
Abstract
Lithium ion batteries are encountering ever-growing demand for further increases in energy density. Li-rich layered oxides are considered a feasible solution to meet this demand because their specific capacities often surpass 200 mAh g−1 due to the additional lithium occupation in the transition metal layers. However, this lithium arrangement, in turn, triggers cation mixing with the transition metals, causing phase transitions during cycling and loss of reversible capacity. Here we report a Li-rich layered surface bearing a consistent framework with the host, in which nickel is regularly arranged between the transition metal layers. This surface structure mitigates unwanted phase transitions, improving the cycling stability. This surface modification enables a reversible capacity of 218.3 mAh g−1 at 1C (250 mA g−1) with improved cycle retention (94.1% after 100 cycles). The present surface design can be applied to various battery electrodes that suffer from structural degradations propagating from the surface.
Similar content being viewed by others


Introduction
The limited specific capacity of cathode materials is one of the main obstacles to increasing the energy densities of current lithium-ion batteries1,2,3,4,5,6,7. In this regard, Li-rich layered oxides with the chemical formula of xLi2MnO3·(1−x)LiMO2 (M=3d and/or 4d transition metal; TM)8,9,10,11,12 have attracted significant attention because their specific capacities usually exceed 200 mAh g−1 at high operating voltages over 3.5 V versus Li+/Li, in contrast with their conventional layered counterparts, that is, LiCoO2 (∼145 mAh g−1) and LiCo1/3Mn1/3Ni1/3O2 (∼165 mAh g−1)13. Despite the resulting high energy density, most of these materials suffer from voltage drop and capacity fading during cycling, limiting their use in practical cells.
To achieve a high specific capacity, Li-rich layered oxides must be activated in the first charge involving Li ion extraction and oxygen evolution. During this process, TMs are liable to migrating to the neighbouring Li slabs (Fig. 1a)8,14,15,16,17, driving a phase transition of the overall crystal framework to spinel and/or rocksalt structures. This spontaneous phase transition continues with cycling and causes a drop in the operation voltage and specific capacity18,19,20, which undermines the original advantage of the Li-rich layered phase.

(a) Without any surface modifications. (b) With conventional surface coating. (c) With surface modification in which Ni is regularly positioned between the face-to-face Li sites of the superlattice Mn layers in the Li-rich layered surface structure.
A number of approaches have been introduced to mitigate this unwanted phase transition. Surface modification has been one of the primary remedies because the given phase transition typically begins from the surface of the particle21,22,23, and well-designed surface structures can suppress this transition process from the initial stage24,25. In this direction, various surface-coating materials, namely, metal oxides26,27, metal phosphates28, spinel-type materials29,30 and olivine-type materials20, have been reported (Fig. 1b). Despite the substantial contributions of these coating materials towards cycle life enhancement, it is still desirable to find a new surface structure with further improved properties. To this end, one can raise a fundamental question: what would be the ideal surface structure for Li-rich layered oxides? In answering this question, a good surface structure would have a homogenous crystal structure at the atomic level with the host Li-rich layered phase so that the surface structure can be uniformly formed over the entire particle surface during synthesis31,32 and can also remain integrated with the host phase during repeated charge–discharge cycles. In this line, the previous surface modifications with different crystal structures have lattice mismatches and are therefore expected to suffer from disintegration during prolonged cycling, as well as non-uniform integration in the pristine state. More critically, a good surface structure of Li-rich layered oxides should be able to suppress the aforementioned transitions. A good surface structure is also desired to be electrochemically active with decent ionic/electronic conductivity so that it does not sacrifice the high specific capacity of the host Li-rich material.
In this work, we develop a surface structure for Li-rich layered cathodes. The surface structure addresses the issues of Li-rich layered oxides and thus improves their structural stability during repeated charge–discharge cycles. In particular, in an effort to minimize the difference in interfacial free energy (thus, formation energy)33,34 with the host material (Li-rich layered phase) and preserve the high electrochemical properties at the surface, Li2MnO3 with a consistent crystal framework to the host is adopted. Moreover, the Li2MnO3 surface phase is further modified in such a way that nickel (Ni) is regularly arranged between the face-to-face Li sites in the superlattice manganese (Mn) layers of Li2MnO3 (Fig. 1c). The presence of Ni in the 3 nm-thick surface layer effectively suppresses phase transition to spinel and/or rocksalt structures, leading to improved cycling and rate performance.
