
Abstract
Gas hydrates stored on continental shelves are susceptible to dissociation triggered by environmental changes. Knowledge of the timescales of gas hydrate dissociation and subsequent methane release are critical in understanding the impact of marine gas hydrates on the ocean–atmosphere system. Here we report a methane efflux chronology from five sites, at depths of 220–400 m, in the southwest Barents and Norwegian seas where grounded ice sheets led to thickening of the gas hydrate stability zone during the last glaciation. The onset of methane release was coincident with deglaciation-induced pressure release and thinning of the hydrate stability zone. Methane efflux continued for 7–10 kyr, tracking hydrate stability changes controlled by relative sea-level rise, bottom water warming and fluid pathway evolution in response to changing stress fields. The protracted nature of seafloor methane emissions probably attenuated the impact of hydrate dissociation on the climate system.
Similar content being viewed by others
Introduction
Large quantities of methane exist as free gas, dissolved in pore fluids and solid gas hydrates within marine sediments1. Gas hydrates are found in a variety of geological settings2 but are most common on continental margins where the combination of appropriate pressure–temperature conditions and the presence of organic-rich sediments are conducive to abundant methane generation. The stability of the marine gas hydrate reservoir is primarily affected by changes in temperature and hydrostatic pressure. Consequently, changes in the thickness of the gas hydrate stability zone (GHSZ) are likely to occur along glaciated continental margins where pressure regimes are governed by alternating glacial loading and unloading cycles, and gas hydrate dissociation may be amplified by the influx of warmer waters following ice retreat2,3,4,5. If bottom water warming and/or hydrostatic pressure decrease is large enough, gas hydrate dissociation and release of a significant amount of methane are possible4,6,7,8,9,10. The transfer of this methane to the water column has the potential to amplify ocean acidification and de-oxygenation and, possibly, atmospheric greenhouse gas concentrations11,12. The rates and timescales over which gas hydrates dissociate control the impact of hydrate-derived methane on the global climate system.
Methane released by the dissociation of marine gas hydrates has been inferred to be the cause of a number of negative δ13C excursions in the geological record6,7. Large magnitude gas hydrate dissociation events are also proposed to have occurred in the more recent past, as evidenced by major fluid flow structures in sedimentary basins, such as pockmarks4,13. However, the role of marine gas hydrate releases in influencing atmospheric methane concentrations during the Quaternary remains uncertain. For example, the radiocarbon and hydrogen isotopic composition of methane from Greenland ice cores suggests that atmospheric methane rise at 11.6 and 38 ka can be predominantly accounted for through wetland emissions14,15. Although it appears that Quaternary marine gas hydrate dissociation events did not have a significant impact on the climate system, recent estimates of the gas hydrate reservoirs stored beneath the grounded Antarctic ice sheet suggest that methane release resulting from accelerated ice wastage has the potential to act as a positive feedback on future climate warming16.
Methane is consumed at a variety of stages during transport through the sediment and overlying water column. On a global scale, >80% of methane migrating within the sedimentary package can be microbially consumed at the sedimentary sulphate–methane transition zone by the anaerobic oxidation of methane (AOM)11,17. However, where gas supply is high and flow through the sediment is advective, the capacity of this benthic microbial filter can be outstripped, resulting in release of methane into the overlying water column18. A proportion (up to 100%) of methane within the water column is consumed via dissolution of free gas bubbles and methanotrophic microbial activity19,20 before it reaches the ocean surface and escapes into the atmosphere. Consequently, the short residence time of methane in the oceans and atmosphere (∼10 years), combined with the processes that control consumption in the sediment and water column, requires a rapid (decadal) release and transfer of large volumes of methane to achieve a significant impact on ocean/atmosphere chemistry.
The processes that influence gas hydrate stability in continental margins act on timescales ranging from near instantaneous (for example, slope failure) to 102–103 years (ice sheet collapse), whereas significant sea-level and bottom water temperature variations operate over 103 years21,22. The transfer of methane from the seabed into the water column is typically detected by water column acoustic gas flares, with observations restricted to timescales of less than a few decades20,23. However, assessments of the timescales over which gas hydrate systems respond to the processes that drive changes in pressure and temperature in the subsurface are limited. Studies from GHSZ pinch outs indicate rapid response to temperature change in the immediate subsurface over a few decades8,22. Modelling results show that heat transfer and temperature re-equilibration of ∼100 m of marine sediments would take 102–103 years8,24, and that gas propagated from such depth may take an additional thousand years to vent at the seafloor24. However, empirical constraints on methane efflux triggered by hydrate dissociation resulting from glacial unloading of a continental margin, both past and present are, as yet, lacking.
Direct information on the timing of past methane release events can be obtained through U-Th dating of methane-derived authigenic carbonates25,26, which form at the sedimentary sulphate–methane transition zone, due to elevated carbonate alkalinity resulting from AOM in areas experiencing relatively intense methane fluxes over 102–103 years25. To date, U-Th dating of methane-derived carbonates has mostly focused on samples from mid-latitudes and was used to assess the periodicity of fluid migration along fault systems27,28 and the effects of long-term changes in sea-level and bottom water temperature on methane efflux and/or gas hydrate dissociation26,29,30. Conversely, U-Th dating of carbonates from methane seeps off Svalbard indicates that the stability of gas hydrates at the updip of the GHSZ at high-latitude sites is sensitive to seasonal bottom water temperature fluctuations21.
