
Abstract
Bi2Se3 initially emerged as a particularly promising host of topological physics. However, in actual materials, several issues have been uncovered including strong surface band bending and potential fluctuations. To investigate these concerns, we study nominally stoichiometric Bi2Se3 using scanning tunnelling microscopy. Here we identify two distinct distributions of BiSe antisites that act as nanometer-scale sensors for the surface band-bending field. To confirm this, we examine bulk Cu-doped Bi2Se3 and demonstrate a significantly reduced surface band-bending field. In addition, we find that in the case of unintentionally doped Bi2Se3, lateral fluctuations of the Dirac point can be directly correlated with specific near-surface point defects, namely Se vacancies.
Similar content being viewed by others
Introduction
Bi2Se3 has emerged as a particularly interesting host of topological physics due to its non-trivial surface state1,2,3. Initial studies indicated that theoretical predictions were largely true—topological protection reduces scattering and carriers are helical Dirac-like fermions4,5,6,7. However, further investigation raised several issues with its physical and structural properties in actual materials: BiSe antisites may allow scattering from the surface to the bulk8,9, strong band bending creates an additional, topologically trivial, surface state10,11,12,13, and deep, unobservable charges may cause fluctuations in the Dirac point4.
To address these concerns, we conducted scanning tunnelling microscopy (STM) studies on nominally stoichiometric Bi2Se3. We have identified two distinct BiSe antisite distributions using spectroscopic imaging and ab initio simulations; their energy states are sensitive to the local valence band position and operate as depth-dependent potential sensors. Using these sensors, we generate nanometer-scale measurements of a strong surface band-bending (SBB) field that is tunable with bulk dopants, which we demonstrate with measurements on CuxBi2Se3. In addition by mapping the lateral fluctuations of the Dirac point with dI/dV imaging, we show spatial correlation of the potential fluctuations with specific near-surface point defects. Disparate influences are observed for different defects: Se vacancies cause Dirac point shifts, whereas BiSe defects follow the Dirac point shifts.
Results
Band bending
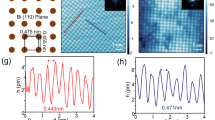
Early STM reports of the surface of Bi2Se3 revealed two primary forms of native point defects: triangular topographic features and purely electronic defect states14. The triangular defects have been identified as Se vacancies, which n-dope the sample15, and are prominently visible in Fig. 1a. The second defect type is the BiSe antisite (where a Bi atom replaces a Se lattice site) and is best observed near or below the Dirac point by dI/dV imaging (Fig. 1b), which provides a map proportional to the local density of states at a given bias. The peculiar features observed for BiSe, like those shown in the atomic resolution topograph in Fig. 1c, has led some recent studies to attribute them to surface-bulk scattering near the Dirac point8—however, angle-resolved photoemission spectroscopy (ARPES) results have not observed a side lobe to the valence band, necessary for this interpretation, and the feature geometry does not change at different biases (Supplementary Figs S1 and S2), which is inconsistent with the proposed scattering states. We therefore attribute the defect features to impurity states unassociated with bulk-surface scattering.

(a) Topography showing Se vacancies and corresponding dI/dV image (b) showing BiSe defects in Bi2Se3 at −380 meV, I=500 pA. (c) 2.9 × 3.5 nm atomic resolution imaging of a BiSe defect at −500 meV and ab initio STM simulations for a BiSe defect in the 5th (d) and 6th (e) layer at the same scale. (f) Contours from d overlaid on c. (g) Atomic structure of Bi2Se3, where hexagons separate the quintuple layers. (h) dI/dV spectroscopy on two defect types, taken on their lobes: the dim defect type, labelled with a circle in b, and a bright defect type, labelled with a square in b, as well as spectroscopy taken away from any defects. The bright and dim defects show a significant difference in their energy position on undoped Bi2Se3. To physically distinguish the defects, the lobes on the dim defects are slightly further from the centre, more likely corresponding to a deeper defect as per the simulation in e, whereas the bright defect is more compact, better corresponding to d. The scale bar in a and b is 30 nm.
