
Abstract
There is considerable debate whether Alzheimer’s disease (AD) originates in basal forebrain or entorhinal cortex. Here we examined whether longitudinal decreases in basal forebrain and entorhinal cortex grey matter volume were interdependent and sequential. In a large cohort of age-matched older adults ranging from cognitively normal to AD, we demonstrate that basal forebrain volume predicts longitudinal entorhinal degeneration. Models of parallel degeneration or entorhinal origin received negligible support. We then integrated volumetric measures with an amyloid biomarker sensitive to pre-symptomatic AD pathology. Comparison between cognitively matched normal adult subgroups, delineated according to the amyloid biomarker, revealed abnormal degeneration in basal forebrain, but not entorhinal cortex. Abnormal degeneration in both basal forebrain and entorhinal cortex was only observed among prodromal (mildly amnestic) individuals. We provide evidence that basal forebrain pathology precedes and predicts both entorhinal pathology and memory impairment, challenging the widely held belief that AD has a cortical origin.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by distributed amyloid and tau pathophysiology throughout the brain. Recent breakthroughs in molecular genetics have identified a trans-synaptic mechanism by which these pathologies spread across anatomically and functionally linked cortical regions1,2,3,4 within a large-scale brain network5,6,7. These findings have potential for novel biomarkers and therapeutic strategies aimed at identifying the earliest signs of pathology and preventing its spread, before the onset of clinical AD.
However, the initial stages of AD pathophysiology remain ill defined8,9, preventing a clear picture of what regions to target as the earliest points of spread. The prevailing model suggests that amyloid and tau deposition first appear within the transentorhinal and entorhinal cortex (EC)10,11,12,13. This model has been called into question by histological14,15,16,17,18 and in vivo structural imaging evidence19,20 of early pathological change to the nucleus basalis of Meynert (NbM) in the basal forebrain. The cholinergic cells of the NbM and their cholinoreceptive targets in EC exhibit particular sensitivity to neurofibrillary degeneration in the early stages of AD14,15,16,17,18, possibly even before the onset of cognitive symptomatology14,19. One possible explanation for these competing findings is that the early emergence of pathology in NbM and EC occurs in parallel. A second unexplored possibility points to pathological spread from one structure to the other, indicating that NbM may constitute an earlier target of AD.
Both the NbM and EC are components of the basolateral strip, an uninterrupted band of core limbic cell groups that also includes the hippocampus, amygdala and pyriform cortex14. The EC receives projections from NbM and adjacent diagonal band of Broca in the primate21,22 and human23 brain. This primarily cholinergic innervation forms a functional pathway24,25 involved in the encoding of novel information26, possibly by enhancing perceptual discrimination of sensory input27,28. Recently, a paradoxical phenomenon of increased memory recall for task-incidental information in older adults has been linked to altered attentional modulation of sensory input at early stages of encoding29,30,31,32, possibly arising from the loss of central cholinergic integrity31,32. Clarification of whether pathophysaiology in these regions manifests at the same time, or in a predictive sequence, is therefore crucial to our understanding of the early anatomical staging of AD11 and of how this pathway may influence cognitive decline.
Given the early degeneration of NbM neurons in AD, as well as the anatomical and functional organization of the NbM—EC pathway, we predicted that NbM structural integrity would selectively determine downstream atrophy in EC. Neuroimaging biomarkers such as subregional anatomical changes in grey matter (GM) volume33 are very highly correlated with the pathophysiological lesions of AD34,35,36. To our knowledge, measures of GM volume have not been used to track whether longitudinal changes in different regions are interdependent. In the present study, we evaluated the hypothesis of predictive pathological spread first by examining whether degeneration in NbM and EC over time exhibits interdependence and directionality. However, by itself, such a relationship does not indicate that an underlying pathology drives the interregional degenerative cascade. We therefore integrated our volumetric measures, in the same individuals, with a molecular biomarker of neuronal amyloid deposition that is extremely sensitive to AD pathophysiology at early presymptomatic stages of disease37,38. This strategy enabled us to test the second and critical hypothesis regarding the predictive sequence of NbM and EC degeneration across individuals at different stages of disease. Specifically, if pathology arises in NbB before spreading to EC, then degeneration in NbM and EC should dissociate at early pre-symptomatic stages of AD.
To interrogate these hypotheses, we performed longitudinal voxel-based morphometry (VBM) analyses on three high-resolution anatomical magnetic resonance imaging (MRI) brain volumes acquired over a 2-year period: T1 (baseline), T2 (1-year interval) and T3 (2-year interval) from a large, age-matched older adult sample (N=434) from the Alzheimer’s Disease Neuroimaging Initiative (ADNI)39. In addition to healthy controls (HCs; n=150), the sample consisted of three groups characterized by different stages of AD as follows: mild cognitive impairment (MCI) individuals who did not progress to AD status from T1 to T3 (MCI-NP; n=103), MCI individuals who progressed to AD status at T3 (MCI-AD; n=84) and individuals classified as AD throughout (AD; n=97). A priori regions of interest (ROIs) were specified from probabilistic anatomical maps of the EC, the Ch4 region of the basal forebrain (the magnocellular group corresponding to NbM) and a control region in the primary somatosensory cortex (PSC; see Fig. 1).

(a) Basal forebrain NbM; area Ch4 (b) EC and (c) PSC. The ROIs are displayed in Montreal Neurological Institute (MNI) space on coronal (left column) and sagittal (right column) slices.
We demonstrate that baseline Ch4 volumes predicted longitudinal decreases in EC volume. By contrast, baseline EC volumes did not predict longitudinal decreases in Ch4 volume, ruling out the alternative explanations that EC precedes Ch4 degeneration, or that EC and Ch4 degeneration occurred in parallel (are mutually predictive). The predictive relationship of Ch4 volume was specific to EC: no such relationship was detected between Ch4 and the control PSC region. We next confirmed that the observed Ch4→EC predictive relationship was driven by a sequential staging of AD pathology. To do so, we used concentrations of cerebrospinal amyloid (Aβ1–42) to distinguish, within the cognitively healthy older adult sample, individuals expressing Aβ1–42 levels diagnostic of pre-symptomatic AD. We isolated an abnormal pattern of degeneration in Ch4, but not in EC, among cognitively healthy adults expressing Aβ1–42 levels of pre-symptomatic AD. Abnormal degeneration of both Ch4 and EC was only detected at later stages of disease—among (MCI-NP) individuals expressing AD-diagnostic levels of Aβ1–42—when short-term memory impairment was clinically detectable. Regression-based mediation and conditional process models revealed that EC degeneration mediates the relationship between Ch4 integrity and memory impairment, and that AD pathology (Aβ1–42) moderates this mediation effect. Our results show that in the staging of AD, Ch4 degeneration precedes and predicts EC degeneration, with memory impairment emerging only after pathology spreads from Ch4 to EC. Moreover, our results suggest that abnormal Ch4 degeneration is either clinically invisible, or that the neuropsychological tests currently in widest use are not sensitive to this early subcortical stage of AD.
