repeats and TEs and by reducing the length of introns, the genome has been streamlined to the minimum yet reported for an insect. Our interpretation is, therefore, that the small midge genome is the result of genome adaptation via fixation of strongly selected mutations that overcame the opposing action of genetic drift inherent in small populations.
Methods
Biological sample
The single fourth instar larva used for sequencing was collected near Palmer Station, Antarctica (64°46′S, 64°04′W) in January 2011. It was held in its natural substrate at 2 °C and shipped to the Denlinger home laboratory at The Ohio State University, where it was held at 4 °C, under a daily 16:8 h light:dark regime until it was used for sequencing. A single individual was used to reduce assembly issues introduced by genetic heterozygosity.
DNA library preparation and sequencing
DNA was extracted using the Qiagen DNeasy Blood and Tissue Kit and sheared using a Covaris E220 (Woburn, MA) (duty cycle 10, intensity 5, cycles/burst 200 and time 180 s) to ~400 base pair (bp). Sheared genomic DNA was gel purified and used as input for Illumina sequencing library prep, with end-repair using the NEBNext end-repair kit (E6050L), and A-tailing with Taq polymerase. Ligation to Illumina paired-end adapters (PE‐102-1003) was done using Ultrapure ligase (L603-HC-L) from Enzymatics, and amplification with iProof (Bio-Rad). Agencourt Ampure XP beads were used for clean-up and size selection at each step. The resulting DNA library was sequenced on a single lane of an Illumina HiSeq2000 at the Stanford Center for Genomics and Personalized Medicine.
PacBio library preparation and sequencing
Genomic DNA was sheared using a G-Tube (Covaris Inc.) to generate 10-kb fragments. The sheared DNA was converted into a SMRTbell library using the SMRT bell Template Preparation Kit 1.0 (Pacific Biosciences, Menlo Park, CA). SMRTbell library templates were sequenced using standard SMRT sequencing with a P4 DNA polymerase on the Pacific Biosciences (PacBio) RSII system according to the manufacturer’s protocol in the Genomics Core at the Washington State University, Pullman, WA.
De novo genome assembly
Genome size determinations from flow cytometry were produced following procedures described in Hare and Johnston41 (see also Supplementary Methods). The assembly individual was sequenced to over 100-fold coverage using one lane of Illumina HiSeq2000 sequencing technology with a 400-bp insert paired-end sequencing library. Filtered sequence reads were assembled using Velvet de novo15. Two iterations of ERANGE42 were used to scaffold the assembled contigs with RNA-sequencing data from pooled larvae10 (Supplementary Methods). Filtered sequence reads were mapped back to the reference genome using Burrows-Wheeler Aligner (bwa) with default parameters43. PacBio scaffolding was accomplished using PBJelly (Jelly 14.1.14 with blasr 1.3.1)44. Mapped sequence reads to the assembled genome were also used to estimate the percent PCR duplicates in the sequencing library using Picard MarkDuplicates tool45 and to estimate coverage using BamTools46. To assess assembly quality, RNA-sequencing data from pooled larvae10 was mapped to the assembled genome using Bowtie with default parameters47.
Repeat annotation
Repeat annotation was accomplished using RepeatMaker48 and T-lex2 de novo pipeline49,50. The T-lex2 de novo pipeline uses reads mapped to the reference and identifies read pairs for which only one read is mapped successfully, called one-end anchored (OEA) pairs, as well as reads that are partially mapped, called split reads. To discriminate TE sequences from other insertions or reference repeated sequences, the unmapped reads from OEA pairs and split reads are BLATed against a library of TE sequences. Finally, a clustering step defines the TE insertion breakpoint on the reference sequence (here our assembly). To avoid bias of the TE discovery owing to the quality of the TE library, we launched RepeatMasker and the T-lex2 de novo pipeline using three previously curated TE libraries from: (i) the Drosophila, (ii) the An. gambiae TE libraries and (iii) a dipteran repeat library constructed from the 12 Drosophila genomes and An. gambiae (RepBase version 17.08 09-01-2012, http://www.fruitfly.org/data/p_disrupt/datasets/ASHBURNER/D_mel_transposon_sequence_set.fasta version 9.41)51. Species-specific TE reconstruction was attempted with ReAs52.
