
Abstract
To utilize biofilms for chemical transformations in biorefineries they need to be controlled and replaced. Previously, we engineered the global regulator Hha and cyclic diguanylate-binding BdcA to create proteins that enable biofilm dispersal. Here we report a biofilm circuit that utilizes these two dispersal proteins along with a population-driven quorum-sensing switch. With this synthetic circuit, in a novel microfluidic device, we form an initial colonizer biofilm, introduce a second cell type (dispersers) into this existing biofilm, form a robust dual-species biofilm and displace the initial colonizer cells in the biofilm with an extracellular signal from the disperser cells. We also remove the disperser biofilm with a chemically induced switch, and the consortial population could tune. Therefore, for the first time, cells have been engineered that are able to displace an existing biofilm and then be removed on command allowing one to control consortial biofilm formation for various applications.
Similar content being viewed by others

Introduction
Biofilms are groups of cells at an interface cemented together by polysaccharides, protein, DNA and lipids1. Biofilms are related to most bacterial chronic inflammatory and infectious diseases2 as well as involved in biocorrosion3 and biofouling4 in diverse areas. They also may be used for beneficial applications such as bioremediation and hold much potential for chemical transformations in biorefineries5. For these applications, compared with monocultures, mixed populations have the advantage of being able to perform more complex transformations (for example, those requiring multiple steps), and they are more resistant to environmental stress6. For these reasons, consortia have been heralded as the new frontier in synthetic biology6. However, to date, it has not been possible to control consortial biofilm formation.
Based on an understanding of signals and regulatory networks during biofilm development7, biofilms have been engineered by manipulating extracellular/intercellular signals and regulators5. The first engineered biofilm was a consortium where Bacillus subtilis was engineered to secrete the peptide antimicrobials indolicidin and bactenecin to inhibit the growth of sulfate-reducing bacteria and thereby decrease corrosion8. Also, the first synthetic signalling circuit to control biofilm formation was developed for Escherichia coli and Pseudomonas fluorescens by manipulating the extracellular concentration of the signal indole produced by E. coli9; indole is a biofilm inhibitor for E. coli. In addition, using directed evolution, the quorum-sensing (QS) regulator SdiA was reconfigured to decrease biofilm formation by increasing indole10, and the global regulator H-NS was evolved to decrease biofilm formation via prophage excision and cell death11.
To remove existing biofilms, T7 bacteriophage was engineered to produce dispersin B of Actinobacillus actinomycetemcomitans to disrupt the glycosidic linkages of polymeric β-1,6-N-acetyl-D-glucosamine found in the biofilm matrix during bacteriophage infection12. In addition, the global transcriptional regulator Hha of E. coli, which reduces hemolysin production13 and decreases biofilm formation14, was engineered using protein engineering (aa changes D22V, L40R, V42I and D48A) to enhance biofilm dispersal primarily by inducing protease HslV and cell lysis15, and BdcA, which increases biofilm dispersal by decreasing the concentration of the second messenger cyclic diguanylate (c-di-GMP) by binding it, was engineered (aa change E50Q) for nearly complete dispersal of biofilms16. Therefore, new genetic modules are available for manipulating biofilms5.
Synthetic biology is an emerging field to develop biological systems that perform novel functions by assembling genetic modules17. The genetic modules include switches, cascades, pulse generators, time-delayed circuits, oscillators, spatial patterning and logic formulas, and they can be utilized to control transcription, translation and post-translational operations in order to tune gene expression, protein production, metabolism and cell–cell communication18. Among these genetic modules, bacterial QS systems are becoming important components of a wide variety of engineered biological devices19, as autoinducers are useful as input signals because most are small, diffuse freely in aqueous media and are easily imported by cells20. As the engineered cells synthesize their own QS signals, they are able to monitor their cell density and modulate their activities21 accordingly without supervision. Hence, QS-based circuits have a wide range of potential engineering applications such as production of biochemicals, tissue engineering and mixed-species fermentations as well as developing biosensors and controlling biofouling20. For example, LuxI from Vibrio fischeri, which produces N-(3-oxo-hexanoyl)-L-homoserine lactone (3oC6HSL) and AiiA from B. thuringiensis, which degrades 3oC6HSL, were utilized to generate synchronized oscillations22. Also, the LuxI/LuxR QS system was coupled to the production of a toxin protein CcdB to induce cell death at high cell densities23, and applied to create programmable cell behaviour that synthesizes a target protein when the cell population reaches a critical density24.
The two best-characterized QS systems of P. aeruginosa are the LasI/LasR and RhlI/RhlR systems, which regulate biofilm formation, virulence, swarming motility and antibiotic efflux pumps25. LasI produces autoinducer molecule, N-(3-oxo-dodecanoyl)-L-homoserine lactone (3oC12HSL), which is sensed by LasR26. Likewise, RhlI produces N-butyryl-L-homoserine lactone (C4HSL) that is sensed by RhlR26. The LasI/LasR and RhlI/RhlR QS systems have been used to engineer bidirectional communication27, and the LasI/LasR QS system was used to both construct a predator–prey ecosystem28 and create a synthetic ecosystem in E. coli29. Furthermore, the RhlI/RhlR QS system was utilized to demonstrate roles for self-organization and aggregation in a synthetic biofilm consortium30. Hence, synthetic QS circuit systems have potential in that population-driven QS switches may be utilized to develop synthetic genetic networks for a variety of applications.
As biofilm formation and dispersal are ultimately genetic processes, they may be manipulated like other genetic systems5 using the tools of synthetic biology18 and directed evolution. In this work, our goal was to control biofilm displacement via a population-driven QS switch coupled to engineered biofilm dispersal proteins. Controlling biofilm dispersal creates a synthetic biological platform for sophisticated patterning of biofilms for engineering applications. The LasI/LasR QS module of P. aeruginosa was combined with our engineered Hha15 and BdcA16 biofilm dispersal proteins, and the system was utilized to selectively remove one type of cell from an existing biofilm, and then remove the second biofilm to create a surface ready for additional biofilms. Constructs were also created that allow the consortial population to be tuned. Therefore, for the first time, a quorum sensing circuit has been devised that allows biofilm formation to be controlled to the extent each cell type may be dispersed allowing one to control consortial biofilm formation for various applications.