Results
GM volume changes as a function of diagnostic group
We first examined whether and how GM volume (T1–T3) changed over the study period as a function of diagnostic group (HC, MCI-NP, MCI-AD and AD) and ROI (Ch4, EC and PSC). See Supplementary Table 1 for a complete list of ADNI individuals included, Fig. 1 for ROIs and Methods for GM volumetry). The Group and ROI factors were entered into a 4 × 3 repeated-measures analysis of variance (ANOVA), which included age, sex, education, head size and longitudinal changes (T1–T3) in whole-brain GM volume as covariates. We detected a significant main effect of Group (F3,425=32.2148, P<0.001) and Group × ROI interaction (F6,850=13.53, P<0.001; see Fig. 2a). We therefore decomposed the full factorial model into separate ANOVA models, one for each ROI, to assess the effect of clinical diagnosis on GM degeneration in each brain region. Magnitudes of GM degeneration in both Ch4 and EC were significantly impacted by diagnosis (Ch4: F3,425=5.33, P=0.001; EC: F3,425=53.39, P<0.001), which increased with clinical progression of AD. By contrast, magnitudes of GM degeneration in the PSC control region did not significantly differ as a function of diagnosis (F3,425=2.10, P=0.10). Controlling for the same covariates, we observed a highly similar pattern of results when comparing baseline GM volume (T1) as a function of diagnostic group (HC, MCI-NP, MCI-AD and AD) and ROI (Ch4, EC and PSC). Specifically, the 4 × 3 ANOVA revealed a significant main effect of Group (F3,425=49.32, P<0.001) and Group × ROI interaction (F6,850=18.02, P<0.001), with clinical progression to AD affecting baseline Ch4 and EC volumes more strongly than PSC (see Fig. 2b). Taken together, these initial findings confirm that our MRI measures are sensitive to local subregional, as opposed to global, anatomical changes in GM volume33 and, moreover, that these changes increase with the clinical progression to AD.

Volumetric group differences (diagnosis) in a priori ROIs for the EC (blue), basal forebrain NbM (Ch4; magenta) and PSC (green). (a) Magnitudes of GM degeneration from baseline (T1) to 2 years post baseline (T3) in each diagnostic group: HCs (n=150), MCI-NP (n=103), MCI-AD (n=84) and probable AD individuals (AD; n=97). (b) GM volume at baseline in each diagnostic group. Error bars are s.e.m.
Parallel versus predictive spread of degeneration in Ch4 and EC
We next assessed the competing hypotheses of parallel versus predictive spread of degeneration. To begin with, we made no assumptions about AD pathology in terms of diagnosis (group status) and thus used multiple regression analyses inclusive of the entire sample (N=434). At baseline (T1), degeneration will already vary significantly across individuals. Smaller T1 GM volumes in EC and Ch4 should therefore reflect more advanced stages of GM degeneration before the baseline scan, after accounting for individual differences in age, sex, education, head size and whole-brain GM volume. Moreover, between time points T1 and T3 of the study period, pre-baseline degeneration is expected to progress in both EC and Ch4, but at a rate dependent on a given individual’s baseline status40. Hence, T1 GM volume should predict the magnitude of post-baseline degeneration between time points at both 1-year (T1–T2) and 2-year (T1–T3) intervals.
According to a parallel staging model, pre-baseline degeneration in either region should predict post-baseline degeneration in the other, because both regions are affected by the same pathology at the same time. In Fig. 3a, parallel spread would be represented by superimposed degenerative trajectories in Ch4 and EC (not pictured). By contrast, according to the predictive staging model, pre-baseline degeneration in one region should only predict post-baseline degeneration in the other due to the spread of pathology over time. We contrasted the hypothesized Ch4→EC model against a competing EC→Ch4 model, where pathology originates in EC and spreads to subcortical areas of the interconnected basolateral strip (see Fig. 3a,b). In either of these scenarios, smaller T1 volumes (higher pre-baseline degeneration) in the source region should predict larger magnitudes of volumetric decrease (higher post-baseline degeneration) in the target region, yielding a negative relationship (see Fig. 3c). Based on existing models of AD progression41,42,43, we predicted that the rate of change in GM degeneration would follow a nonlinear sigmoid shape. In prior volumetric MRI work, rates of GM degeneration have been shown to accelerate as patients approach clinical dementia44,45. A sigmoid shape as a function of time thus indicates that GM degeneration is not constant, but rather varies over the course of disease progression.

(a,b) Hypothesized volumetric decrease (y axes) due to the spread of pathology over time (x axes) for EC (blue) and Ch4 (purple). Parallel spread model: both regions are affected by pathology at the same time (superimposed trajectories, not pictured). Predictive spread model: one region is affected by pathology before the other (offset trajectories), in either the (a) EC→Ch4 or (b) Ch4→EC model. (c) Pre- to post-baseline degeneration. Smaller T1 volumes in the source region of hypothetical subjects (s1—s3) predict larger volumetric decreases (T1–T3) in the target region (red lines). (d,e) The Ch4→EC model was significant at 1-year (middle) and 2-year intervals (bottom) across the entire sample (n=434). (f) Direct comparisons of the predictive models at one and 2-year intervals (y axis range reflects the horizontal refence lines in d,e). Regression slopes are presented with the 95% CIs and are adjusted relative to covariates of non-interest. **Two-tailed P<0.01; ***two-tailed P<0.001.