The R1 and R2 TE reconstruction attempt was a multi-approach attempt. Known R1 and R2 elements were downloaded from NCBI, and BLAST was used to map B. antarctica raw reads to the available R1 and R2 element sequences. Few reads mapped to the rDNA from other species, and with no significant evidence for rDNA sequence. Moreover, we undertook a targeted reassembly of the rDNA region using rDNA sequence from D. melanogaster and An. gambiae. Raw reads were mapped to the D. melanogaster and An. gambiae 18S and 28S rDNA sequence. All reads were used in a targeted reassembly of the region, which was compared with the reference assembly and an assembly from RNA-seq data obtained from Teets et al.10 using Trinity de novo53. All genomic reads were then mapped to the rDNA region using bwa43 and visually inspected for evidence of insertions or reads with split-mappings of OEA mappings that would be indicative of TE insertion.
Gene annotation
Gene annotation was accomplished using the MAKER annotation pipeline54 to map protein homology data, expressed sequence tag evidence and ab initio gene predictions to the draft genome. Protein homology data were provided by protein databases of D. melanogaster and Ae. aegypti obtained from FlyBase55 and VectorBase56, respectively. To avoid spurious matches to repetitive regions of the genome, RepeatMasker was used to mask low-complexity regions48. In addition to the included libraries, a custom repeat library for use with RepeatMasker was created with RepeatModeler, RECON, RepeatScout and TRF57. Filtered RNA-sequencing reads from Teets et al.10 were mapped to the repeat-masked genome with Bowtie and TopHat and putative transcripts were assembled with Cufflinks58. The putative transcripts were used in MAKER as expressed sequence tag evidence. An iterative approach with four rounds of training was used with MAKER and the training of the ab initio predictors SNAP59 and AUGUSTUS60. The benefit of the iterative approach is that with each round, the gene models improve and consequently training the ab initio predictors improves. For the first round, SNAP was not used and the included ‘fly’ hidden Markov model was used in AUGUSTUS. In subsequent rounds, gene models predicted in the previous round of MAKER were used to generate hidden Markov models for SNAP and AUGUSTUS. Functional annotation was accomplished with Blast2GO29,61. Transcripts predicted by MAKER were compared using BLAST with the SwissProt database62 (E<10−10) and the non-redundant database (E<10−10).
Transcripts predicted by MAKER and the top 19 BLAST hits (blastx, NR, e-value <0.0001) were loaded into Blast2GO. GO terms were assigned based on BLAST hits and InterProScan results63. The set of core eukaryotic genes in the assembled genome was identified using CEGMA64. Nuclear transfer RNA were predicted using tRNAscan-SE 1.3.1 (ref. 65)65 using options -H and -y and Aragorn 1.2.34 (ref. 66)66 using options -w -t -i116 -l -d (Supplementary Data 3).
Comparative analyses
For comparative analyses, we used the first annotated isoforms for each locus from the annotations of An. gambiae (12,669)22,67, Ae. aegypti (15,996)21, C. quinquefasciatus (18,954)23 and D. melanogaster (13,492)55. Clusters of orthologous genes between the five species were identified using the OrthoMCL package27. Effective number of codons, for codon usage estimates, was calculated using ENCPrime using the implementation of Nc’ that accounts for background nucleotide composition68. Sizes of introns and exons were calculated using in-house scripts.
Functional enrichment analysis
We conducted GO enrichment analysis to determine whether any functional groups of genes were enriched in B. antarctica relative to An. gambiae. GO annotations for B. antarctica and An. gambiae were obtained via Blast2GO29,61, and compared with the built-in Fisher’s exact test (false discovery rate<0.05).