We observed that BiSe defects show tunnelling amplitude maxima at vastly different voltages—for example, Fig. 1h shows dI/dV spectroscopy for two defects whose tunnelling maxima are separated by ~150 meV. By analyzing the dI/dV spectra from numerous BiSe defects, a histogram analysis shows that there is a bimodal distribution (Fig. 2a). Therefore, contrary to previous reports14, these nearly identical features are due to BiSe defects at two separate Se layers. From ab initio calculations, we can rule out the middle layer (BiSe2) due to its high energy of formation16, which means that the defects are in structurally equivalent locations (that is, the top or bottom of a quintuple layer—these positions are symmetric). Structurally equivalent defects at different depths should be energetically degenerate. However, in this case, the degeneracy is broken by surface band-bending mapping the defect energies at different depths maps the local band bending. On the basis of our ab initio simulations of STM topographs for these defects (Fig. 1d,e), we attribute the higher amplitude defect (square in Fig. 1b) to BiSe on the 5th layer and the lower amplitude defect (circle in Fig. 1b) to BiSe on the 6th atomic layer, which correspond to the bottom of the 1st quintuple layer and the top of the 2nd quintuple layer, respectively (see Fig. 1g and Supplementary Fig. S3). The n-shifted defect has a higher tunnelling amplitude, meaning that it is closer to the surface/STM tip, demonstrating downward SBB of the bulk conduction and valence bands. Downward SBB is consistent with ARPES observations10, and is possibly due to more Se vacancies forming at the sample–vacuum interface following the ultra high vacuum (UHV) sample cleave.

(a) Histograms of the BiSe defect energies in CuxBi2Se3, extracted from multipass dI/dV images, show a decreasing gap between the peaks as the Cu content is increased. (b) Schematic of the proposed band-bending mechanism responsible for the energy difference between the defects; the orange profile represents the BiSe defect’s observable states on the Bi2Se3 surface. Cross-sectional dI/dV linescans from two different defects on Bi2Se3 are shown for direct comparison, as well. Colour scale ranges in b: left, 1.4 × 10−11 pA meV−1, right, 5.0 × 10−12 pA meV−1.
These defects should retain their position relative to the valence band, so adding an n-type dopant, like incorporating Cu into the bulk, should n-shift the entire electronic structure including the BiSe distributions, without altering the nature of the BiSe defect itself (see Supplementary Fig. S4 for more information). We have observed several sub-surface Cu defect types in the topography (Fig. 3a), but they do not appear to have changed the shape of the BiSe defects in a significant way, as shown in the dI/dV images in Fig. 3b–f. The identification of these Cu defects varies in the literature17,18, so the implications of their structural configurations within the lattice are left to future studies. Regardless of the defects’ identities, the observed Cu defect density does not suggest that significant structural changes have occurred, allowing us to consider them, simply, as dopants. Upon adding Cu, we observed the two BiSe distributions shift closer together (Fig. 2a). From the band-bending picture, this means that the SBB is reduced (Fig. 2b). As more carriers are introduced with higher n-type doping (more of the bulk conduction band is below the Fermi level), we believe screening effects are causing the reduction in the band-bending potential field.

Cu doping introduces a number of new defects, as shown in the topograph in a. However, as shown in the dI/dV data in b–f, the distinct BiSe defect is still present. Indeed, it still shows up at different biases depending on its layer, as highlighted by the circled and squared defects. Scale bar in b is 10 nm.
We have considered the possibility that the band bending was induced by the electric field from the STM tip, an effect known as tip-induced band bending (TIBB). We performed spatial spectroscopic imaging at a variety of tunnelling set points (150–500 pA) and observed less than a 5-meV shift in the impurity peak position (Supplementary Fig. S5), indicating that TIBB is not the dominant source of the energy shift. Presumably, as the surface is already n-doped even in the case of zero Cu doping, the available free carrier concentration is high enough to screen out the tip-induced field.
To quantitatively approximate the magnitude of SBB as a function of doping (Fig. 4a), the energy separation of the defect distributions is divided by the approximate separation between the Se layers terminating adjacent quintuple layers, as determined by local-density approximation energy relaxation calculations. We note that intercalation of Cu increases the separation between layers18, and any TIBB effects would further amplify the observed bending. Therefore, the values we have measured are upper bounds.

(a) Band bending as a function of doping level. To numerically determine the band bending, we used the distance determined by ab initio simulations of 0.254 nm. (b) Schematic showing the surface potential dependence on doping. In Cu-doped samples, more of the bulk conduction band is below the Fermi energy, providing enough carriers to screen the band bending.