We evaluated these competing inter-regional hypotheses over both 1- and 2-year intervals. The EC→Ch4 regression model revealed virtually no relationship between pre-baseline degeneration in EC and post-baseline degeneration in Ch4 at 1-year (t429<1, r=−0.03, P=0.50) and 2-year intervals (t429<1, r=−0.02, P=0.69), after accounting for age, sex, education, head size and longitudinal changes in whole-brain GM volume (see Fig. 3d). By contrast, the Ch4→EC regression model yielded a significant negative relationship between pre-baseline degeneration in Ch4 and post-baseline degeneration in EC at 1-year (t429=−4.78, r=−0.21, P<0.001) and 2-year intervals (t429=−5.26, r=−0.25, P<0.001; see Fig. 3e). Taken separately, the coefficients produced by the EC→Ch4 and Ch4→EC staging models strongly favour the Ch4→EC model, but they do not provide a direct quantitative comparison between the two models. We therefore computed a test of the equality of these two coefficients (see Methods). We found that the negative relationship observed in the Ch4→EC model was significantly stronger than the EC→Ch4 model at both 1-year (z=2.77, P=0.003) and 2-year intervals (z=3.77, P<0.001; see Fig. 3f).
We next explored the possibility that Ch4 predicts a general pattern of degeneration in neocortex, irrespective of focal susceptibility to AD or anatomical connectivity. If this were the case, pre-baseline degeneration in Ch4 should predict post-baseline degeneration even in cortical sites relatively spared by AD pathology, such as somatosensory cortex (PSC ROI). As with the direct model comparisons between Ch4 and EC (Fig. 3f), we first directly compared the Ch4→PSC model against the reverse PSC→Ch4 model, to determine whether pre-baseline degeneration in one region preferentially predicts post-baseline degeneration in the other. However, no differences were detected between these models at either 1-year (z=0.076, Ptwo-tailed=0.94) or 2-year intervals (z=−1.27, Ptwo-tailed=0.20). We next directly compared the Ch4→PSC model against the observed Ch4→EC model. To do so, we computed a test of the equality of the two coefficients produced by the Ch4→EC and Ch4→PSC models (see Methods). The negative relationship produced by the Ch4→EC model was significantly stronger than the Ch4→PSC model at both 1-year (z=−2.22, Ptwo-tailed=0.03) and 2-year intervals (z=−2.37, Ptwo-tailed=0.02). These results suggest that Ch4 does not predict a general pattern of neocortical degeneration, but rather is selective to anatomically connected cortical targets known to be affected in the early pathological staging of AD. Together, the results from our multiple regression analyses support the hypothesis that changes in GM volume between Ch4 and EC are interdependent rather than coincidental, with pre-baseline Ch4 degeneration selectively predicting the downstream degenerative trajectory in EC.
Amyloid staging of AD confirms Ch4 precedes EC degeneration
Although our regression models establish evidence for a predictive sequence of GM degeneration from Ch4 to EC, by themselves they do not reveal how this pattern is influenced by AD pathology. We therefore next turned to the critical question of whether the observed pattern of Ch4→EC spread constitutes a previously unknown early link in the predictive pathological staging of AD. To do so, we obtained cerebrospinal fluid (CSF) measures of the Amyloid-β1 to 42 peptide (Aβ1–42), which were available in a subset of our sample (N=244). Data from the ADNI Cores were recently integrated to generate a model for the temporal ordering of AD biomarkers38,46, which indicates that Aβ1–42 is the first biomarker to become abnormal, followed by changes in other AD biomarkers (CSF tau, F-18 fluorodeoxyglucose-positron emission tomography) and, lastly, the onset of clinical symptoms. Crucially, receiver operating curve analysis of autopsy-confirmed AD cases versus normal controls has provided a cutpoint for CSF Aβ1–42 concentration at which diagnostic sensitivity and specificity to AD is maximal (192 pg ml−1), yielding correct detection of 96.4% (concentrations below 192 pg ml−1) and correct rejection of 95.2% (concentrations above 192 pg ml−1)37. We therefore first partitioned our sample into normal Aβ (individuals who fell above the 192 pg ml−1 Aβ1–42 cutpoint) and all individuals below this cutpoint expressing AD neuropathology (Aβ+). Individuals presenting abnormal cognitive impairment but normal CSF Aβ levels were excluded from all forthcoming analyses, as their cognitive symptoms are likely to be caused by non-AD pathology, for example, vascular dementia and hippocampal sclerosis. Of these individuals, 19 were MCI-NP, 4 were MCI-AD and 5 were AD. This left a total remaining sample size of 216: HCs with normal Aβ (HCNAβ: n=52) and Aβ+ individuals (n=164). We next further partitioned the Aβ+ group according to clinical diagnoses. This yielded the following four subgroups: individuals in the clinically silent phase of AD (HCAβ+: n=28), MCI non-progressors (MCI-NPAβ+: n=39), MCI-AD (MCI-ADAβ+: n=41) and AD subgroups (ADAβ+: n=56). This analysis strategy enabled us to explore the predictive sequence of Ch4 and EC degeneration due to AD neuropathology across individuals at difference clinical stages of AD.
The five subgroups (HCNAβ, HCAβ+, MCI-NPAβ+, MCI-ADAβ+ and ADAβ+) were first submitted to a 5 × 3 (ROI) repeated-measures ANOVA, which included age, sex, education, head size and longitudinal changes in whole-brain GM volume as covariates. This model revealed a significant Group × ROI interaction (F8,412=6.15, P<0.001). This interaction was not dependent on GM degeneration in the PSC control region, as confirmed by a follow-up 5 × 2 ANOVA, which excluded this ROI (F4,206=6.98, P<0.001). We therefore focused next on how Ch4 and EC degeneration differentially increased as a function of AD neuropathology and clinical diagnosis.
If AD neuropathology in Ch4 precedes and predicts EC, as proposed by the Ch4→EC model of predictive pathological spread, then Ch4 and EC degeneration should dissociate at early stages of disease (Fig. 4a). Consistent with this model, an independent samples t-test comparing the Ch4 ROI between the HCNAβ and HCAβ+ subgroups revealed significantly larger magnitudes of GM degeneration in the clinically silent HCAβ+ subgroup (t78=1.9, P1-tail=0.03, d=0.50). No between-group differences were detected in EC (t78=0.19, P1-tail=0.85, d=0.05) or in PSC (t78=1.07, P1-tail=0.16, d=0.33; see Fig. 4b). Crucially, this pattern of Ch4 degeneration was clinically silent: no differences in cognitive function between HCNAβ and HCAβ+ subgroups were detected on any measure collected in the ADNI neuropsychological battery, even when averaging scores across study time points to produce the most stable estimates of cognitive function (see Table 1). These findings thus reveal a striking anatomical dissociation between Ch4 and EC degeneration in the clinically silent HCAβ+ subgroup, suggesting that EC is relatively spared alongside Ch4 atrophy within cognitively normal individuals expressing the Aβ biomarker of probable AD.