Polymorphism detection
We identified SNPs in the individual used for the genome assembly. Putative SNPs were identified by mapping trimmed sequencing reads back to the assembled genome using bwa43. We then applied the Genome Analysis Toolkit69 to the mapped reads for PCR duplicate removal, base quality score recalibration and indel realignment. SNP discovery as well as genotyping was performed using standard hard filtering parameters70.
Demography from a single genome
We estimated the demographic history of the single individual using the pairwise sequentially Markovian coalescent method31. This method uses sequence data mapped to the reference genome from a diploid individual to infer ancestral effective population sizes at time points in the past determined by the rate of lineage coalescence at that time. The pairwise sequentially Markovian coalescent method was run on data mapped to contigs larger than 10 kb, with parameters -d 58 and -D 354 limits for coverage and bootstrap sampling was executed 100 times.
Additional information
Accession codes: Data for the Belgica antarctica genome has been deposited in GenBank/EMBL/DDBJ BioProject database under the accession code PRJNA172148. Raw sequence data has been deposited in GenBank/EMBL/DDBJ sequence read archive under the accession code SRA057118.
How to cite this article: Kelley, J. L. et al. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat. Commun. 5:4611 doi: 10.1038/ncomms5611 (2014).
Accession codes
References
Livermore, R., Eagles, G., Morris, P. & Maldonado, A. Shackleton fracture zone: no barrier to early circumpolar ocean circulation. Geology 32, 797–800 (2004).
Convey, P. & Block, W. Antarctic diptera: ecology, physiology and distribution. Eur. J. Entomol. 93, 1–13 (1996).
Sugg, P., Edwards, J. S. & Baust, J. Phenology and life history of Belgica antarctica, an Antarctic midge (Diptera: Chironomidae). Ecol. Entomol. 8, 105–113 (1983).
Peckham, V. Notes on the chironomid midge Belgica antarctica Jacobs at Anvers Island in the maritime Antarctic. Pac. Insects Monogr. 25, 145–166 (1971).
Teets, N. M. & Denlinger, D. L. Surviving in a frozen desert: Environmental stress physiology of terrestrial Antarctic arthropods. J. Exp. Biol. 217, 84–93 (2014).
Elnitsky, M. A., Benoit, J. B., Lopez-Martinez, G., Denlinger, D. L. & Lee, R. E. Jr. Osmoregulation and salinity tolerance in the Antarctic midge, Belgica antarctica: seawater exposure confers enhanced tolerance to freezing and dehydration. J. Exp. Biol. 212, 2864–2871 (2009).
Lopez-Martinez, G., Elnitsky, M. A., Benoit, J. B., Lee, R. E. Jr. & Denlinger, D. L. High resistance to oxidative damage in the Antarctic midge Belgica antarctica, and developmentally linked expression of genes encoding superoxide dismutase, catalase and heat shock proteins. Insect. Biochem. Mol. Biol. 38, 796–804 (2008).
Rinehart, J. P. et al. Continuous up-regulation of heat shock proteins in larvae, but not adults, of a polar insect. Proc. Natl Acad. Sci. USA 103, 14223–14227 (2006).
Lopez-Martinez, G. et al. Dehydration, rehydration, and overhydration alter patterns of gene expression in the Antarctic midge, Belgica antarctica. J. Comp. Physiol. B 179, 481–491 (2009).
Teets, N. M. et al. Gene expression changes governing extreme dehydration tolerance in an Antarctic insect. Proc. Natl. Acad. Sci. USA 109, 20744–20749 (2012).
Goto, S. G. et al. Functional characterization of an aquaporin in the Antarctic midge Belgica antarctica. J. Insect. Physiol. 57, 1106–1114 (2011).
Buckley, B. A., Place, S. P. & Hofmann, G. E. Regulation of heat shock genes in isolated hepatocytes from an Antarctic fish, Trematomus bernacchii. J. Exp. Biol. 207, 3649–3656 (2004).