Results
Microfluidic biofilm engineering circuit
The microfluidic biofilm engineering (μBE) signalling circuit was constructed in E. coli using two engineered biofilm-dispersing proteins, Hha13D6 (ref. 15) and BdcAE50Q16, along with the P. aeruginosa LasI/LasR QS system (Fig. 1a) for use in the novel microfluidic device (Fig. 1b). E. coli hha31 was used as the host as deletion of hha increases biofilm formation14 and provides a background in which there is no wild-type Hha. Lactococcal promoter CP2532 was used as the strong constitutive promoter for two of the three proteins on each plasmid. To obtain high concentrations of intercellular signal 3oC12HSL and regulator LasR, a synthetic ribosomal-binding site (RBS II)32 was first utilized. However, high expression of lasI or lasR was deleterious; thus, we used the native RBS of these genes. All the cloned genes for the two cell types were placed on a single plasmid (pCA24N derivative, Fig. 2a,b) to avoid plasmid instability and so that a single antibiotic could be used to maintain the key plasmid during growth of the consortia.

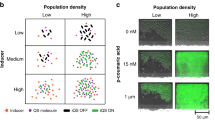
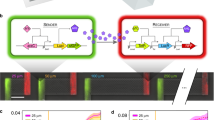
(a) The two E. coli cell types communicate by using the LasI/LasR QS module of P. aeruginosa. In the disperser cell, the LasI protein (autoinducer synthase) is constitutively produced and synthesizes the QS signal 3oC12HSL. 3oC12HSL freely diffuses into the initial colonizer cell and makes a complex with LasR (LuxR family transcriptional regulator), and the 3oC12HSL+LasR complex induces biofilm dispersal protein BdcAE50Q by activating the lasI promoter. BdcAE50Q disperses biofilms by binding cyclic diguanylate. The biofilm dispersal protein Hha13D6 in the disperser cell is induced upon adding IPTG. Hha13D6 disperses biofilms by activating proteases. (b) The novel microfluidic device is shown with its two PDMS layers, a bottom layer with a diffusive mixer and eight microchambers, and a top layer containing a second diffusive mixer and the pneumatic elements to control microvalves. The diffusive mixer in the bottom layer was used to generate different concentrations of dispersal signals (for example, IPTG for removing disperser cells and 3oC12HSL for dispersing initial colonizer cells) and to perfuse growth media into the biofilm microchambers. The mixer in the top layer was used to introduce bacteria into the microchambers at different cell densities.

(a) pHha13D6-gfp-lasI with hha13D6 under control of the T5-lac promoter, and gfp and lasI under control of the constitutive CP25 promoter. (b) pBdcAE50Q-rfp-lasR with bdcAE50Q under control of the lasI promoter, and rfp and lasR under control of the constitutive CP25 promoter. cat encodes chloramphenicol acetyltransferase, lacI^q encodes a repressor mutant of the lac operator and rrnB-T1 indicates the rrnB T1 transcription termination sequence.
In the μBE circuit, disperser cells (lasI+, hha13D6+, gfp+ via E. coli hha/pHha13D6-gfp-lasI) produce constitutively green fluorescent protein (GFP) and the QS signal 3oC12HSL, and have hha13D6 induced upon addition of isopropyl-β-D-thiogalactopyranoside (IPTG) (Fig. 1a). The initial colonizer cells (lasR+, bdcAE50Q+, rfp+ via E. coli hha/pBdcAE50Q-rfp-lasR) produce constitutively red fluorescent protein (RFP) and regulator LasR, the receptor of 3oC12HSL. The initial colonizer cells also have bdcAE50Q under the control of the lasI promoter, which is activated via the 3oC12HSL+LasR complex33 (Fig. 1a). Thus, disperser cells produce the signalling molecule 3oC12HSL, and the initial colonizer biofilm-forming cells sense it and disperse when the disperser cells reach a quorum.
Disperser cells grow more slowly than the initial colonizer cells
As we desire the disperser cells to supplant the initial colonizer cells, we checked the specific growth rates of the two strains to see if they are comparable; the disperser cells grew 14% slower than the initial colonizer cells in rich medium (Luria-Bertani (LB)-glucose, μdisperser=1.13±0.08 h−1 and μinitial colonizer=1.31±0.05 h−1, respectively). The slower growth of the disperser cells is due to somewhat leaky expression of toxin hha13D6 rom the T5-lac promoter34, as a strain with a plasmid with the araBAD promoter to better repress hha13D6 in the absence of arabinose (pPBAD-hha13D6-gfp-lasI) increased the growth of the disperser cells by 12% (μdisperser with araBAD promoter=1.27±0.23 h−1) (Supplementary Methods). Corroborating this difference in cell growth, disperser cells formed biofilms more slowly compared with initial colonizer cells: the biomass of initial colonizer cells after 9 h was 5.7±0.1 μm3 μm−2 (Fig. 3a), while the biomass of the disperser cells after 9 h was 4.1±0.1 μm3 μm−2 (Fig. 3b).

(a) Biomass of initial colonizer biofilms (BdcAE50Q+ and LasR+; E. coli hha/pBdcAE50Q-rfp-lasR) with 500 μM of 3oC12HSL for 10 h. (b) Biomass of disperser biofilms (Hha13D6+ and LasI+; E. coli hha/pHha13D6-gfp-lasI) with 2 mM of IPTG for 10 h. (c) Biomass after 19 h for the initial colonizer biofilms with different concentrations of 3oC12HSL (0, 71, 143, 214, 286, 357, 429 and 500 μM for 10 h). (d) Biomass after 19 h for the disperser biofilms with different concentrations of IPTG (0, 0.3, 0.6, 0.9, 1.1, 1.4, 1.7 and 2.0 mM for 10 h). Robust biofilms at 9 h were formed by seeding the initial colonizer or the disperser cells into microchambers for (a), (b), (c) and (d). Three independent cultures were tested, and error bars indicate the s.d. from two different positions in one of the three sets. Biomass was determined by COMSTAT analysis.