(a) A model of predictive pathological staging in which Ch4 and EC degeneration (y axis) dissociate at early stages of pathological progression (x axis), with Ch4 degeneration emerging at stages before amnestic symptoms (HCs expressing the CSF biomarker of AD; HCAβ+) and degeneration in both Ch4 and EC at prodromal amnestic stages of AD (MCI non-progressors; MCI-NPAβ+). (b) Subgroups were delineated according to the CSF Aβ1–42 cutpoint diagnostic of AD pathophysiology and then further delineated according to clinical diagnosis, yielding five subgroups: HCs with normal Aβ levels (HCNAβ; n=52), versus HCAβ+ (n=28), MCI-NPAβ+ (n=39), MCI-ADAβ+ (n=41) and probable ADAβ+ individuals (n=56). Abnormal degeneration in the HCAβ+ subgroup was isolated to Ch4. Abnormal degeneration in the MCI-NPAβ+ subgroup was observed in both Ch4 and EC. *One-tailed P<0.05. Error bars are s.e.m.
As expected, we observed impairment in memory in the MCI-NPAβ+ compared with the HCAβ+ subgroup, which was most pronounced for delayed recall performance on the Logical Memory Test (see Table 2). Predictive pathological spread in the Ch4→EC pathway was thus predicted to be at a more advanced stage among MCI-NPAβ+ individuals, with evidence of spread to EC (Fig. 4a). Consistent with this hypothesis, independent samples t-test confirmed significantly larger magnitudes of EC GM degeneration in the MCI-NPAβ+ subgroup compared with the HCAβ+ subgroup (t65=2.05, P1–tail=0.02, d=0.51). No between-group difference was detected in Ch4 (t89=0.29, P1-tail=0.39, d=0.07), due to the abnormal elevation of Ch4 GM degeneration in both of the HCAβ+ and MCI-NPAβ+ subgroups. Moreover, no between-group difference was detected in PSC (t89=0.04, P1-tail=0.48, d=0.01). In sum, compared with the isolated Ch4 pathology observed in HCAβ+ subgroup, we observed both Ch4 and EC pathology in the MCI-NPAβ+ subgroup (Fig. 4b). Taken together, these two patterns of degeneration in the Ch4→EC pathway distinguish the clinically silent and early prodromal phases of AD, strongly supporting the sequential staging of AD pathology from Ch4 to EC. By integrating volumetric and molecular biomarkers from the ADNI, structural MRI can thus potentially be leveraged beyond its established sensitivity to early AD pathology.
At increasingly advanced stages of AD, abnormal Ch4 and EC degeneration becomes more difficult to detect against the background of global cortical degeneration. Comparing between the MCI-ADAβ+ and ADAβ+ subgroups, magnitudes of GM degeneration were indistinguishable in both Ch4 (t95=0.19, P1-tail=0.85, d=0.04) and EC (t95=0.80, P1-tail=0.43, d=0.19), despite being substantially elevated relative to the HCNAβ, HCAβ+ and MCI-NPAβ+ subgroups (Fig. 4b). This observation serves to highlight the importance of both EC and Ch4 as important early indicators of pathological change, which lose their viability as biomarkers with the current clinical tools used to detect AD progression.
A hypothesized subcortical–cortical pathway to AD
Thus far, we have shown evidence that GM integrity in Ch4 predicts subsequent GM degeneration in EC, and that abnormal Ch4 GM degeneration precedes both memory impairment and abnormal EC degeneration in the pathological staging of AD. These findings suggest a subcortical–cortical pathway to AD, with the spread of pathology from Ch4 to EC inducing selective impairments in memory recall for novel information. We therefore next examined with mediation analyses47 how the regression-based evidence of Ch4→EC predictive pathological spread may relate to memory dysfunction. Critically, we then used conditional process analysis to determine whether these relationships are dependent on the presence of AD neuropathology (see Methods).
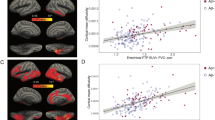
To do so, we first used multiple linear regression analysis of the entire study sample (N=434) to determine whether a relationship existed between pre-baseline degeneration in Ch4 (T1 volume) and memory recall performance. The latter was indexed from the Logical Memory Test delayed recall score, given its observed sensitivity to early impairments in the MCI-NPAβ+ group (see Table 2). We found a significant positive relationship (r=0.33, t=7.30, P<0.001), whereby smaller Ch4 GM volumes predicted lower recall performance, after accounting for age, sex, education, head size and longitudinal changes in whole-brain GM volume. However, our findings from the Aβ1–42 staging of Ch4 and EC degeneration indicate that deficits in memory recall manifest only once pathological spread from Ch4 to EC has occurred. If this is the case, then the observed relationship between Ch4 atrophy and impaired recall should be better accounted for by modelling the Ch4→EC pathway. We tested this hypothesis using a mediation analysis, which included post-baseline EC degeneration (T1–T3) as an indirect pathway between Ch4 and delayed recall. Path a in the mediation model thus replicates the prior Ch4→EC regression model. The same nuisance covariates used in the linear regression were included in the mediation model (see Methods). The bootstrapped unstandardized indirect effect was 6.76, with the 95% confidence interval (95% CI) ranging from 4.22 to 9.68, indicating a significant mediation. We further confirmed the significance of this mediation effect using the Sobel test (z=4.73, P<0.001). We also confirmed the anatomical specificity of this mediation effect by performing a second control analysis, substituting EC with PSC. This single alteration to the model abolished the mediation effect (unstandardized indirect effect=0.26, 95% CI (−0.19 to 1.33); Sobel z=0.75, P=0.46).