Results
Material synthesis and structure characterization
In our experiment, Li2MnO3 was first coated on a Li-rich layered oxide, 0.5Li2MnO3–0.5LiNi0.44Mn0.32Co0.24O2. The modification of the surface structure to position Ni in the Li layers was achieved by inter-diffusion of Ni from the host during a heat treatment in the coating process. See the Methods section for details and Supplementary Note 1 for additional description of the bulk and surface materials used, including their crystal structures (Supplementary Fig. 1), X-ray diffraction patterns (Supplementary Fig. 2a), and transmission electron microscopy (TEM; Supplementary Fig. 2b,c) characterization. The diffraction patterns were indexed based on a hexagonal unit cell with the space group  (No. 166)17. With increasing Li2MnO3 content, the normalized intensity of the peak at 20.83°, which is reflective of the superlattice, increases gradually (Fig. 2a, Supplementary Fig. 3 and Supplementary Table 1). This superlattice is associated with the repeated 2:1 atomic arrangement between TM and Li in the TM layers10. As neither impurity phases nor significant peak shifts were detected for the modified sample, the increased intensity of the superlattice peak indicates the formation of a Li2MnO3-like phase on the surface without perturbing the host framework. The lattice parameters before and after the surface modification were calculated to be ahex=2.802(3) Å, chex=14.174(1) Å and ahex=2.801(1) Å, chex=14.171(7)Å, respectively, verifying that the surface phase was structurally well linked to the host framework. The surface layers were coated over all of the host particles in a uniform fashion with well-controlled thickness around 3 nm according to our TEM characterization with multiple particles (Supplementary Fig. 4).
(No. 166)17. With increasing Li2MnO3 content, the normalized intensity of the peak at 20.83°, which is reflective of the superlattice, increases gradually (Fig. 2a, Supplementary Fig. 3 and Supplementary Table 1). This superlattice is associated with the repeated 2:1 atomic arrangement between TM and Li in the TM layers10. As neither impurity phases nor significant peak shifts were detected for the modified sample, the increased intensity of the superlattice peak indicates the formation of a Li2MnO3-like phase on the surface without perturbing the host framework. The lattice parameters before and after the surface modification were calculated to be ahex=2.802(3) Å, chex=14.174(1) Å and ahex=2.801(1) Å, chex=14.171(7)Å, respectively, verifying that the surface phase was structurally well linked to the host framework. The surface layers were coated over all of the host particles in a uniform fashion with well-controlled thickness around 3 nm according to our TEM characterization with multiple particles (Supplementary Fig. 4).

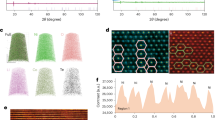
(a) X-ray diffraction patterns of the pristine electrode and the surface-modified electrode with 5 wt% Li2MnO3. (b,c) HAADF-STEM images of (b) the pristine electrode and (c) the surface-modified electrode with 5 wt% Li2MnO3. b,c were obtained along the rhombohedral [100] and monoclinic [010] directions, respectively. The red, blue and orange circles represent TM (M=Ni, Co and Mn), Ni and Mn, respectively. Scale bars, 5 Å.
High-angle annular dark field-scanning TEM (HAADF-STEM) images of both the pristine and surface-modified electrodes display typical layered atomic columns (red circles in Fig. 2b and orange circles in Fig. 2c and Supplementary Fig. 5, respectively) based on their Z-contrast35. Interestingly, in the case of the surface-modified electrode, additional atomic columns (blue circles and yellow arrows) were observed in the Li layers of the surface-modified region. Since light elements, such as Li and O, are invisible in HAADF imaging36,37, the bright contrasts between the TM layers indicate the existence of heavy TMs, as the given modified material consists solely of TMs, Li and O. In addition to STEM characterization, energy-dispersive X-ray spectroscopy (EDX) analyses showed that there was a high Ni content across the surface region of the modified electrode (Supplementary Fig. 6). Thus, these combined analyses suggest the diffusion of Ni atoms from the host to the surface during the heat treatment. The distance (∼3 nm) from the surface to the far edge, where the interlayer Ni begins to be found, agrees well with the thickness of the Ni layer in the EDX analysis (Supplementary Fig. 6) and the coating thickness in the low-magnification TEM images (Supplementary Fig. 4). Given the fact that at this outermost region the primary phase is Li2MnO3 and a high Ni content is found, it can be concluded that the observed orange and blue atomic columns in the TM and Li layers are Mn and Ni, respectively. The well-known high diffusivity of Ni during heat treatment38,39 is also in keeping with Ni diffusion from the host region. The atomic resolution STEM images exhibited weaker contrast of Ni atoms compared with that of Mn atoms (Fig. 2c). Since the HAADF mode provides the contrast in proportion to the atomic density and the atomic number, the weaker contrast of the Ni atoms indicates their lower occupancy. On the other hand, the crystallinity of the modified surface at the pristine state was relatively lower, and therefore the superlattice arrangement between the Mn and Li atoms in the TM layers was difficult to observe in the atomic resolution. However, the given superlattice arrangement was clearly detected from the X-ray diffraction results (Fig. 2a, Supplementary Fig. 3 and Supplementary Table 1). According to X-ray photoelectron spectroscopy spectra (Supplementary Fig. 7), Mn3+ was detected, indicating that some Mn in Li2MnO3 was reduced in the modified surface because of the Ni migration that supplies additional positive charge. See Supplementary Note 2 for detailed description on the Ni diffusion. With this substantial presence of Ni, the blue columns are unlikely to represent Mn because the structure would have then undergone serious Mn migration to the Li slabs, and this TM mixing would have resulted in a phase transition to a spinel-like structure40,41. Because of the low Co content in the EDX profile (Supplementary Fig. 6), the blue columns are also unlikely to represent Co. Additional structural description of the surface-modified electrode is provided in Supplementary Note 2.