Disperser cells produce 3oC12HSL
To confirm the disperser μBE circuit synthesizes 3oC12HSL, we measured the 3oC12HSL concentration of the disperser cells (E. coli hha/pHha13D6-gfp-lasI) in the biofilm using a lacZ reporter (lasB–lacZ translational fusion) that is activated by 3oC12HSL (Supplementary Methods)35. In flow cells, disperser cells in biofilms produced 14-fold higher concentrations of 3oC12HSL compared with the planktonic cells in the effluent (6.7±2.1 μM versus 0.5±0.2 μM), and produced 51-fold higher concentrations of 3oC12HSL compared with planktonic cells in shake flasks (0.1±0.1 μM). The negative control (no lasI) had no detectable 3oC12HSL. These results confirm that autoinducer concentrations in biofilms are higher than in planktonic cultures36 and compare well with levels of 3oC12HSL produced in P. aeruginosa biofilms (137–600 μM36). As maximum activity of the lasI promoter is obtained with 0.1 μM of 3oC12HSL and LasR33, 3oC12HSL production in the disperser biofilms should induce the lasI promoter in the LasR-producing initial colonizer cells to express bdcAE50Q to disperse the initial colonizer biofilms. Moreover, as 3oC12HSL diffusion is significantly slower compared with C4HSL diffusion38, local concentrations of 3oC12HSL in biofilms may be much higher than the 3oC12HSL concentration measured here.
3oC12HSL disperses the initial colonizer biofilm
To demonstrate that 3oC12HSL disperses biofilms produced by the initial colonizer cells (E. coli hha/pBdcAE50Q-rfp-lasR) by binding LasR and inducing bdcAE50Q, exogenous 3oC12HSL at different concentrations was added to biofilms formed by the initial colonizer cells in microfluidic channels. As expected, the initial colonizer biofilms were dispersed upon adding 3oC12HSL in a dose-dependent manner (Fig. 3c; Supplementary Fig. S1); near complete biofilm dispersal was obtained at 500 μM of 3oC12HSL, and at lower 3oC12HSL concentrations, dispersal of the initial colonizer biofilms was reduced (Supplementary Fig. S1). In contrast, there was no dispersal in the absence of 3oC12HSL (Supplementary Fig. S1), and the initial colonizer cells formed thick biofilms (10.8±0.6 μm3 μm−2) (Fig. 3c). Hence, initial colonizer cells recognize 3oC12HSL and this signal may be used to disperse initial colonizer biofilms.
IPTG removes the disperser biofilm
To demonstrate that IPTG disperses biofilms produced by disperser cells by inducing hha13D6, exogenous IPTG at different concentrations was added to biofilms formed by disperser cells in microfluidic channels. As expected, disperser biofilms were dispersed upon adding IPTG in a dose-dependent manner (Fig. 3d) with near complete biofilm dispersal at 2 mM IPTG (Supplementary Fig. S2); hence, we used 2 mM IPTG in subsequent experiments. In contrast, there was no dispersal in the absence of IPTG (Supplementary Fig. S2). Thus, the disperser cell has active hha13D6 to disperse its own biofilm upon IPTG addition.
Engineered BdcA and Hha are necessary for biofilm dispersal
To confirm that the biofilm dispersal upon addition of 3oC12HSL and IPTG is the result of production of the engineered biofilm dispersal proteins, we performed dispersal experiments of initial colonizer cells that lack bdcAE50Q and disperser cells that lack hha13D6. As expected, initial colonizer biofilms formed without bdcAE50Q (via E. coli hha/pRFP-lasR) did not disperse in the presence of 3oC12HSL (Fig. 4a), while initial colonizer biofilms formed with bdcAE50Q dispersed with 3oC12HSL (Supplementary Fig. S1). Similarly, disperser biofilms formed without hha13D6 (via E. coli hha/pGFP-lasI) did not disperse upon addition of IPTG (Fig. 4b), while disperser biofilms formed with hha13D6 dispersed with IPTG (Supplementary Fig. S2). Hence, BdcAE50Q and Hha13D6 are necessary to disperse the initial colonizer and disperser biofilms, respectively. Taken together, both disperser and initial colonizer cells were constructed to allow us to manipulate biofilm dispersal using a population-driven switch.

(a) Initial colonizer biofilms that lack BdcA (BdcAE50Q− and LasR+ via E. coli hha/pRFP-lasR) with 500 μM of 3oC12HSL for 10 h. (b) Disperser biofilms that lack Hha (Hha13D6− and LasI+ via E. coli hha/pGFP-lasI) with 2 mM of IPTG for 10 h. Robust biofilms of initial colonizer or disperser cells were developed for 9 h in each microchamber. Scale bar indicates 20 μm. Three independent cultures were tested, and representative images are shown.
Disperser cells displace initial colonizer biofilms
Having verified the disperser and initial colonizer cell elements of the μBE signalling circuit, we combined both cell types to form a consortial biofilm and investigated whether the disperser cells could displace the initial colonizer cells. First, robust biofilms of initial colonizer cells were developed for 9 h after seeding, and then disperser cells were added to the initial colonizer biofilms for 5 h to form the biofilm consortium (Fig. 5a; Supplementary Movie 1). As disperser cells synthesize 3oC12HSL constitutively, 3oC12HSL should bind to LasR when the concentration of 3oC12HSL is increased as the disperser biofilms mature. Then, the 3oC12HSL+LasR complex should induce dispersal of initial colonizer biofilms by switching on bdcAE50Q under control of the lasI promoter. As expected, the initial colonizer biofilms were displaced from the surface as the disperser cells grew (Fig. 5a; Supplementary Movie 1). After 44 h, 80% of the maximum initial colonizer biofilm formed was removed (Fig. 5a,b; Supplementary Movie 1). The displacement of the initial colonizer cells by the disperser cells was accomplished by the production of 3oC12HSL from the disperser biofilms, not by shear force, as the disperser biofilms that lack LasI did not reduce initial colonizer biofilms; that is, both no lasI disperser and initial colonizer biofilms grew when 3oC12HSL was not produced (Fig. 5c), and the biofilm became essentially that of the faster-growing initial colonizer cells after 40 h (Fig. 5c,d). Hence, the disperser cells completely displaced the initial colonizer biofilm via the population-driven synthetic μBE system.