Our Ch4→EC regression and mediation results demonstrate that pathology originating in Ch4 gives rise to memory dysfunction through predictive spread to EC. To confirm that the mediation effect is indeed dependent on AD neuropathology, we returned to the subsample of individuals in whom CSF Aβ measures were available (N=216). We first confirmed that the regression and mediation findings observed in the whole sample held in the subsample (regression model: r=0.32, t=4.84, P<0.001; mediation model: unstandardized indirect effect=8.60, 95% CI (4.93 to 12.99), Sobel z=3.86, P<0.001; PSC control mediation model: unstandardized indirect effect=0.67, 95% CI (−0.21 to 3.01); Sobel z=0.93, P=0.35; see Fig. 5a,b). We next re-partitioned the subsample according to those who fell below or above the 192 pg ml−1 Aβ1–42 cutpoint at which AD diagnostic accuracy is maximal37, that is, the HCNAβ (N=52) and Aβ+ (N=164) groups. The two groups were coded as a dichotomous moderator variable. We were thus able to determine whether the observed Ch4→EC→Recall mediation effect was moderated by the presence of AD neuropathology. Specifically, we hypothesized that the Aβ+ group would drive the mediation effect by increasing the strength of the relationships on both the Ch4→EC path (a) and the EC→Recall path (b). As before, we included age, sex, education, head size and longitudinal changes in whole-brain GM volume as covariates. Consistent with our hypotheses, a significant mediation effect was detected in the Aβ+ subgroup (unstandardized indirect effect=6.11, 95% CI (2.49 to 10.48)). The mediation effect was abolished altogether in the HCNAβ subgroup (standardized indirect effect=−1.90, 95% CI (−6.78 to 0.30)). A direct test of the equality between these conditional indirect effects in each subgroup confirmed that they differed significantly (unstandardized moderation effect=−8.01, 95% CI (−13.81 to −3.46); see Fig. 5c). We therefore provide evidence of a conditional process model for early predictive pathological staging in the Ch4→EC pathway. This model links the Ch4→EC degenerative sequence to memory dysfunction and reveals that this process is dependent on AD neuropathology.

(a) Multiple linear regression analysis confirmed that a relationship exists between smaller Ch4 volume and lower delayed recall performance (n=216). (b) Mediation analysis revealed that Ch4 volume better predicts recall performance when accounting for longitudinal EC degeneration, that is, the direct relationship (path c’) is suppressed. (c) Conditional process analysis demonstrated that the observed mediation effect was moderated by the CSF Aβ biomarker of AD neuropathology: the Ch4→EC→Recall mediation effect was significant for the Aβ+ individuals (n=164), that is, path c’ is suppressed, but not for individuals with normal Aβ (n=52). All path coefficients were significant two-tailed P<0.001. Error terms for the mediation (em) and criterion variables (ey) are denoted with grey hashed arrows.
Discussion
In sum, our findings build on a varied body of histological evidence, which collectively point to both Ch4 and EC as early targets of AD pathology8. The cell groups constituting these structures are among the first to express intraneuronal neurofibrilliary tangles and Aβ-containing plaques in cognitively normal older adults. They are also among the cell groups most devastated by tangles and plaques in MCI and AD8,11,14. The proliferation of tangles and plaques in these structures leads to depletion of axons and cell loss, both of which contribute to microstructural decreases in volume8,15,17,18. Our in vivo structural data are sensitive to these volumetric changes, which we confirmed through regression analyses with the CSF Aβ1–42 biomarker of amyloid pathology. Strikingly, by leveraging the biomarker data to identify cognitively normal individuals expressing AD neuropathology, we show that abnormal changes in Ch4 GM volume were apparent even in clinically silent stages of probable AD.
Several lines of evidence have diminished the role of cholinergic degradation in the cognitive dysfunctions of AD9,48,49,50, which appears at odds with the histological evidence emphasizing a cholinergic lesion in AD pathology. Our findings reconcile the contention that Ch4 is at once a central pathological target of AD, but also that Ch4 pathology by itself does not account for the memory impairments observed in AD9. We isolated abnormal degeneration to Ch4 in the clinically silent stage (HCAβ+), before cognitive symptoms were detectable on any of the ADNI neuropsychological measures. Memory dysfunction manifested only later in the progression of AD, among MCI-NPAB+ individuals expressing abnormal degeneration in both Ch4 and EC. Hence, it is a lesion of the Ch4→EC pathway, causally induced by predictive pathological spread, which gives rise to the memory dysfunctions observed in early AD. This interpretation is consistent with animal work showing that neurotoxic lesions to either Ch4 or EC yield only moderate impairments in memory, whereas lesions to both structures yields a dramatic deficit in the ability to acquire new memories and cause behavioural disturbances that mimic the restlessness and wandering observed in AD51. In a separate line of neurophysiological research, cholinergic Ch4 projections have been proposed to tune the oscillatory dynamics of EC neurons during memory encoding24,25. Confirming this hypothesis, selective lesions to the cholinergic innervations of EC were subsequently shown to impair working memory for novel but not familiar stimuli26. Memory encoding thus depends on the functional integrity of both Ch4 and EC. Damage to both of these structures, or their connections, yields a selective memory impairment highly consistent with the anterograde amnesia observed in prodromal stages of AD52 (see Table 2).
One question raised by the present study is whether the clinically silent phase of AD, during which pathophysiology is restricted to Ch4, is indeed clinically silent. An emerging pattern in the cognitive ageing literature indicates that some healthy, older adults more than others exhibit unique deficits of feature-selective attention—that is, the capacity to suppress unattended features of sensory input—yielding a greater susceptibility to incidental encoding of stimuli such as visual distractors29,30,31,32. Population neural coding of sensory information in the visual cortex is strongly linked to the basal forebrain cholinergic system, in both human53,54,55,56 and animal27,28,57,58,59 research. The stimulus-driven pattern of encoding in older adulthood may therefore constitute an early pathological sign of AD due to loss of cortical cholinergic neurons in the basal forebrain. Precise behavioural measures of feature-selective attention, sensitive to early cholinergic dysfunction in older adults, have the potential to provide clinical measures sensitive to early stages of AD that precede memory impairment.
The present study demonstrates that changes in Ch4 GM volume predict and precede both memory impairment and longitudinal changes in EC GM volume, and critically that these relationships are dependent on AD neuropathology. Among individuals expressing the Aβ1–42 CSF biomarker of probable AD, patterns of abnormal degeneration in the Ch4→EC pathway can dissociate those with memory impairments from those who appear cognitively normal. Specifically, a pattern of isolated abnormal Ch4 degeneration was observed among clinically silent probable AD individuals (HCAβ+), with selective memory impairment emerging only once this pattern of abnormal degeneration affected both Ch4 and EC (MCI-NPAβ+). One limitation of this study is the relatively small number of HCAβ+ individuals available through ADNI, whose measures of CSF Aβ can be integrated with longitudinal structural MRI and neuropsychological data (n=28). Our findings indicate that future large-scale research initiatives on AD would benefit from a multimodal biomarker strategy including, at a minimum, CSF Aβ and longitudinal structural MRI, focused on cognitively healthy adults. Nevertheless, our findings strongly suggest that a subcortical–cortical pathological spread from Ch4 to EC defines the earliest link in the predictive pathological staging of AD.