Several groups have observed bulk two-dimensional electron gas (2DEG) states appearing on samples left in vacuum, and the ARPES data suggest band bending is the culprit10,11,12,13. STM is surface sensitive, so this calls into question the results of all STM studies above the conduction band minimum on this material: is the measured surface state really of topological origin, or simply due to a traditional bulk 2DEG? If a 2DEG exists, it can be eliminated by reducing the SBB. To ensure that future STM studies are truly investigating the topological surface state in Bi2Se3, control of the surface potential will be essential—bulk n-type doping provides one such mechanism, as shown schematically in Fig. 4b.
Correlating potential fluctuations
The Dirac point shifts rigidly with bulk band fluctuations4 (Fig. 5a) and the dI/dV data ~200 meV below the Dirac point are effectively linear with bias, allowing us to map the dI/dV value below the Dirac point to the fluctuations of the Dirac point. This technique has been successfully applied to graphene19, and we confirmed the validity of this technique for Bi2Se3 from line spectroscopy data, as shown in Fig. 5b,c. However, dI/dV images can contain additional information that could be interpreted as quasiparticle scattering20 or variations in the apparent barrier height21, so we quantitatively demonstrated that above the Dirac point (Fig. 5f), the dI/dV image is a reversal of the dI/dV image below Dirac point with a cross-correlation coefficient of −0.5. Therefore, the dI/dV images in Fig. 5d,e are best described as maps of Dirac point shifts.

(a) Two dI/dV curves taken from different regions on Bi2Se3 show how the Dirac point shift can be correlated, to first order, with the intensity shift in dI/dV intensity—this allows single-pass dI/dV scans to map the Dirac point fluctuations. (b) A scatter plot determined from line spectroscopy data across 300 nm of Bi2Se3 shows the first-order relation between the Dirac point and the lock-in signal at −540 meV; R2=0.72. (c) Seven-point moving average from the line spectroscopy visually shows the correlation from b. A 350 × 50 nm region of Bi2Se3 is then investigated with this technique: topography (d) at −200 meV shows Se vacancies, and the dI/dV image of the same region taken at −540 meV shows fluctuations in the Dirac point (e). In e, the red circles represent bright triangular defects, determined from d. (f) dI/dV image at −270 meV showing that the dI/dV map gives an inverse image of e above the Dirac point. The cross-correlation coefficient is −0.5, quantitatively confirming the contrast reversal despite the higher noise level in f. A visualization of the deviations from the distribution mean for dim-type BiSe defects is shown in g, where red points indicate a BiSe defect with an energy state that is more positive than the distribution mean, and blue points indicate a BiSe defect below the distribution mean (see inset). The dI/dV image at −350 meV (h) shows both dim and bright type defects. The fluctuations in the BiSe defect energy scale correlates directly with the fluctuations in the Dirac point; a few extreme examples are highlighted in boxes in e–h. Colour scales: d, 40 pm; e, 30 meV; h, 10 pA/V.
Figure 5d and e shows the topography and corresponding map of Dirac point fluctuations, respectively, in nominally stoichiometric Bi2Se3. Se vacancies observed in Fig. 5d, indicated with red circles in Fig. 5e, can be correlated with n-shifts in the Dirac point. The energy of formation for Se vacancies increases as the sample is n-doped16—so they would preferentially form in more positively shifted regions. Therefore, their correlation with negatively shifted regions suggests they are causing the n-type fluctuations. It is also apparent that these defects are the origin of the fluctuations (responsible for the finite width of the peak energy distributions in Fig. 2a) in the BiSe distributions: negatively shifted BiSe defects are on negatively shifted regions of the sample and positively shifted BiSe defects are found on positively shifted regions of the sample (Fig. 5e–h). Significantly, while these BiSe defects do have strong electronic interactions, they do not appear to have strong effects on the position of the Dirac point, rather, they appear to follow the bulk fluctuations.
Discussion
Our re-evaluation of the BiSe defect as an impurity state suggests that bulk-surface interactions are not directly evident. Furthermore, this classification allows the band bending to be directly visualized, highlighting the concerns that non-topological surface states may be inadvertently formed on Bi2Se3. To prevent the formation of such a state, it will be important to deliberately tailor the band bending using advanced synthesis techniques such as molecular beam epitaxy. Furthermore, by correlating observable defects with the fluctuations in the Dirac point through dI/dV imaging, we have shown that different defects influence the Dirac point disparately—as different preparation techniques produce different characteristic defects, an understanding of the interplay between specific defects and the local electronic structure is critical. We expect that such atomic-scale insights will enable application-optimal doping and growth schemes. Ultimately, Bi2Se3 remains a model system for exploring topological physics.