(a) An initial colonizer biofilm (red, BdcAE50Q+ and LasR+; E. coli hha/pBdcAE50Q-rfp-lasR) was developed for 9 h, then disperser cells (green, Hha13D6+ and LasI+; E. coli hha/pHha13D6-gfp-lasI) were seeded for 5 h to form both initial colonizer and disperser biofilms. After 44 h, 2 mM of IPTG was added for an additional 18 h to remove the disperser biofilm. (b) Biomass of the initial colonizer (BdcAE50Q+ and LasR+) and disperser (Hha13D6+ and LasI+) biofilms as determined by COMSTAT analysis. (c) Initial colonizer biofilms (BdcAE50Q+ and LasR+) was developed for 9 h, then control disperser cells that lack LasI (green, Hha13D6+ and LasI−; E. coli hha/pHha13D6-gfp) were seeded for 5 h to form both initial colonizer and control disperser biofilms. After 40 h, 2 mM of IPTG was introduced for an additional 20 h to try to disperse the control disperser biofilms, which lack lasI. (d) Biomass of the initial colonizer (BdcAE50Q+ and LasR+) and the no LasI disperser control (Hha13D6+ and LasI−) biofilms as determined by COMSTAT analysis. Scale bar indicates 20 μm. Three independent cultures were tested, and representative images are shown for (a) and (c). Error bars indicate the s.d. from two different positions in one of the three sets for (b) and (d).
The second key element of our design was the removal of the disperser biofilm; we found, we could remove the disperser biofilm by inducing Hha13D6 with IPTG (Fig. 5a). After 62 h (18 h with 2 mM IPTG), 92% of the maximum disperser biofilm was removed (Fig. 5b). Note that the 3oC12HSL signal is still made while IPTG is added so the small number of remaining colonizer cells are not able to form a biofilm while the disperser cells are being removed.
Fine-tuning consortial populations using the araBAD promoter
To fine-tune the consortial composition, a plasmid with the araBAD promoter instead of the lasI promoter for bdcAE50Q was constructed so that BdcAE50Q may be produced in the initial colonizer cells by adding arabinose. We confirmed the activity of the araBAD promoter in the new construct by testing the swimming motility of the new colonizer strain (E. coli hha/pPBAD-bdcAE50Q-rfp-lasR), as BdcAE50Q decreases the concentration of c-di-GMP by binding it and thereby increases swimming motility (Supplementary Methods)16. As expected, swimming motility of the araBAD promoter-controlled initial colonizer strain was increased 2.2±0.3-fold by adding arabinose (halo diameter 6.2±0.1 cm with 0.2% arabinose versus 2.8±0.1 cm with no arabinose), whereas the swimming motility of the old initial colonizer strain (E. coli hha/pBdcAE50Q-rfp-lasR) was not changed by arabinose addition (Supplementary Fig. S3). Hence, the araBAD promoter controls bdcAE50Q expression as a function of arabinose concentration.
To disperse the new initial colonizer cells (E. coli hha/pPBAD-bdcAE50Q-rfp-lasR, red, initial colonizer-2), we added arabinose (1 wt%) after both the initial colonizer-2 and the disperser biofilms were formed in the microfluidic channel. Dispersal of the initial colonizer-2 biofilm increased with production of BdcAE50Q via arabinose addition (Fig. 6a,b). Therefore, the initial colonizer-2 cells could be dispersed upon adding arabinose.

(a) Initial colonizer-2 biofilms (E. coli hha/pPBAD-bdcAE50Q-rfp-lasR, red) were developed in the microfluidic device for 10 h, then disperser cells (E. coli hha/pHha13D6-gfp-lasI, green) were seeded for 5 h to form both initial colonizer-2 and disperser biofilms in LB-glucose medium. After 21 h, 1% arabinose in tryptone medium was added for an additional 26 h to disperse the initial colonizer-2 biofilm. Three independent cultures were tested, and representative images of biofilms from channel 2 at each time point are shown. Scale bar indicates 20 μm. (b) Biomass (μm3 μm−2) of initial colonizer-2 and disperser biofilms at each time point in channel 2. Error bars indicate the s.d. from two different positions.
To vary the consortial biofilm population in the microfluidic device using the araBAD promoter and the initial colonizer-2 strain so that the population could be controlled by arabinose addition, a gradient of initial colonizer-2 biofilms was developed across the channels 1 through 8 of the microfluidic device. In this setup, the maximum initial colonizer-2 biofilm is formed in channel 8, then a uniform concentration of disperser cells (E. coli hha/pHha13D6-gfp-lasI, green) were seeded in all eight channels to form both initial colonizer-2 and disperser biofilms at different biofilm ratios across the eight channels at 21 h (Fig. 7a). The initial colonizer-2 biofilms were dispersed to various degrees from 62% dispersal in channel 2 to 26% dispersal in channel 6 at 37 h, while the disperser biofilms were not affected (Fig. 7b). Thus, the consortial population of initial colonizer and disperser cells may be controlled.

(a) A gradient of initial colonizer-2 biofilms (E. coli hha/pPBAD-bdcAE50Q-rfp-lasR, red) was developed across channels 1 through 8 in the microfluidic device for 10 h such that the maximum initial colonizer-2 biofilm was formed in channel 8, then a uniform concentration of disperser cells (E. coli hha/pHha13D6-gfp-lasI, green) were seeded for 5 h in all eight channels to form both initial colonizer-2 and disperser biofilms in LB-glucose medium at different biofilm ratios across the eight channels. After 21 h, 1% arabinose in tryptone medium was added to disperse the initial colonizer-2 biofilm. Three independent cultures were tested, and representative images of biofilms at time 21 h and 37 h are shown. Scale bar indicates 20 μm. (b) Biomass (μm3 μm−2) of initial colonizer-2 and disperser biofilms at time 21 h and 37 h in the eight channels. Error bars indicate the s.d. from two different positions.