Although our imaging results complement recent breakthroughs in molecular genetics showing that AD spreads via a trans-synaptic mechanism1,2,3,4, they also necessitate reconsideration of EC as the origin point of disease. Molecular genetics holds promise for developing therapeutic strategies to prevent the spread of pathology at stages of AD preceding even the earliest memory impairments. Our evidence strongly indicates that these efforts will be more effective if they target the basal forebrain rather than EC.
Methods
ADNI participants and MRI acquisition
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, M.D. The primary goal of ADNI has been to test whether serial MRI, positron emission tomography, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians, to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials. Subjects were recruited from 50 sites in the United States and Canada. Written informed consent was obtained from all participants before protocol-specific procedures were performed. All data acquired as part of this study are publicly available. For up-to-date information, see www.adni-info.org.
In the present study, we included ADNI1 participants (N=434, 187 women, see Supplementary Table 1 and Table 3, downloaded 5 October 2012) with three time points, separated by at least 12 months, and no more than 30 months. Participants were divided into four groups: HCs, MCI-NP to AD and MCI patients who did progress to AD (MCI-AD) and probable AD patients. To assess brain change covering the transition from MCI to AD, we anchored the longitudinal analysis to the scan in which MCI participants progressed to AD. Therefore, MCI-AD patients always transitioned to AD at time point 2. We then defined the study window as 12 months (minimum 6 months) before progression and 12 months (maximum 18 months) post progression. This 24-month study window was replicated for the other three cohorts from the baseline scan and the two subsequent annual follow-up scans (12 months and 24 months). Healthy participants who subsequently progressed to AD or MCI were excluded (n=10). All participant neuroimage ID numbers are in Supplementary Table 1. Baseline diagnostic status was assessed with the Mini-Mental Status Examination (Table 3), Wechsler Memory Scale (Logical Memory subtest), Clinical Dementia Rating Scores, in addition to subjective reports. A probable AD diagnosis was made following NINCDS/ADRDA criteria. Information on recruitment and diagnostic criteria can be found on the ADNI website: www.adni-info.org.
MRI data were collected according to a standardized protocol (Jack et al.60). This protocol included a high-resolution T1-weighted, rapid gradient echo sequence on a 1.5 T scanner. The ADNI MRI Core optimized acquisition parameters of the neuroimage sequences for each scanner make and model. Sample high-resolution T1-weighted, rapid gradient echo acquisition parameters for one platform (Siemens Magnetom Sonata syngo MR 2004 A) was as follows: T1=1,000 ms, TR=2,400 ms, TE=minimum, flip angle=88, bandwidth 180 Hz per pixel, FOV=240 mm, matrix size=192 × 192, 60 slices and slice thickness=1.2 mm. All data correction and neuroimage quality-control procedures were performed at the Mayo Clinic. Neuroimage quality control included inspection for protocol compliance, clinically significant medical abnormalities and neuroimage quality. To enhance standardization across ADNI sites, post-acquisition correction of neuroimage artefacts was also implemented. This included corrections in geometry for gradient nonlinearity, intensity non-uniformity due to non-uniform receiver coil sensitivity or additional causes60. Consistent with the formulation of standardized data sets61, participant scans were included in the current study if the MRI of one of the two T1 anatomical scans passed the quality-control process.
MRI data preprocessing
All neuroimages were preprocessed in SPM8 using the diffeomorphic anatomical registration through exponentiated lie algebra (DARTEL)62 and longitudinal VBM8 toolboxes (http://dbm.neuro.uni-jena.de/vbm8/). Anatomical images were segmented into the GM, white matter, cerebral spinal fluid, bone and soft tissue. GM neuroimages were realigned within subject, then normalized to a population template in Montreal Neurological Institute space. All neuroimages were then subjected to non-linear modulation that plotted the absolute amount of brain tissue, corrected for participant head size in VBM8. Neuroimages were then sampled with a resulting voxel size 1.5 mm3. Total intracranial volume (that is, head size) was computed as the sum of the GM, white matter and CSF volumes derived from non-normalized segmented images.
ROI analysis
ROIs for NbM and EC were defined from probabilistic maps using the SPM Anatomy Toolbox63, to ensure anatomical precision and replicability (see Fig. 1 and Methods). The basal forebrain is composed of distinct magnocellular cholinergic cell groups, defined histologically in non-human primates as Ch1–Ch6 (ref. 22), with Ch4 corresponding to NbM. A stereotaxic probabilistic anatomical map of Ch4 was recently obtained in humans from postmortem brains64. We refer to the NbM ROI as Ch4 henceforth, to reflect this anatomical parcellation. The EC ROI was similarly obtained from an existing stereotaxic probabilistic anatomical map65. We also obtained measures of GM volume from a third ROI in the PSC, which we used as a control to confirm the anatomical specificity of predictive pathological staging. Alzheimer’s is characterized by a relative sparing of PSC and a lack of somatosensory symptomatology12,66. The PSC was derived from a stereotaxic probabilistic anatomical map of somatosensory area 3a, which lies at the fundus of the central sulcus67.
Probability maps were calculated from postmortem histological analyses based on a sample of ten brains. Each map describes the relative frequency at which the same area (for example, Ch4 EC or PSC) was represented in each voxel of the reference space. The ROIs were created by applying a threshold of 50% to the corresponding probability map. Thus, for the ROIs, only those voxels were considered that were present in more than five postmortem brains. The ROIs were linearly coregistered with the modulated GM images in Montreal Neurological Institute space. To produce indices of longitudinal degeneration, for each participant we subtracted their unsmoothed modulated GM images at T2 (short interval) or at T3 (long interval) from their unsmoothed modulated GM image at T1 using custom Matlab scripts. Within each ROI, values for mean GM volume and longitudinal degeneration were extracted using the Marsbar toolbox68 and custom Matlab scripts.