Electrochemical properties
The surface-modified electrode exhibited good electrochemical performance in various aspects, such as specific capacity, rate capability and cycle life. Detailed cell preparation and measurement conditions are described in the Methods section. From the testing of various contents of Li2MnO3 (Supplementary Fig. 8), 5 wt% was chosen as a main surface-modified electrode in the current investigation. When galvanostatically tested in the voltage range of 2.0–4.8 V versus Li+/Li at 0.1C (25 mA g−1), the surface-modified electrode exhibited higher first charging and discharging capacities than the pristine electrode (Fig. 3a). In the first charge, the surface-modified and pristine electrodes delivered 329.4 and 315.0 mAh g−1, respectively, whereas in the first discharge, they showed 292.7 and 284.2 mAh g−1, respectively. The larger charge capacity of the surface-modified electrode is ascribed to its higher content of a Li2MnO3-like phase with a high specific capacity. The magnified discharge profile (Supplementary Fig. 9) of the surface-modified electrode exhibited weakened voltage fading around 3.0 V, which implies that the suppressed phase transition contributes to the increased discharge capacity. By contrast, most of the previous approaches involving other substitutional elements that are indiscreetly distributed in the Li interlayer (often referred to pillar atoms) impair Li ion migration and consequently sacrifice the intrinsic electrochemical performance of the host Li-rich material42,43,44.

(a), The first charge–discharge voltage profiles of the pristine electrode and the surface-modified electrode with 5 wt% Li2MnO3 in the range of 2.0–4.8 V at 0.1C (25 mA g−1). (b) Comparative rate capabilities of both electrodes measured at various C-rates after the first activating cycle at 0.1C (25 mA g−1). (c,d) Cycling performance of both electrodes at (c) 1C (250 mA g−1) and (d) 3C (750 mA g−1) after the first activating cycle at 0.1C (25 mA g−1).
The surface-modified electrode also displayed improved rate capability (Fig. 3b, and Supplementary Figs 8 and 10). After the activation in the first cycle, as the C-rate increased by 5, 10, 30, 50 and 100 times from 0.1C (1C=250 mA g−1), the surface-modified electrode retained 85.8%, 77.9%, 63.3%, 55.4% and 43.1% of the capacity (281.2 mAh g−1) in the second cycle, respectively. By contrast, with the same C-rate variations, the pristine electrode retained 85.8%, 76.1%, 54.8%, 44.4% and 32.0%, even though its capacity (268.4 mAh g−1) in the second cycle was lower.
More critically, the surface-modified electrode showed better cycling performance than the pristine electrode. When measured at 0.1C (25 mA g−1), the discharge capacity of the surface-modified electrode was 292.3 mAh g−1 in the second cycle, and this value dropped only to 259.7 mAh g−1 after 45 cycles, corresponding to an 88.8% capacity retention (Supplementary Fig. 11). After the same number of cycles, however, the pristine electrode retained only 80.9% with respect to the capacity (279.7 mAh g−1) in the second cycle. When measured at higher rates of 1C (250 mA g−1) and 3C (750 mA g−1), 94.1 and 98.2% of the capacities (218.3 and 176.0 mAh g−1) in the second cycle were preserved for the surface-modified electrode after 100 cycles (Fig. 3c,d). These values are in contrast with those (80.1 and 75.3%) of the pristine counterpart. Remarkably, unlike the pristine electrode, the discharge capacity of the surface-modified electrode became saturated even after several cycles. These capacity retentions are quite noticeable, as they are better than those of other nano-structured45,46, cation-doped47,48,49 and surface-coated20,26,27,28,29,30,50 Li-rich layered oxide electrodes reported to date. Consistent with the first cycle, the improved capacity retentions over the prolonged cycles are attributed to the suppressed phase transition, as verified by long-term cycling (Supplementary Figs 12 and 13) and differential capacity results (Supplementary Fig. 14). More electrochemical results are provided in Supplementary Note 3.