Discussion
We developed a synthetic μBE system by combining a QS signalling module with two of our engineered biofilm dispersal proteins. With this synthetic circuit, in a microfluidic channel, we formed an initial colonizer biofilm with cells tagged red, introduced a second cell type (dispersers, tagged green) into this existing biofilm, created a means of communication between the two cell types and formed a robust biofilm with the disperser cells in an existing initial colonizer biofilm. We then displaced the initial colonizer cells in the biofilm with a QS signal from the disperser cells, and removed the disperser cells with a chemically induced switch. Our work demonstrates that biofilms can be formed, that new cells may be engineered to integrate and then replace the initial colonizer biofilm, and that both cell types may be removed, which is a promising strategy for applications requiring different kinds of engineered cells such as creating a biorefinery.
Although some of the biofilms may be dispersed naturally upon changes in environmental conditions (for example, nutrition level and oxygen depletion)39, it is a significant challenge to remove biofilms40,41 as cells in biofilms are cemented in place by the secreted polymer matrix consisting of polysaccharide, protein, DNA and lipids1. The matrix holds bacterial cells together and forms a protective barrier that confers resistance to killing by nonspecific and specific host defenses during infection and that confers tolerance to various antimicrobial agents such as disinfectants and antibiotics1. Thus, the defensive nature of the biofilm colony makes most biofilms difficult or impossible to eradicate39; hence, our demonstration that both the initial colonizer and disperser biofilms may be nearly completely removed is significant.
To preferentially remove one type of cell in a biofilm, our system requires that the second cell type elicits robust growth such that it can attach to the existing biofilm and propagate, that it flourishes, that it communicates to the other cell type via a QS signal, and that it displaces the existing biofilm without itself being displaced so that it instead forms a strong biofilm. Here, we produced the QS signal in the biofilm itself to remove the initial colonizer cells. As the signal accumulated, the engineered BdcA in the initial colonizer cells reduces c-di-GMP levels, which results in a cascade of events, such as an increase in motility and reduction in adhesion production, that allows the initial colonizer cells to disperse16.
As the initial colonizer cells disperse, the disperser cells must form a robust biofilm. After the disperser biofilm is formed, the engineered Hha protein, once induced, causes dispersal by inducing cell lysis15. Therefore, our synthetic μBE system provides a useful platform for the removal of existing deleterious biofilms via generating signalling molecules in situ. In addition, as the disperser cells grow more slowly than the initial colonizer ones, the disperser cells cannot displace the initial colonizer biofilm based on a difference in growth rates. This clearly demonstrates that a QS circuit was required to complete this feat of progressive biofilm development/dispersal. As several biofilm dispersal signals have been identified including the auto-inducing peptide of the agr QS system of Staphylococcus aureus42, changes in carbon sources43, reduction in the concentration of c-di-GMP16 (as utilized here with BdcA), surfactant44, cis-2-decenoic acid45, as well as D-amino acids46, we envision that other biofilm dispersal mechanisms may also be utilized to control biofilms.
The μBE device described here offers several advantages over the commercially available BioFlux device developed by Benoit et al.47 and other microfluidic devices used for biofilm study48. With our device, we can precisely control the development of biofilm by intermittent flow of nutrients, completely isolate the biofilm from the media inlet and gradient-generating channels using the pneumatic valves, and sequentially introduce different cell types into the biofilm chamber. Of course, the ability to study a range of concentrations simultaneously with the eight channels (for example, Fig. 3c,d) was instrumental in analysing the effect of various concentrations of 3oC12HSL and IPTG.
Bacterial QS systems have the attractive design features that they utilize diffusible signals20. Here we show, for the first time, that a QS system may be utilized with biofilm dispersal proteins to control consortial biofilm formation; that is, that an existing biofilm may be formed and then replaced by another biofilm, which then may be removed. These types of synthetic QS circuits may be used to pattern biofilms by facilitating the reuse of platforms and to create sophisticated reactor systems that will be used to form biorefineries. Although it may not be needed in practice to remove one biofilm with another, we chose to show this may be accomplished in order to show in principle that biofilms may be controlled; that is, that biofilms may be dispersed and that consortia populations may be controlled such that complex synthetic biocatalysis may be performed. Furthermore, these systems may be adopted in industrial and clinical processing as an alternative strategy to overcome the current limitations of biofilm control.
Methods
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1 and were cultured at 37 °C. LB49 with 0.2% glucose (LB-glucose) was used in all of the non-microfluidic experiments, and M949 supplemented with 0.2% glucose (M9-glucose), LB-glucose, and tryptone medium (10 g of tryptone and 2.5 g of NaCl per litre) were used in the microfluidic device. Kanamycin (50 μg ml−1) was used for overnight cultures, chloramphenicol (100 μg ml−1) was used for maintaining the pCA24N-based plasmids and erythromycin (300 μg ml−1) was used for maintaining the pCM18-based plasmids.
Plasmid construction
All primers used for cloning are listed in Supplementary Table S2. Plasmid pHha13D6-gfp-lasI (Fig. 2a) contains hha13D6 (ref. 15) under the control of the IPTG-inducible T5-lac promoter, as well as gfp and lasI under the control of constitutive CP25 promoter. To form this plasmid, gfp was amplified by three rounds of PCR: the first PCR with primers gfp-F3 and gfp-R and template pCM18 (ref. 32) was to amplify gfp with the same RBS of rfp, and the second PCR with primers gfp-F2 and gfp-R using the first PCR product as a template, and the third PCR with primers gfp-F1 and gfp-R using the second PCR product as a template were performed to include the constitutive CP25 promoter of pCM18. pHha13D6-gfp was constructed by cloning the third PCR product into pCA24N-hha13D6 (ref. 15) using the NotI and BlpI restriction sites after hha13D6 sequence. The final construct pHha13D6-gfp-lasI was formed by cloning lasI with its native RBS from the P. aeruginosa PAO1 chromosome by using the lasI-F and lasI-R primers; the PCR product was cloned into pHha13D6-gfp using the BlpI restriction site. As a control plasmid for producing GFP and LasI but not producing Hha13D6, pGFP-lasI was constructed by inserting lasI into pCM18 using the lasI-F and lasI-R primers.