Regression analysis
All multiple linear regression analyses employed robust estimation, thereby minimizing potential outlier effects. Statistical tests of the equality between two dependent correlations with no variables in common were performed as follows: first, each correlation coefficient was converted into a z-score using Fisher’s r-to-z transformation. Then, equations (2) and (11) from Steiger69 were used to compute the asymptotic covariance of the estimates. These quantities were used in an asymptotic z-test. Statistical tests of the equality between two dependent correlations with one variable in common were performed in the same manner, except that equations (3) and (10) from Steiger69 were used to compute the asymptotic covariance of the estimates.
The mediation and conditional process analyses employed a regression-based path analytic framework for estimating direct and indirect effects47,70. The dependent measure, Logical Memory Test delayed recall, was first averaged within subjects across the three study time points to produce the most reliable estimate of performance. For the mediation model, the standardized indirect effect was determined by multiplying paths a(−0.309) and b(−0.436), yielding 0.135. For both the mediation and conditional process models, inference of statistical significance for the conditional indirect effects was determined using bias-corrected bootstrapping procedures. Specifically, unstandardized indirect effects were computed for each of 10,000 bootstrapped samples and the 95% CI was computed by determining the indirect effects at the 2.5th and 97.5th percentiles.
Data availability
All data used in this study are available from the ADNI database (adni.loni.usc.edu).
Additional information
How to cite this article: Schmitz, T. W. et al. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 7, 13249 doi: 10.1038/ncomms13249 (2016).
References
Clavaguera, F. et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 (2009).
de Calignon, A. et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697 (2012).
Khan, U. A. et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 17, 304–311 (2014).
Liu, L. et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7, e31302 (2012).
Buckner, R. L. et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 25, 7709–7717 (2005).
Seeley, W. W., Crawford, R. K., Zhou, J., Miller, B. L. & Greicius, M. D. Neurodegenerative diseases target large-scale human brain networks. Neuron 62, 42–52 (2009).
Spreng, R. N. & Turner, G. R. Structural covariance of the default network in healthy and pathological aging. J. Neurosci. 33, 15226–15234 (2013).
Arendt, T., Bruckner, M. K., Morawski, M., Jager, C. & Gertz, H. J. Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol. Commun. 3, 10 (2015).
Mesulam, M. The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn. Mem. 11, 43–49 (2004).
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H. & Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 (2006).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991).
Corder, E. H. et al. Density profiles of Alzheimer disease regional brain pathology for the Huddinge brain bank: pattern recognition emulates and expands upon Braak staging. Exp. Gerontol. 35, 851–864 (2000).
Duyckaerts, C. et al. Rating of the lesions in senile dementia of the Alzheimer type: concordance between laboratories. A European multicenter study under the auspices of EURAGE. J. Neurol. Sci. 97, 295–323 (1990).
Mesulam, M., Shaw, P., Mash, D. & Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 55, 815–828 (2004).
Geula, C. & Mesulam, M. M. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer's disease. Cereb. Cortex 6, 165–177 (1996).
Sassin, I. et al. Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta. Neuropathol. 100, 259–269 (2000).
Schliebs, R. & Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural. Transm. 113, 1625–1644 (2006).
Schliebs, R. & Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563 (2011).
Grothe, M., Heinsen, H. & Teipel, S. Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer’s disease. Neurobiol. Aging 34, 1210–1220 (2013).
Grothe, M., Heinsen, H. & Teipel, S. J. Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol. Psychiatry 71, 805–813 (2012).
Alonso, J. R. & Amaral, D. G. Cholinergic innervation of the primate hippocampal formation. I. Distribution of choline acetyltransferase immunoreactivity in the Macaca fascicularis and Macaca mulatta monkeys. J. Comp. Neurol. 355, 135–170 (1995).
Mesulam, M. M., Mufson, E. J., Levey, A. I. & Wainer, B. H. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol. 214, 170–197 (1983).
De Lacalle, S. et al. Cholinergic innervation in the human hippocampal formation including the entorhinal cortex. J. Comp. Neurol. 345, 321–344 (1994).
Egorov, A. V., Hamam, B. N., Fransen, E., Hasselmo, M. E. & Alonso, A. A. Graded persistent activity in entorhinal cortex neurons. Nature 420, 173–178 (2002).
Klink, R. & Alonso, A. Muscarinic modulation of the oscillatory and repetitive firing properties of entorhinal cortex layer II neurons. J. Neurophysiol. 77, 1813–1828 (1997).
McGaughy, J., Koene, R. A., Eichenbaum, H. & Hasselmo, M. E. Cholinergic deafferentation of the entorhinal cortex in rats impairs encoding of novel but not familiar stimuli in a delayed nonmatch-to-sample task. J. Neurosci. 25, 10273–10281 (2005).
Goard, M. & Dan, Y. Basal forebrain activation enhances cortical coding of natural scenes. Nat. Neurosci. 12, 1444–1449 (2009).
Pinto, L. et al. Fast modulation of visual perception by basal forebrain cholinergic neurons. Nat. Neurosci. 16, 1857–1863 (2013).
Campbell, K. L., Hasher, L. & Thomas, R. C. Hyper-binding: a unique age effect. Psychol. Sci. 21, 399–405 (2010).
Quigley, C., Andersen, S. K., Schulze, L., Grunwald, M. & Muller, M. M. Feature-selective attention: evidence for a decline in old age. Neurosci. Lett. 474, 5–8 (2010).
Schmitz, T. W., Cheng, F. H. & De Rosa, E. Failing to ignore: paradoxical neural effects of perceptual load on early attentional selection in normal aging. J. Neurosci. 30, 14750–14758 (2010).
Schmitz, T. W., Dixon, M. L., Anderson, A. K. & De Rosa, E. Distinguishing attentional gain and tuning in young and older adults. Neurobiol. Aging 35, 2514–2525 (2014).
Holland, D., Brewer, J. B., Hagler, D. J., Fennema-Notestine, C. & Dale, A. M. Subregional neuroanatomical change as a biomarker for Alzheimer’s disease. Proc. Natl Acad. Sci. USA 106, 20954–20959 (2009).
Jack, C. R. Jr. et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 58, 750–757 (2002).
Vemuri, P. et al. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage 42, 559–567 (2008).
Whitwell, J. L. et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology 71, 743–749 (2008).
Shaw, L. M. et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413 (2009).
Trojanowski, J. Q. et al. Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimers Dement. 6, 230–238 (2010).