Hofmann, G. E., Buckley, B. A., Airaksinen, S., Keen, J. E. & Somero, G. N. Heat-shock protein expression is absent in the antarctic fish Trematomus bernacchii (family Nototheniidae). J. Exp. Biol. 203, 2331–2339 (2000).
Atchley, W. R. & Davis, B. L. Chromosomal variability in the Antarctic insect, Belgica antarctica (Diptera: Chironomidae). Ann. Entomol. Soc. Am. 72, 246–252 (1979).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Johnston, J. S., Ross, L. D., Beani, L., Hughes, D. P. & Kathirithamby, J. Tiny genomes and endoreduplication in Strepsiptera. Insect. Mol. Biol. 13, 581–585 (2004).
Kirkness, E. F. et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl. Acad. Sci. USA 107, 12168–12173 (2010).
Hessen, D. O., Daufresne, M. & Leinaas, H. P. Temperature-size relations from the cellular-genomic perspective. Biol. Rev. Camb. Philos. Soc. 88, 476–489 (2013).
Schmidt-Ott, U., Rafiqi, A. M., Sander, K. & Johnston, J. S. Extremely small genomes in two unrelated dipteran insects with shared early developmental traits. Dev. Genes Evol. 219, 207–210 (2009).
Biemont, C. Genome size evolution: within-species variation in genome size. Heredity (Edinb) 101, 297–298 (2008).
Nene, V. et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316, 1718–1723 (2007).
Sharakhova, M. V. et al. Update of the Anopheles gambiae PEST genome assembly. Genome Biol. 8, R5 (2007).
Arensburger, P. et al. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science 330, 86–88 (2010).
Quesneville, H. et al. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 1, 166–175 (2005).
Adams, M. D. et al. The genome sequence of Drosophila melanogaster. Science 287, 2185–2195 (2000).
Jakubczak, J. L., Burke, W. D. & Eickbush, T. H. Retrotransposable elements R1 and R2 interrupt the rRNA genes of most insects. Proc. Natl Acad. Sci. USA 88, 3295–3299 (1991).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Brennecke, J. et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103 (2007).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Haag-Liautard, C. et al. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445, 82–85 (2007).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Allegrucci, G., Carchini, G., Todisco, V., Convey, P. & Sbordoni, V. A molecular phylogeny of Antarctic Chironomidae and its implications for biogeographical history. Polar Biol. 29, 320–326 (2006).
Ibarra-Laclette, E. et al. Architecture and evolution of a minute plant genome. Nature 498, 94–98 (2013).
Roest Crollius, H. et al. Characterization and repeat analysis of the compact genome of the freshwater pufferfish Tetraodon nigroviridis. Genome Res. 10, 939–949 (2000).
Cridland, J. M., Macdonald, S. J., Long, A. D. & Thornton, K. R. Abundance and distribution of transposable elements in two Drosophila QTL mapping resources. Mol. Biol. Evol. 30, 2311–2327 (2013).
Robertson, H. M. in:Mobile DNA II eds Craig N. L., Craigie R., Gellert M., Lambowitz A. M. ASM (2002).
Robertson, H. M. & Lampe, D. J. Distribution of transposable elements in arthropods. Annu. Rev. Entomol. 40, 333–357 (1995).
Specchia, V. et al. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463, 662–665 (2010).
Cornette, R. et al. Identification of anhydrobiosis-related genes from an expressed sequence tag database in the cryptobiotic midge Polypedilum vanderplanki (Diptera; Chironomidae). J. Biol. Chem. 285, 35889–35899 (2010).
Lynch, M. The origins of eukaryotic gene structure. Mol. Biol. Evol. 23, 450–468 (2006).
Hare, E. E. & Johnston, J. S. Genome size determination using flow cytometry of propidium iodide-stained nuclei. Methods Mol. Biol. 772, 3–12 (2011).