Reaction mechanism
The origin of the improved electrochemical stability by the surface modification was revealed in detail by HAADF-STEM analysis. The HAADF-STEM image of the pristine electrode after the first charge displays rocksalt-like and spinel-like phases sequentially from the surface (Fig. 4a), indicating that spontaneous transition to these phases progresses quite a bit even in the early period of cycling. By contrast, in the same charged state, the surface region of the modified electrode kept the original layered structure (Fig. 4b). In particular, the image of the outermost region (the area above the yellow dotted line) provides the atomic-scale information on its crystal structure on charge. As opposed to the pristine electrode, once again, the surface region preserves the layered framework all the way to the edge, indicating its structural robustness. In many regions, the repeated 2:1 superlattice arrangement of the Mn and Li atoms in the TM layers was clearly detected; one blank spot reflective of the original Li sites (now emptied on charge) was observed after every two orange circles. Also, similar to the state before cycling, Ni atoms were observed in the Li layers, and a majority of the Ni atoms are located right next to the face-to-face Li sites located in the TM layers (Fig. 4b). On the whole, it appears that the atoms in the surface region are ordered in the superlattice configuration, presumably because, on Li ion extraction, the Ni atoms in the Li layers migrate and settle at the positions next to the face-to-face Li sites. This atomic rearrangement reflects the enhanced crystallinity of the surface layer during charge, although analyses to identify detailed mechanism is underway. This trend was confirmed from multiple spots along the surface and also from multiple particles (Supplementary Fig. 15).

(a,b) HAADF-STEM images of (a) the pristine electrode and (b) the surface-modified electrode with 5 wt% Li2MnO3 after the first charge. a,b were obtained along the rhombohedral [100] (=cubic spinel [110]) and monoclinic [100] directions, respectively. (c) Nyquist plots of both electrodes after the tenth and hundredth cycles. (d) Graphical illustration of the effect of the surface modification. The green, purple, red, orange and blue circles represent Li, TM (M=Ni, Co and Mn), O, Mn and Ni, respectively. Scale bars, 5 Å.
By the assistance of this robust layered surface structure incorporating Ni atoms specifically located at the 2c sites, the inner region (the area below the yellow dotted line) preserves the original layered framework (purple circle) with no cation mixing. In Li-rich layered oxides, it is known22,51 that Li extraction is accompanied with the oxidation of oxygen atoms to O(2-δ)−, which can lead to the oxygen loss. The oxidation of oxygen perturbs its stability via delocalization of the bonding electrons52,53. It was reported54 that the structural stability is deteriorated more significantly by the 4i oxygen vacancy than the 8j oxygen vacancy. Once the 4i oxygen atoms are destabilized, the TM migration through the adjacent tetrahedral sites would be accelerated. In this regard, the improved structural stability is credited to the Ni coordination (2c sites) to the adjacent TM layers because the Ni atoms form strong bonding with the adjacent oxygen atoms (4i sites) and suppress the TM migration through the adjacent tetrahedral sites based on high electrostatic repulsion between the TMs and Ni atoms. The decreased interlayer distance (4.4 Å) in the modified surface compared with that (4.6 Å) in the host directly reflects the strong bonding between TMs and oxygen atoms in the surface region (Fig. 4b). Thus, it can be concluded that the robust superlattice surface structure enabled by the well-defined Ni coordination (2c sites) plays a crucial role in inhibiting the structural changes that would otherwise propagate from the surface. It is well known8,14,15,16,17 that in Li-rich layered oxides cation mixing is a spontaneous process at the very beginning of the first charging, and even the migration of a small portion of TMs can initiate structural disordering sufficient for eventual phase transition to spinel and/or rocksalt phases. In this sense, the observed layered surface structure with high atomic regularity is remarkable. The structural stability of the surface-modified electrode was also verified by ex situ X-ray diffraction analysis (Supplementary Fig. 16).