Methods
Sample growth
Ternary CuxBi2Se3 bulk crystals were grown by the vertical Bridgman technique using a three-zone high-temperature furnace. The temperature profiles in the growth zone were designed to produce a 3–5 °C axial gradient. The starting materials used for crystal growth (6 N Bi and 6 N Se) were deoxidized and outgassed under vacuum directly before use. Stoichiometric amounts of Bi and Se along with 6 N purity deoxidized Cu were enclosed in a graphitized fused quartz ampoule, evacuated for several hours and sealed off under a dynamic vacuum of ~10−8 torr. The pre-loaded charge was then placed in the middle zone of the furnace. After the preliminary reaction was completed at 400 °C, the charge was slowly heated up to 770 °C for ~6 h and then the melt was allowed to homogenize for 24 h under a high linear temperature gradient to promote mixing. The melt was solidified with a rate of about 1.5 mm h−1 at a small linear gradient of 3–5 °C cm−1.
Ab initio calculations
Our calculations are based on density functional theory with the Perdew-Burke-Ernzerhof approximation22. Interactions between ion cores and valence electrons are described by the projector augmented wave method23,24, as implemented in the VASP package25,26. Plane-waves with a kinetic energy cutoff of 270 eV were used as the basis set. We used a (7 × 7) double quintuple slab containing 196 Bi and 294 Se atoms with gamma point sampling of the Brillouin zone. All atoms were allowed to relax with a convergence criterion for the structural relaxations of 0.04 eV Å−1. To compare with experiment, we simulate the STM images based on the theory of Tersoff and Hamann27. When simulating STM images for comparison with experimental images, care has to be taken with regard to the location of the experimental Fermi energy. To correspond to the observed images at a −500 meV bias, the Fermi energy window used to construct the charge density for the simulated images range from conduction band minimum +300 meV to the valence band maximum +100 meV. This window is chosen relative to the band edges because the use of only two slabs increases the theoretical gap.
STM measurements
Samples were cleaved in an UHV with a base pressure <10−10 torr, then immediately transferred to the STM at liquid nitrogen temperature. The STM is a Pan-type STM scan head from RHK in a home-built UHV chamber. Electrochemically etched tungsten STM tips were cleaned by e-beam irradiation under UHV. STS data were taken using the built-in lock-in amplifier on a Nanonis STM controller at 780 Hz and 10-mV modulation amplitude. STM topographs were taken in constant current mode and the bias was applied to the sample. BiSe peak positions were determined from multipass scan data at constant current, unless otherwise noted, and were extracted from the lobes of the defects; the background spectra, averaged on either side of the given lobes, were subtracted from the impurity peak spectra to remove systematic offsets due to the shape of the nearby dI/dV spectra. More information is provided with Supplementary Fig. S6.
Additional information
How to cite this article: Mann, C. et al. Mapping the 3D surface potential in Bi2Se3. Nat. Commun. 4:2277 doi: 10.1038/ncomms3277 (2013).
References
Hasan, M. & Kane, C. Colloquium: topological insulators. Rev. Mod. Phys. 82, 3045–3067 (2010).
Moore, J. E. The birth of topological insulators. Nature 464, 194–198 (2010).
Qi, X.-L. & Zhang, S.-C. The quantum spin Hall effect and topological insulators. Phys. Today 63, 33–38 (2010).
Beidenkopf, H. et al. Spatial fluctuations of helical Dirac fermions on the surface of topological insulators. Nat. Phys. 7, 939–943 (2011).
Wang, Y. et al. Observation of a warped helical spin texture in Bi2Se3 from circular dichroism angle-resolved photoemission spectroscopy. Phys. Rev. Lett. 107, 207602 (2011).
Zhao, Y., Hu, Y., Liu, L., Zhu, Y. & Guo, H. Helical states of topological insulator Bi2Se3 . Nano Lett. 11, 2088–2091 (2011).
Hanaguri, T., Igarashi, K., Kawamura, M., Takagi, H. & Sasagawa, T. Momentum-resolved Landau-level spectroscopy of Dirac surface state in Bi2Se3 . Phys. Rev. B 82, 081305 (2010).
Kim, S. et al. Surface scattering via bulk continuum states in the 3D topological insulator Bi2Se3 . Phys. Rev. Lett. 107, 056803 (2011).