Mortazavi, A. et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20, 1740–1747 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Picard. http://picard.sourceforge.net (2013).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Stromberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-3.0 http://www.repeatmasker.org (1996–2010).
Fiston-Lavier, A. S., Vejnar, C. E. & Quesneville, H. Transposable sequence evolution is driven by gene context. Preprint at arXiv:12090176 [q-bioGN] (2012).
Fiston-Lavier, A. S., Carrigan, M., Petrov, D. A. & Gonzalez, J. T-lex: a program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 39, e36 (2011).
Smith, C. D. et al. Improved repeat identification and masking in Dipterans. Gene 389, 1–9 (2007).
Li, R. et al. ReAS: recovery of ancestral sequences for transposable elements from the unassembled reads of a whole genome shotgun. PLoS Comput. Biol. 1, e43 (2005).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
McQuilton, P. St, Pierre, S. E. & Thurmond, J. Consortium F. FlyBase 101–the basics of navigating FlyBase. Nucleic Acids Res. 40, D706–D714 (2012).
Megy, K. et al. VectorBase: improvements to a bioinformatics resource for invertebrate vector genomics. Nucleic Acids Res. 40, D729–D734 (2012).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0 http://www.repeatmasker.org (2008–2010).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Conesa, A. & Gotz, S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 619832 (2008).
Consortium TU. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75 (2012).
Burge, S. et al. Manual GO annotation of predictive protein signatures: the InterPro approach to GO curation. Database (Oxford) 2012, bar068 (2012).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004).
Holt, R. A. et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science 298, 129–149 (2002).
Novembre, J. A. Accounting for background nucleotide composition when measuring codon usage bias. Mol. Biol. Evol. 19, 1390–1394 (2002).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Acknowledgements
This work was funded by NSF OPP-ANT-0837613 and ANT-0837559 to D.L.D. and R.E.L. and NIH NRSA GM087069 to J.L.K. We thank Marc Mangel for making this collaboration possible through a timely introduction. We thank Jeffrey D. Jensen for useful discussions.
Author information
Authors and Affiliations
Contributions
J.L.K. and D.L.D. conceived the study. J.T.P. and A.-S.F.-L. are joint second authors. N.M.T., R.E.L. and D.L.D. collected the samples in Antarctica; J.L.K., M.-C.Y. and C.D.B. coordinated the assembly; J.T.P. and N.M.T. coordinated the annotation; A.-S.F.-L. analysed the repeat elements; J.S.J. completed the flow cytometry; J.L.K. performed comparative analyses; J.L.K., J.T.P., A.-S.F.-L., N.M.T., J.S.J., C.D.B. and D.L.D. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Figures, Tables, Methods and References
Supplementary Figures 1-9, Supplementary Tables 1-12, Supplementary Methods and Supplementary References (PDF 1054 kb)
Supplementary Data 1
Annotated Transposable elements (XLSX 93 kb)
Supplementary Data 2
GO Enrichment, comparison of Gene Ontology terms between B. antarctica and An. gambiae. (XLSX 45 kb)
Supplementary Data 3
tRNA annotations (XLSX 58 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kelley, J., Peyton, J., Fiston-Lavier, AS. et al. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat Commun 5, 4611 (2014). https://doi.org/10.1038/ncomms5611
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5611
This article is cited by
-
Genome-wide signatures of adaptation to extreme environments in red algae
Nature Communications (2023)
-
Advances in genome sequencing reveal changes in gene content that contribute to arthropod macroevolution
Development Genes and Evolution (2023)
-
Annotation of transposable elements in the transcriptome of the Neotropical brown stink bug Euschistus heros and its chromosomal distribution
Molecular Genetics and Genomics (2023)
-
Multi-omics for studying and understanding polar life
Nature Communications (2023)
-
Gene family expansions in Antarctic winged midge as a strategy for adaptation to cold environments
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