Electrochemical impedance spectroscopy (EIS) analyses also reveal the more stable electrode/electrolyte interface of the surface-modified electrode for 100 cycles compared with the pristine electrode (Fig. 4c and Supplementary Fig. 17); the semi-circle of the surface-modified electrode in the EIS curve stays smaller and more steady throughout cycling. Detailed EIS results are summarized in Supplementary Table 2. The lower interfacial resistance of the surface-modified electrode can be explained by its superlattice layered structure with facile Li ion diffusion (Supplementary Fig. 18 and Supplementary Table 3), as well as mitigated electrolyte side reactions due to the stable electrode surface. In the same line, the excessive and indiscreet Ni arrangement in the Li interlayer could hinder the Li ion diffusion42, sacrificing the electrochemical properties of the host material. The effect of the modified surface on the structural and interfacial stability is graphically summarized in Fig. 4d and is further described in Supplementary Note 4.
Discussion
We have demonstrated a robust surface structure for Li-rich layered oxide cathodes. The Li-rich layered framework of the modified surface with atomic-level structural consistency with the host phase allows the surface region to remain seamlessly connected during battery operation. Moreover, in this modified surface, Ni atoms are regularly positioned between the TM layers and maintain the overall Li-rich layered framework. To the best of our knowledge, the prepared Li-rich surface structure, in which TMs are located specifically between the face-to-face Li sites in the TM layers, has never been reported previously. This unique atomic coordination significantly suppresses a detrimental transition to spinel and/or rocksalt phases by mitigating TM mixing, a main trigger step of the phase transition.
The present surface structure is also clearly distinct from a number of surface-coating approaches to date because the majority of the reported surface materials adopted different structures from the host materials and therefore impaired the specific capacities and electronic and/or ionic conductivities of the host materials. Although the Ni atoms at the 2c sites do not block the channels for Li ion diffusion, the high Ni content would decrease the Li content, which could affect the Li diffusivity and the reversible capacity.
Complementing the structural stability and electrochemical performance, the surface structure achieved in this study was created through simple dip-dry and heat-treatment processes, which neither perturb the host framework nor generate any impurities. Taking the present work as the first step, detailed properties (thickness, Ni content and so on) related to the surface structure can be further optimized by tuning the surface-to-bulk ratio and heat treatment condition. Although a surface structure based on NiO rocksalt was reported to stabilize Ni-rich layered cathodes42, the given approach is not appropriate to form Li-rich surface phases as in the present case. Highly important is the fact that this study provides a general design principle on how to build the surface structure, targeting a variety of battery electrode materials that suffer from structural collapse starting heavily from their surfaces: creating a surface structure with the same crystal structure as the host phase, followed by a heat treatment to stimulate atomic interaction with the host structure. During the interaction, the surface structure would modify itself and transform to a more stable phase, as the present material demonstrated the inter-diffusion of highly diffusive Ni from the host phase to the specific crystal sites that can play a critical role in keeping the framework during Li ion extraction.
Methods
Synthesis
Commercial 0.5Li2MnO3–0.5LiNi0.44Mn0.32Co0.24O2 (BASF SE)55,56 was used for both the pristine and surface-modified electrodes. First, 1 g of 0.5Li2MnO3–0.5LiNi0.44Mn0.32Co0.24O2 was dispersed in 10 ml of anhydrous ethanol by ultrasonic stirring for 12 h. All of the chemicals were purchased from Sigma-Aldrich and used without any purification. Lithium acetate (LiCOOCH3·2H2O) and manganese acetate (Mn(COOCH3)2·4H2O) were added in a 2:1 molar ratio to the host material suspension, and this suspension was vigorously stirred for 12 h. The suspension was then heated at 70 °C until its solvent evaporated. The remaining powder was calcined at 600 °C for 6 h to obtain the final surface-modified sample. The Li2MnO3 content was controlled to be 1, 3, 5, 10 and 20 wt%.
Characterization
The crystal structure was investigated by X-ray diffraction (Rigaku) analysis in the 2θ range of 10°–80° with a scan step of 0.02° and an acquisition time of 2 s for each step. The specimens were sealed in a homemade holder inside an Ar-filled glove box to prevent hydration from moisture in air. Morphology and elemental analyses were carried out by TEM (JEM-ARM200F, JEOL) characterization at an acceleration beam voltage of 300 kV and its attached EDX (Quantax 400, Bruker), respectively. HAADF-STEM characterization was performed using a JEM-ARM200F (JEOL) equipped with a cold field-emission gun and double spherical aberration correctors, operated at an acceleration voltage of 200 kV. The probe beam had a convergent semi-angle of 23 mrad. The inner-outer semi-angle of the HAADF detector was 90–370 mrad. The electron probe current was 11 pA. The dwell time per pixel was 38 μs. The size of each pixel was 16.5 × 16.5 pm2. The oxidation states of TMs were examined using X-ray photoelectron spectroscopy (Thermo Scientific Sigma Probe). The impedance was measured using a frequency response analyser (VSP multi potentiostat, BioLogic) over the frequency range of 0.01 Hz–1 MHz with an amplitude of 10 mV.