Plasmid pBdcAE50Q-rfp-lasR (Fig. 2b) contains bdcAE50Q16 under the control of the lasI promoter, as well as rfp and lasR under the control of the constitutive CP25 promoter. pBdcAE50Q was constructed by replacing the T5-lac promoter in pCA24N-bdcAE50Q16 with the lasI promoter from P. aeruginosa using the plasI-F and plasI-R primers; the PCR fragment was cloned into the AvaI and BseRI restriction sites. Plasmid pDsRed-lasR was constructed by inserting lasR and its native RBS into the NotI site downstream of the rfp sequence in pDsRed-Express (Clontech) using the lasR-F and lasR-R primers. As a BlpI restriction site lies within lasR but was required for the next cloning steps, the BlpI site in pDsRed-lasR was disrupted by site-directed mutagenesis15 (5′-GCTGAGC-3′ to 5′-TCTGAGC-3′) using the BlpIX-F and BlpIX-R primers (this mutation did not change the aa sequence), to form pDsRed-BlpIX-lasR. rfp and lasR were amplified from pDsRed-BlpIX-lasR by two rounds of PCR to include the constitutive CP25 promoter of pCM18: the first PCR was performed using the rfp-lasR-F2 and rfp-lasR-R primers, and the second PCR was performed using the rfp-lasR-F1 and rfp-lasR-R primers with the first PCR product. The final construct pBdcAE50Q-rfp-lasR was formed by inserting the rfp and lasR PCR products into the BlpI site downstream of bdcAE50Q in pBdcAE50Q. As a control plasmid for producing RFP and LasR but not producing BdcAE50Q, pRFP-lasR was constructed by inserting rfp and lasR using the rfp-lasR-F3 and rfp-lasR-R primers into pCM18-X, in which gfp was disrupted by introducing a truncation at Y66 of GFP using the gfpX-F and gfpX-R primers in pCM18. All plasmids were confirmed by PCR and DNA sequencing.
Microfluidic device
The poly(dimethyl)siloxane (PDMS)-based μBE device (Fig. 1b) was fabricated in the Materials Characterization Facility at Texas A&M University using conventional soft lithographic techniques50. The μBE device consists of a glass slide and two layers, a bottom layer, with a diffusive mixer and eight microchambers, and a top layer, which contains the pneumatic elements for controlling microvalves and a second diffusive mixer. The diffusive mixer in the bottom layer was used to generate different concentrations of dispersal signals (for example, IPTG for removing disperser cells) and to perfuse growth media into the biofilm microchambers. The mixer in the top layer was used to introduce bacteria into the microchambers at different cell densities (Fig. 1b). The dimensions of the diffusive mixers in both the top and bottom layers were 100 μm (width) ×150 μm (height) and 200 μm (width) ×200 μm (height), respectively, and the biofilm microchambers were 600 μm (width) ×150 μm (height). All pneumatic channels were 200 μm thick. The two layers were fabricated separately, and assembled by sequential oxygen plasma treatment and bonding (100 mTorr, 100 W, 40 s) in a reactive ion etcher. The top pneumatic layer was first aligned and bonded to the bottom diffusive mixer/microchamber membrane layer followed by bonding of the combined PDMS layer to a cover glass (22×50 mm). Tygon tubing (0.01′′ ID×0.03′′ OD, Saint Gobain performance plastics) was used for all fluidic connections. Two PicoPlus 11 syringe pumps (Harvard Apparatus) were used for each experiment to separately control fluid flow rates in the two layers. A temperature-controlled micro-incubator was used to maintain the temperature of the device at 37 °C. Moist air flowed continuously over the device in order to maintain humidity and avoid bubble formation inside the microchambers. The opening and closing of valves were pneumatically controlled by introducing vacuum or compressed air through the solenoid valves. The operation of solenoid valves and syringe pumps were remotely controlled through programs developed in-house for the LabVIEW platform (National Instruments).
Microfluidic biofilm experiments
For mono-species biofilm dispersal experiments, overnight cultures were washed and resuspended in M9 medium supplemented with glucose (0.2%) at a turbidity at 600 nm of ~1.0. The bacterial suspension was introduced into the eight biofilm microchambers through the top layer in the PDMS device (Fig. 1b). During this process, the main inlet valves (Fig. 1b) remained closed to prevent cells from entering and forming biofilm in the gradient-mixing channels, and to ensure proper mixing of dispersal signals before they enter the microchambers. The main outlet valves and seeding valves were then closed, and the culture was maintained without flow for 2 h to enable attachment of bacteria to the glass surface (seeding). After 2 h, both main inlet and outlet valves were opened, unattached cells were removed and the attached bacteria were allowed to grow by flowing LB-glucose at 2 μl min−1. After 3 h, the medium was switched from LB-glucose to M9-glucose for 3 h because we found that a sudden depletion of nutrients promoted rapid development of biofilms. The biofilm was then developed for another 3 h by introducing LB-glucose into the chambers in a semi-batch mode (55-min static and 5-min flow). Thus, within 9 h after seeding, a robust and mature biofilm was formed. To disperse the biofilm, LB-glucose and LB-glucose containing a single concentration of the dispersal signal (IPTG for disperser cells and 3oC12HSL for initial colonizer cells) was introduced through the two media inlets and allowed to mix in the serpentine gradient-generating channels to form eight concentrations of the dispersal signal in LB-glucose medium. Each stream leaving the diffusive mixer was used to perfuse a specific biofilm microchamber for 10 h.