Romanowich, M. et al. Interplay of topological surface and bulk electronic states in Bi2Se3 . Phys. Rev. B 87, 085310 (2013).
Bianchi, M. et al. Coexistence of the topological state and a two-dimensional electron gas on the surface of Bi2Se3 . Nat. Commun. 1, 128 (2010).
Bianchi, M., Hatch, R., Mi, J., Iversen, B. & Hofmann, P. Simultaneous quantization of bulk conduction and valence states through adsorption of nonmagnetic impurities on Bi2Se3 . Phys. Rev. Lett. 107, 086802 (2011).
King, P. et al. Large tunable Rashba spin splitting of a two-dimensional electron gas in Bi2Se3 . Phys. Rev. Lett. 107, 096802 (2011).
Zhu, Z.-H. et al. Rashba spin-splitting control at the surface of the topological insulator Bi2Se3 . Phys. Rev. Lett. 107, 186405 (2011).
Urazhdin, S. et al. Scanning tunneling microscopy of defect states in the semiconductor Bi2Se3 . Phys. Rev. B 66, 161306 (2002).
Hor, Y. et al. p-Type Bi2Se3 for topological insulator and low-temperature thermoelectric applications. Phys. Rev. B 79, 195208 (2009).
West, D., Sun, Y. Y., Wang, H., Bang, J. & Zhang, S. B. Native defects in second-generation topological insulators: effect of spin-orbit interaction on Bi2Se3 . Phys. Rev. B 86, 121201 (2012).
Wang, Y.-L. et al. Structural defects and electronic properties of the Cu-doped topological insulator Bi2Se3 . Phys. Rev. B 84, 075335 (2011).
Hor, Y. S. et al. Superconductivity in CuxBi2Se3 and its implications for pairing in the undoped topological insulator. Phys. Rev. Lett. 104, 057001 (2010).
Zhang, Y., Brar, V. W., Girit, C., Zettl, A. & Crommie, M. F. Origin of spatial charge inhomogeneity in graphene. Nat. Phys. 5, 722–726 (2009).
Alpichshev, Z. et al. STM imaging of impurity resonances on Bi2Se3 . Phys. Rev. Lett. 108, 206402 (2012).
Hamers, R. J. Atomic-resolution surface spectroscopy with the scanning tunneling microscope. Annu. Rev. Phys. Chem. 40, 531–559 (1989).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Physik, T., Wien, T. U. & Hauptstrasse, W. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Tersoff, J. & Hamann, D. R. Theory of the scanning tunneling microscope. Phys. Rev. B 31, 805–813 (1985).
Acknowledgements
C.M. and C.-K.S. acknowlege support from the Welch Foundation grant F-1672, ARO grants W911NF-09-1-0527 and W911NF-12-1-0308, and NSF grant DMR-0955778. The work at Purdue was supported by the DARPA MESO program (Grant No. N66001-11-1-4107). The work at RPI of D.W. and S.Z. was supported by DARPA Grant No. N66001-12-1-4034, and that of S.Z. was also supported by the Department of Energy (DOE) under Grant No. DE-SC0002623. The supercomputer time was provided by the CCNI at RPI and NERSC under DOE Contract No. DE-AC02-05CH11231. C.M. thanks Dr R. Hatch for insightful discussions. Y.P.C. thanks J. Tian and Y. Xu for sample preparation assistance.
Author information
Authors and Affiliations
Contributions
All authors contributed to the intellectual content of the paper. I.M. and Y.P.C. provided the samples. D.W. performed and analysed simulations under the guidance of S.Z. C.M. collected and analysed the STM data and prepared the manuscript, C.-K.S. advised on the experiment and provided input on the data analysis and the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S6 and Supplementary References (PDF 721 kb)
Rights and permissions
About this article
Cite this article
Mann, C., West, D., Miotkowski, I. et al. Mapping the 3D surface potential in Bi2Se3. Nat Commun 4, 2277 (2013). https://doi.org/10.1038/ncomms3277
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms3277
This article is cited by
-
Electrical resistance of individual defects at a topological insulator surface
Nature Communications (2017)
-
Nanoscale electron transport at the surface of a topological insulator
Nature Communications (2016)
-
Topological transport and atomic tunnelling–clustering dynamics for aged Cu-doped Bi2Te3 crystals
Nature Communications (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