Electrochemical test
For electrode fabrication, a slurry consisting of 80 wt% of active material, 10 wt% of super P and 10 wt% of poly(vinylidene fluoride) was first prepared. The slurry was then cast onto aluminum foil using the doctor blade technique, followed by drying at 70 °C for 12 h in a vacuum oven. The mass loading of the active material in each electrode was ∼4.0 mg cm−2. Li foil was used as the counter and reference electrode. The electrolyte solution was 1 M lithium hexafluorophosphate (LiPF6) dissolved in an ethylene carbonate/dimethyl carbonate mixture (1:1 v/v). Polypropylene membranes (2,400, Celgard) were used as separators. These cell components were assembled in the form of CR2032 coin cells in an argon-filled glove box. All of the cells were aged for 6 h before any electrochemical processes to ensure sufficient soaking of the electrolyte into the separator and electrode. The cells were cycled in the voltage range of 2.0–4.8 V (versus Li+/Li) at 25 °C using a battery tester (WBCS3000L, Wonatech). The C-rate in this study is defined based on 1C=250 mA g−1. For the calculation of all of the gravimetric capacities/currents, the mass of the active material only was taken into account.
Data availability
All relevant data are available from the authors on request.
Additional information
How to cite this article: Kim, S. et al. A stable lithium-rich surface structure for lithium-rich layered cathode materials. Nat. Commun. 7, 13598 doi: 10.1038/ncomms13598 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Tarascon, J. M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Whittingham, M. S. Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4301 (2004).
Armstrong, A. R. & Bruce, P. G. Synthesis of layered LiMnO2 as an electrode for rechargeable lithium batteries. Nature 381, 499–500 (1996).
Yamada, A. et al. Room-temperature miscibility gap in LixFePO4 . Nat. Mater. 5, 357–360 (2006).
Sun, Y. K. et al. Nanostructured high-energy cathode materials for advanced lithium batteries. Nat. Mater. 11, 942–947 (2012).
Chung, S. Y., Bloking, J. T. & Chiang, Y. M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater. 1, 123–128 (2002).
Dunn, B., Kamath, H. & Tarascon, J. M. Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011).
Johnson, C. S. et al. The significance of the Li2MnO3 component in ‘composite’ xLi2MnO3·(1-x)LiMn0.5Ni0.5O2 electrodes. Electrochem. Commun. 6, 1085–1091 (2004).
Thackeray, M. M. et al. Advances in manganese-oxide ‘composite’ electrodes for lithium-ion batteries. J. Mater. Chem. 15, 2257–2267 (2005).
Yabuuchi, N. et al. Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3-LiCo1/3Ni1/3Mn1/3O2 . J. Am. Chem. Soc. 133, 4404–4419 (2011).
Lee, J. et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 343, 519–522 (2014).
Sathiya, M. et al. Origin of voltage decay in high-capacity layered oxide electrodes. Nat. Mater. 14, 230–238 (2015).
Chikkannanavar, S. B., Bernardi, D. M. & Liu, L. A review of blended cathode materials for use in Li-ion batteries. J. Power Sources 248, 91–100 (2014).
Lu, Z. H. & Dahn, J. R. The effect of Co substitution for Ni on the structure and electrochemical behavior of T2 and O2 structure Li2/3[COxNi1/3-xMn2/3]O2 . J. Electrochem. Soc. 148, A237–A240 (2001).
Kobayashi, H. et al. Structure and charge/discharge characteristics of new-layered oxides: Li1.8Ru0.6Fe0.6O3 and Li2IrO3 . J. Power Sources 68, 686–691 (1997).
Gallagher, K. G. et al. Correlating hysteresis and voltage fade in lithium- and manganese-rich layered transition-metal oxide electrodes. Electrochem. Commun. 33, 96–98 (2013).
Thackeray, M. M. et al. Li2MnO3-stabilized LiMO2 (M=Mn, Ni, Co) electrodes for lithium-ion batteries. J. Mater. Chem. 17, 3112–3125 (2007).
Johnson, C. S. et al. Synthesis, characterization and electrochemistry of lithium battery rlectrodes: xLi2MnO3·(1-x)LiMn0.333Ni0.333Co0.333O2 (0≤x≤0.7). Chem. Mater. 20, 6095–6106 (2008).
Gu, M. et al. Formation of the spinel phase in the layered composite cathode used in Li-Ion batteries. ACS Nano 7, 760–767 (2013).
Zheng, F. et al. Nanoscale surface modification of lithium-rich layered-oxide composite cathodes for suppressing voltage fade. Angew. Chem. Int. Ed. 54, 13058–13062 (2015).