For dual-species biofilm dispersal experiments using 3oC12HSL signalling, initial colonizer cell biofilms were developed uniformly across all eight microchambers for 9 h as for mono-species biofilms. During this 9-h period, unattached initial colonizer cells were continuously removed from the cell mixer and connecter tubing through the cell outlet by flowing M9 medium at 8 μl min−1. After formation of the initial colonizer biofilm, disperser cells (turbidity at 600 nm of 2.0 in M9-glucose) were continuously perfused into the microchamber for 5 h to allow disperser cells to colonize the initial colonizer biofilm as well as the glass surface in vacant regions. After 5 h, LB-glucose was introduced into the microchamber in the semi-batch mode (55-min static and 5-min flow) for 28–30 h. The static condition ensured biofilm development and build-up of 3oC12HSL needed for induction of the BdcAE50Q dispersal protein in initial colonizer cells. To remove the disperser cell biofilm, LB-glucose containing 2mM IPTG was introduced in semi-batch mode for 18 to 20 h.
For dual-species biofilm dispersal experiments using arabinose, initial colonizer cell biofilms were developed as a gradient across the eight microchambers for 10 h, then a uniform concentration of disperser cells were seeded for 5 h in all eight channels in LB-glucose medium. After 21 h, 1% arabinose in tryptone medium was added for an additional 26 h to disperse the initial colonizer biofilm.
Additional information
How to cite this article: Hong, S.H. et al. Synthetic quorum-sensing circuit to control consortial biofilm formation and dispersal in a microfluidic device. Nat. Commun. 3:613 doi: 10.1038/ncomms1616 (2012).
References
Flemming, H.- C. & Wingender, J. The biofilm matrix. Nat. Rev. Micro. 8, 623–633 (2010).
Sauer, K., Rickard, A. H. & Davies, D. G. Biofilms and biocomplexity. Microbe 2, 347–353 (2007).
Beech, I. B. & Sunner, J. Biocorrosion: towards understanding interactions between biofilms and metals. Curr. Opin. Biotechnol. 15, 181–186 (2004).
Dobretsov, S., Teplitski, M. & Paul, V. Mini-review: quorum sensing in the marine environment and its relationship to biofouling. Biofouling 25, 413–427 (2009).
Wood, T. K., Hong, S. H. & Ma, Q. Engineering biofilm formation and dispersal. Trends Biotechnol. 29, 87–94 (2011).
Brenner, K., You, L. & Arnold, F. H. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol. 26, 483–489 (2008).
Prüß, B. M., Besemann, C., Denton, A. & Wolfe, A. J. A complex transcription network controls the early stages of biofilm development by Escherichia coli. J. Bacteriol. 188, 3731–3739 (2006).
Jayaraman, A., Mansfeld, F. B. & Wood, T. K. Inhibiting sulfate-reducing bacteria in biofilms by expressing the antimicrobial peptides indolicidin and bactenecin. J. Ind. Microbiol. Biotechnol. 22, 167–175 (1999).
Lee, J., Jayaraman, A. & Wood, T. K. Indole is an inter-species biofilm signal mediated by SdiA. BMC Microbiol. 7, 42 (2007).
Lee, J., Maeda, T., Hong, S. H. & Wood, T. K. Reconfiguring the quorum-sensing regulator SdiA of Escherichia coli to control biofilm formation via indole and N-acylhomoserine lactones. Appl. Environ. Microbiol. 75, 1703–1716 (2009).
Hong, S. H., Wang, X. & Wood, T. K. Controlling biofilm formation, prophage excision, and cell death by rewiring global regulator H-NS of Escherichia coli. Microb. Biotechnol. 3, 344–356 (2010).
Lu, T. K. & Collins, J. J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl Acad. Sci. USA 104, 11197–11202 (2007).
Godessart, N., Muñoa, F. J., Regue, M. & Juárez, A. Chromosomal mutations that increase the production of a plasmid-encoded haemolysin in Escherichia coli. J. Gen. Microbiol. 134, 2779–2787 (1988).
García-Contreras, R., Zhang, X.- S., Kim, Y. & Wood, T. K. Protein translation and cell death: the role of rare tRNAs in biofilm formation and in activating dormant phage killer genes. PLoS ONE 3, e2394 (2008).
Hong, S. H., Lee, J. & Wood, T. K. Engineering global regulator Hha of Escherichia coli to control biofilm dispersal. Microb. Biotechnol. 3, 717–728 (2010).
Ma, Q., Yang, Z., Pu, M., Peti, W. & Wood, T. K. Engineering a novel c-di-GMP-binding protein for biofilm dispersal. Environ. Microbiol. 13, 631–642 (2011).
Fu, P. A perspective of synthetic biology: assembling building blocks for novel functions. Biotechnol. J. 1, 690–699 (2006).
Purnick, P. E. M. & Weiss, R. The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 10, 410–422 (2009).
Hooshangi, S. & Bentley, W. E. From unicellular properties to multicellular behavior: bacteria quorum sensing circuitry and applications. Curr. Opin. Biotechnol. 19, 550–555 (2008).
Choudhary, S. & Schmidt-Dannert, C. Applications of quorum sensing in biotechnology. Appl. Microbiol. Biotechnol. 86, 1267–1279 (2010).
Ryan, R. P. & Dow, J. M. Diffusible signals and interspecies communication in bacteria. Microbiology 154, 1845–1858 (2008).
Danino, T., Mondragón-Palomino, O., Tsimring, L. & Hasty, J. A synchronized quorum of genetic clocks. Nature 463, 326–330 (2010).
You, L., Cox, R. S., Weiss, R. & Arnold, F. H. Programmed population control by cell-cell communication and regulated killing. Nature 428, 868–871 (2004).
Kobayashi, H. et al. Programmable cells: interfacing natural and engineered gene networks. Proc. Natl Acad. Sci. USA 101, 8414–8419 (2004).
Williams, P. & Cámara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 12, 182–191 (2009).
Pesci, E. C., Pearson, J. P., Seed, P. C. & Iglewski, B. H. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 179, 3127–3132 (1997).
Brenner, K., Karig, D. K., Weiss, R. & Arnold, F. H. Engineered bidirectional communication mediates a consensus in a microbial biofilm consortium. Proc. Natl Acad. Sci. USA 104, 17300–17304 (2007).