Mueller, S. G. et al. Ways toward an early diagnosis in Alzheimer’s disease: the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Alzheimers Dement. 1, 55–66 (2005).
Apostolova, L. G. et al. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch. Neurol. 63, 693–699 (2006).
Jack, C. R. Jr. et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128 (2010).
Jack, C. R. Jr. et al. Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch. Neurol. 69, 856–867 (2012).
Iturria-Medina, Y., Sotero, R. C., Toussaint, P. J., Mateos-Perez, J. M. & Evans, A. C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 7, 11934 (2016).
Chan, D. et al. Change in rates of cerebral atrophy over time in early-onset Alzheimer’s disease: longitudinal MRI study. Lancet 362, 1121–1122 (2003).
Ridha, B. H. et al. Tracking atrophy progression in familial Alzheimer’s disease: a serial MRI study. Lancet Neurol. 5, 828–834 (2006).
Young, A. L. et al. A data-driven model of biomarker changes in sporadic Alzheimer’s disease. Brain 137, 2564–2577 (2014).
Hayes, A. F. PROCESS: a versatile computational tool for observed variable mediation, moderation, and conditional process modeling (White paper). Retrieved from http://www.afhayes.com/public/process2012.pdf (2012).
Terry, A. V. Jr. & Buccafusco, J. J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 306, 821–827 (2003).
DeKosky, S. T. et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. 51, 145–155 (2002).
Gilmor, M. L. et al. Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J. Comp. Neurol. 411, 693–704 (1999).
Traissard, N. et al. Combined damage to entorhinal cortex and cholinergic basal forebrain neurons, two early neurodegenerative features accompanying Alzheimer’s disease: effects on locomotor activity and memory functions in rats. Neuropsychopharmacology 32, 851–871 (2007).
Flicker, C., Ferris, S. H. & Reisberg, B. Mild cognitive impairment in the elderly: predictors of dementia. Neurology 41, 1006–1009 (1991).
Bauer, M. et al. Cholinergic enhancement of visual attention and neural oscillations in the human brain. Curr. Biol. 22, 397–402 (2012).
Bentley, P., Husain, M. & Dolan, R. J. Effects of cholinergic enhancement on visual stimulation, spatial attention, and spatial working memory. Neuron 41, 969–982 (2004).
Furey, M. L., Pietrini, P. & Haxby, J. V. Cholinergic enhancement and increased selectivity of perceptual processing during working memory. Science 290, 2315–2319 (2000).
Ricciardi, E., Handjaras, G., Bernardi, G., Pietrini, P. & Furey, M. L. Cholinergic enhancement reduces functional connectivity and BOLD variability in visual extrastriate cortex during selective attention. Neuropharmacology 64, 305–313 (2013).
Gill, T. M., Sarter, M. & Givens, B. Sustained visual attention performance-associated prefrontal neuronal activity: evidence for cholinergic modulation. J. Neurosci. 20, 4745–4757 (2000).
Herrero, J. L. et al. Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature 454, 1110–1114 (2008).
Kang, J. I., Groleau, M., Dotigny, F., Giguere, H. & Vaucher, E. Visual training paired with electrical stimulation of the basal forebrain improves orientation-selective visual acuity in the rat. Brain Struct. Funct. 219, 1493–1507 (2014).
Jack, C. R. Jr. et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J. Magn. Reson. Imag. 27, 685–691 (2008).
Wyman, B. T. et al. Standardization of analysis sets for reporting results from ADNI MRI data. Alzheimers Dement. 9, 332–337 (2013).
Ashburner, J. A fast diffeomorphic image registration algorithm. Neuroimage 38, 95–113 (2007).
Eickhoff, S. B. et al. A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25, 1325–1335 (2005).
Zaborszky, L. et al. Stereotaxic probabilistic maps of the magnocellular cell groups in human basal forebrain. Neuroimage 42, 1127–1141 (2008).
Amunts, K. et al. Cytoarchitectonic mapping of the human amygdala, hippocampal region and entorhinal cortex: intersubject variability and probability maps. Anat. Embryol. (Berl) 210, 343–352 (2005).
Jagust, W. J., Davies, P., Tiller-Borcich, J. K. & Reed, B. R. Focal Alzheimer’s disease. Neurology 40, 14–19 (1990).
Geyer, S., Schleicher, A. & Zilles, K. Areas 3a, 3b, and 1 of human primary somatosensory cortex. Neuroimage 10, 63–83 (1999).
Brett, M., Anton, J. L., Valabregue, R. & Poline, J. B. Region of interest analysis using an SPM toolbox (Abstract). 8th International Conference on Functional Mapping of the Human Brain (2002).
Steiger, J. H. Tests for comparing elements of a correlation matrix. Psychol. Bull. 87, 245–251 (1980).
Preacher, K. J. & Hayes, A. F. SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behav. Res. Methods Instrum. Comput. 36, 717–731 (2004).
Acknowledgements
We thank Elizabeth DuPre and Marieke C. Mur for advice and assistance. This work was supported in part by an Alzheimer’s Association grant (NIRG-14-320049) to R.N.S. Data used in preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data, but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Data collection and sharing for this project were funded by the Alzheimer’s Disease Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research and Development, LLC; Johnson & Johnson Pharmaceutical Research and Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer, Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Author information
Authors and Affiliations
Consortia
Contributions
ADNI collected the data. R.N.S. preprocessed the data. T.W.S. conducted the analyses. T.W.S. and R.N.S. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Table 1 (PDF 56 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Schmitz, T., Nathan Spreng, R. & The Alzheimer's Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat Commun 7, 13249 (2016). https://doi.org/10.1038/ncomms13249
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms13249
This article is cited by
-
Atrophy of the cholinergic regions advances from early to late mild cognitive impairment
Neuroradiology (2024)
-
Spatial navigation is associated with subcortical alterations and progression risk in subjective cognitive decline
Alzheimer's Research & Therapy (2023)
-
REM sleep is associated with the volume of the cholinergic basal forebrain in aMCI individuals
Alzheimer's Research & Therapy (2023)
-
An Imbalance in the Pro/mature BDNF Ratio Occurs in Multiple Brain Regions During Normal Ageing in Wild-Type Mice
Journal of Molecular Neuroscience (2023)
-
Turning the Spotlight to Cholinergic Pharmacotherapy of the Human Language System
CNS Drugs (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