Tran, N. et al. Mechanisms associated with the ‘plateau’ observed at high voltage for the overlithiated Li1.12(Ni0.425Mn0.425Co0.15)0.88O2 system. Chem. Mater. 20, 4815–4825 (2008).
Genevois, C. et al. Insight into the atomic structure of cycled lithium-rich layered oxide Li1.20Mn0.54Co0.13Ni0.13O2 using HAADF STEM and electron nanodiffraction. J. Phys. Chem. C 119, 75–83 (2015).
Yan, P. F. et al. Probing the degradation mechanism of Li2MnO3 cathode for Li-ion batteries. Chem. Mater. 27, 975–982 (2015).
Amatucci, G. et al. The elevated temperature performance of the LiMn2O4/C system: failure and solutions. Electrochim. Acta 45, 255–271 (1999).
Hirayama, M. et al. Dynamic structural changes at LiMn2O4/electrolyte interface during lithium battery reaction. J. Am. Chem. Soc. 132, 15268–15276 (2010).
Wu, Y. & Manthiram, A. High capacity, surface-modified layered Li[Li(1-x)/3Mn(2-x)/3Nix/3Cox/3]O2 cathodes with low irreversible capacity loss. Electrochem. Solid State Lett. 9, A221–A224 (2006).
Qiu, B. et al. Enhanced electrochemical performance with surface coating by reactive magnetron sputtering on lithium-rich layered oxide electrodes. ACS Appl. Mater. Interfaces 6, 9185–9193 (2014).
Wang, Q. Y., Liu, J., Murugan, A. V. & Manthiram, A. High capacity double-layer surface modified Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode with improved rate capability. J. Mater. Chem. 19, 4965–4972 (2009).
Wu, F. et al. Spinel/layered heterostructured cathode material for high-capacity and high-rate Li-ion batteries. Adv. Mater. 25, 3722–3726 (2013).
Wu, F. et al. Ultrathin spinel membrane-encapsulated layered lithium-rich cathode material for advanced Li-ion batteries. Nano Lett. 14, 3550–3555 (2014).
Kim, S., Hirayama, M., Taminato, S. & Kanno, R. Epitaxial growth and lithium ion conductivity of lithium-oxide garnet for an all solid-state battery electrolyte. Dalton Trans. 42, 13112–13117 (2013).
Kim, S., Hirayama, M., Suzuki, K. & Kanno, R. Hetero-epitaxial growth of Li0.17La0.61TiO3 solid electrolyte on LiMn2O4 electrode for all solid-state batteries. Solid State Ionics 262, 578–581 (2014).
Cahn, J. W. & Hilliard, J. E. Free energy of a nonuniform system. I. Interfacial free energy. J. Chem. Phys. 28, 258–267 (1958).
Auer, S. & Frenkel, D. Suppression of crystal nucleation in polydisperse colloids due to increase of the surface free energy. Nature 413, 711–713 (2001).
Pennycook, S. J. & Jesson, D. E. High-resolution Z-contrast imaging of crystals. Ultramicroscopy 37, 14–38 (1991).
Okunishi, E. et al. Visualization of light elements at ultrahigh resolution by STEM annular bright field microscopy. Microsc. Microanal. 15, 164–165 (2009).
Oshima, Y. et al. Direct imaging of lithium atoms in LiV2O4 by spherical aberration-corrected electron microscopy. J. Electron Microsc. 59, 457–461 (2010).
Gu, M. et al. Conflicting roles of nickel in controlling cathode performance in lithium ion batteries. Nano Lett. 12, 5186–5191 (2012).
Zheng, J. et al. Mitigating voltage fade in cathode materials by improving the atomic level uniformity of elemental distribution. Nano Lett. 14, 2628–2635 (2014).
Reed, J., Ceder, G. & Van der Ven, A. Layered-to-spinel phase transition in LixMnO2 . Electrochem. Solid State Lett. 4, A78–A81 (2001).
Kim, S. et al. Direct observation of an anomalous spinel-to-layered phase transition mediated by crystal water intercalation. Angew. Chem. Int. Ed. 54, 15094–15099 (2015).
Cho, Y., Oh, P. & Cho, J. A new type of protective surface layer for high-capacity Ni-based cathode materials: nanoscaled surface pillaring layer. Nano Lett. 13, 1145–1152 (2013).
Liu, W. et al. Countering voltage decay and capacity fading of lithium-rich cathode material at 60 °C by hybrid surface protection layers. Adv. Energy Mater. 5, 1500274 (2015).