Balagaddé, F. K. et al. A synthetic Escherichia coli predator-prey ecosystem. Mol. Syst. Biol. 4, 187 (2008).
Kambam, P. K. R., Henson, M. A. & Sun, L. Design and mathematical modelling of a synthetic symbiotic ecosystem. IET Syst. Biol. 2, 33–38 (2008).
Brenner, K. & Arnold, F. H. Self-organization, layered structure, and aggregation enhance persistence of a synthetic biofilm consortium. PLoS ONE 6, e16791 (2011).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006).
Hansen, M. C., Palmer, R. J. Jr, Udsen, C., White, D. C. & Molin, S. Assessment of GFP fluorescence in cells of Streptococcus gordonii under conditions of low pH and low oxygen concentration. Microbiology 147, 1383–1391 (2001).
Seed, P., Passador, L. & Iglewski, B. Activation of the Pseudomonas aeruginosa lasI gene by LasR and the Pseudomonas autoinducer PAI: an autoinduction regulatory hierarchy. J. Bacteriol. 177, 654–659 (1995).
Kitagawa, M. et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299 (2005).
Pearson, J. P. et al. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl Acad. Sci. USA 91, 197–201 (1994).
Charlton, T. S. et al. A novel and sensitive method for the quantification of N-3-oxoacyl homoserine lactones using gas chromatography–mass spectrometry: application to a model bacterial biofilm. Environ. Microbiol. 2, 530–541 (2000).
Flickinger, S. T. et al. Quorum sensing between Pseudomonas aeruginosa biofilms accelerates cell growth. J. Am. Chem. Soc. 133, 5966–5975 (2011).
Pearson, J. P., Van Delden, C. & Iglewski, B. H. Active efflux and diffusion are involved in transport of Pseudomonas aeruginosa cell-to-cell signals. J. Bacteriol. 181, 1203–1210 (1999).
Kaplan, J. B. Biofilm dispersal: mechanisms, clinical implications, and potential therapeutic uses. J. Dent. Res. 89, 205–218 (2010).
del Pozo, J. L. & Patel, R. The challenge of treating biofilm-associated bacterial infections. Clin. Pharmacol. Ther. 82, 204–209 (2007).
Bardouniotis, E., Huddleston, W., Ceri, H. & Olson, M. E. Characterization of biofilm growth and biocide susceptibility testing of Mycobacterium phlei using the MBEC™ assay system. FEMS Microbiol. Lett. 203, 263–267 (2001).
Boles, B. R. & Horswill, A. R. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 4, e1000052 (2008).
Sauer, K. et al. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 186, 7312–7326 (2004).
Boles, B. R., Thoendel, M. & Singh, P. K. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol. Microbiol. 57, 1210–1223 (2005).
Davies, D. G. & Marques, C. N. H. A fatty acid messenger is responsible for inducing dispersion in microbial biofilms. J. Bacteriol. 191, 1393–1403 (2009).
Kolodkin-Gal, I. et al. D-amino acids trigger biofilm disassembly. Science 328, 627–629 (2010).
Benoit, M. R., Conant, C. G., Ionescu-Zanetti, C., Schwartz, M. & Matin, A. New device for high-throughput viability screening of flow biofilms. Appl. Environ. Microbiol. 76, 4136–4142 (2010).
Lee, J.- H., Kaplan, J. & Lee, W. Microfluidic devices for studying growth and detachment of Staphylococcus epidermidis biofilms. Biomed. Microdevices 10, 489–498 (2008).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning, A Laboratory Manual 2nd edn (Cold Spring Harbor Laboratory Press, 1989).
Kim, J., Hegde, M. & Jayaraman, A. Co-culture of epithelial cells and bacteria for investigating host-pathogen interactions. Lab Chip 10, 43–50 (2010).
Acknowledgements
This work was supported by the National Institutes of Health (R01 GM089999) and by the National Science Foundation (CBET 0846453). We thank the National Institute of Genetics in Japan for providing the Keio and ASKA strains as well as Prof. Barbara Iglewski for providing the 3oC12HSL reporter strain.
Author information
Authors and Affiliations
Contributions
T.K.W. and A.J. conceived the experiments with T.K.W. primarily supervising the synthetic biology experiments and A.J. supervising the microfluidic experiments. S.H.H. constructed the strains and performed the assays, M.H. and J.K. performed the microfluidic experiments and evaluated the biofilm compositions, and X.W. evaluated some of the bacterial strains. All authors helped write the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Movie 1
Biofilm dispersal using a synthetic quorum sensing circuit. An initial colonizer biofilm (red, BdcAE50Q+, LasR+; E. coli hha/pBdcAE50Q-rfp-lasR) was developed for 9 h, then disperser cells (green, Hha13D6+, LasI+; E. coli hha/pHha13D6-gfp-lasI) were seeded for 5 h to form a dual-species biofilm. As the disperser cells increased in density, the initial colonizer biofilm dispersed gradually based on production and detection of the quorum sensing signal 3oC12HSL. After 44 h, 2 mM IPTG was added for an additional 18 h to remove the disperser biofilm as well. The time lapse movie is from images taken at one hour intervals. (AVI 3142 kb)
Supplementary Information
Supplementary Figures S1-S4, Supplementary Tables S1-S2, Supplementary Methods and Supplementary References. (PDF 709 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hong, S., Hegde, M., Kim, J. et al. Synthetic quorum-sensing circuit to control consortial biofilm formation and dispersal in a microfluidic device. Nat Commun 3, 613 (2012). https://doi.org/10.1038/ncomms1616
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms1616
This article is cited by
-
De novo engineering of a bacterial lifestyle program
Nature Chemical Biology (2023)
-
Microfluidic hotspots in bacteria research: A review of soil and related advances
Soil Ecology Letters (2023)
-
The Application Potential of Synthetic Biology in Microbial Communication
Current Clinical Microbiology Reports (2023)
-
Zooming in to acquire micro-reaction: Application of microfluidics on soil microbiome
Soil Ecology Letters (2022)
-
Emerging Molecular Tools for Engineering Phytomicrobiome
Indian Journal of Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