Kim, H. et al. A new coating method for alleviating surface degradation of LiNi0.6Co0.2Mn0.2O2 cathode material: nanoscale surface treatment of primary particles. Nano Lett. 15, 2111–2119 (2015).
Zhang, L. et al. Rod-like hierarchical nano/micro Li1.2Ni0.2Mn0.6O2 as high performance cathode materials for lithium-ion batteries. J. Power Sources 240, 644–652 (2013).
Chen, L. et al. Hierarchical Li1.2Ni0.2Mn0.6O2 nanoplates with exposed {010} planes as high-performance cathode material for lithium-ion batteries. Adv. Mater. 26, 6756–6760 (2014).
Park, S. H. & Sun, Y.-K. Synthesis and electrochemical properties of layered Li[Li0.15Ni(0.275−x/2)AlxMn(0.575−x/2)]O2 materials prepared by sol-gel method. J. Power Sources 119–121, 161–165 (2003).
Kang, S. et al. Preparation and electrochemical performance of yttrium-doped Li[Li0.20Mn0.534Ni0.133Co0.133]O2 as cathode material for lithium-ion batteries. Electrochim. Acta 144, 22–30 (2014).
Qing, R.-P. et al. Enhancing the kinetics of Li-rich cathode materials through the pinning effects of gradient surface Na+ doping. Adv. Energy Mater. 6, 1501914 (2016).
Sun, Y.-K. et al. The role of AlF3 coatings in improving electrochemical cycling of Li-enriched nickel-manganese oxide electrodes for Li-ion batteries. Adv. Mater. 24, 1192–1196 (2012).
Koyama, Y., Tanaka, I., Nagao, M. & Kanno, R. First-principles study on lithium removal from Li2MnO3 . J. Power Sources 189, 798–801 (2009).
Xiao, P., Deng, Z. Q., Manthiram, A. & Henkelman, G. Calculations of oxygen stability in lithium-rich layered cathodes. J. Phys. Chem. C 116, 23201–23204 (2012).
Boulineau, A. et al. First evidence of manganese-nickel segregation and densification upon cycling in Li-rich layered oxides for lithium batteries. Nano Lett. 13, 3857–3863 (2013).
Okamoto, Y. Ambivalent effect of oxygen vacancies on Li2MnO3: a first-principles study. J. Electrochem. Soc. 159, A152–A157 (2012).
Lanz, P., Villevieille, C. & Novák, P. Ex situ and in situ Raman microscopic investigation of the differences between stoichiometric LiMO2 and high-energy xLi2MnO3·(1−x)LiMO2 (M=Ni, Co, Mn). Electrochim. Acta 130, 206–212 (2014).
Castel, E. et al. Differential electrochemical mass spectrometry study of the interface of xLi2MnO3·(1−x)LiMO2 (M=Ni, Co, and Mn) material as a positive electrode in Li-Ion batteries. Chem. Mater. 26, 5051–5057 (2014).
Acknowledgements
J.W.C. acknowledges the support by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF-2014R1A4A1003712 and NRF-2015R1A2A1A05001737) and KAIST Institute NanoCentury. A part of this work was conducted in JAIST, supported by Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. S.K. acknowledges the postdoc fellowship from the KAIST Institute NanoCentury.
Author information
Authors and Affiliations
Contributions
S.K., Y.O. and J.W.C. designed the research; S.K. carried out the synthesis, electrochemical measurements and data analyses; W.C. assisted in the TEM analyses; S.K. and X.Z. conducted the STEM characterization; S.K., Y.O. and J.W.C. wrote the manuscript; Y.O. and J.W.C supervised the research. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1 - 18, Supplementary Tables 1- 3, Supplementary Notes 1 - 4 and Supplementary References (PDF 9657 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kim, S., Cho, W., Zhang, X. et al. A stable lithium-rich surface structure for lithium-rich layered cathode materials. Nat Commun 7, 13598 (2016). https://doi.org/10.1038/ncomms13598
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms13598
This article is cited by
-
AlF3 coating improves cycle and voltage decay of Li-rich manganese oxides
Journal of Materials Science (2023)
-
Electrodes for Li-ion batteries: From high-voltage LiCoO2 to Co-reduced/Co-free layered oxides with potential anodes
Nano Research (2023)
-
In situ analysis of dynamic evolution of the additive-regulated cathode processes in quasi-solid-state lithium-metal batteries
Science China Chemistry (2023)
-
Challenges and strategies of lithium-rich layered oxides for Li-ion batteries
Nano Research (2023)
-
Cerium-doped lithium-rich Li1.2Mn0.56Ni0.11Co0.13O2 as cathode with high performance for lithium-ion batteries
Ionics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