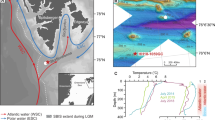
The shelf areas of the Norwegian and Barents Seas offer ideal settings in which to assess the processes governing gas hydrate stability on glaciated continental margins with grounded ice and hence can serve as a palaeo-analogue of the Antarctic gas hydrate reservoir16. Here we report on the timing of methane release in the Barents and Norwegian Seas, where a drop in seabed pressure associated with the retreat of the grounded Scandinavian Ice Sheet (SIS) has been inferred as the driving force behind the dissociation of a significant amount of gas hydrates3,13,31 (Fig. 1). We use U-Th geochronology, stable carbon and oxygen isotope analyses and petrographic observations on authigenic carbonate crusts associated with methane seeps to provide constraints on the timescales of past methane fluxes from five locations for that region. Our new U-Th data set, combined with model estimates of the amount of methane potentially stored as gas hydrates before and after the collapse of the SIS offers an opportunity to study the timescales of methane efflux across the seafloor in response to the environmental changes associated with the last deglaciation.

Results
Studied areas
The southwest Barents and Norwegian Sea are active provinces for oil and gas exploration, with extensively documented, focused, hydrocarbon-rich fluid migration through regional fault networks3,32,33. Mega-scale glacial lineations (for example, in the Ingøydjupet depression) indicate that the shelf areas of those seas were covered by the grounded SIS during the last glaciation34,35,36. The pressure exerted by grounded ice on the underlying sediments was associated with a ∼500 m thickening of the GHSZ3,37 and reactivation of faults and fractures as fluid pathways. Age constraints on grounded ice wedges indicate that ice persisted on the southwest Barents Sea shelf at least until 17 ka38,39, and that ice streams extended to the edge of the shelf at least twice34,36. In this area the seafloor is marked by ubiquitous pockmarks, circular depressions resulting from the expulsion of fine-grained sediments in response to fluid seepage3,13,31,40,41. Although some pockmarks coincide with observed water-column gas flares, indicating active seepage, most are inactive and constitute records of widespread fluid escape at the seafloor related to abrupt gas hydrate dissociation after the last deglaciation3,13,31. The cross-cutting relationships between pockmarks and surrounding sediments, coupled with radiocarbon dating, indicate that pockmark formation post-dated the deposition of glaciomarine sediments at ∼15 ka31.
An autonomous underwater vehicle was used to map the seabed at high spatial resolution (<10 cm) in some pockmark areas and areas of known seepage3,42, and revealed the presence of numerous carbonate crusts. Guided by the autonomous underwater vehicle mapping results, carbonate crusts were sampled by a remotely operated underwater vehicle from four sites in the southwest Barents Sea at depths ranging between 320 and 400 m (PR1, PR3, PR4 and PR5; Fig. 1 and Table 1). Additional samples were collected from the Hola site (depth 218 m) off the Vesterålen islands in the Norwegian Sea. Active gas seepage, inferred from gas flares in the water column, occurs at all sites except at PR5.
Carbonate crust characterization
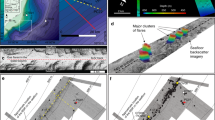
The typical thickness of studied crust samples varies between 10 and 30 cm (Fig. 2). Authigenic carbonates occur as impure early-generation cements, lithifying mud to gravel-size detrital glacial/glaciomarine sediments and as pure detritus-free later-generation botryoidal-layered fillings of centimetre-scale cavities formed within the early-generation carbonate-cemented sediments (Fig. 2b,c). Such cavities are interpreted as conduits generated by a focused fluid flow, which washed out weakly cemented sediments. Microcrystalline and radial fibrous aragonite cements are the dominant authigenic carbonate phases, but minor micritic Mg-calcite (13–15% mol MgCO3) is also present (Supplementary Fig. 1 and Supplementary Table 1). Both aragonite and high Mg-calcite cements contain disseminated framboidal pyrite indicative of active microbial sulfate reduction during carbonate precipitation43. The dominance of aragonite among diagenetic carbonate phases probably indicates precipitation under methane fluxes capable of sustaining high rates of AOM activity in sediments just beneath the seafloor44.

Detailed U-Th ages (in ka±2σ) on representative cross-sections of (a) sample P1210002 and (b) sample P1210017. (c) Scanning electron microscope image showing early-stage cements containing detrital material and detritus-free layer of radial fibrous aragonite cavity fill.
Carbon and oxygen isotope compositions of the sampled carbonate crusts were measured on powders hand-drilled on a millimetre scale, to spatially resolve distinctive carbonate generations. Carbonate δ13C values range from −43.1 to −13.0‰ Vienna Pee Dee Belemnite (VPDB) (Fig. 3 and Table 1, average −32.3±4.3‰ VPDB), indicating that microbial oxidation of methane-rich fluids is the dominant source of carbon. The absence of very 13C-depleted (δ13C <−45‰ VPDB) carbonates in the studied crusts further suggests that the principal source of methane is thermogenic, which is typically less 13C depleted than microbial methane45. A predominantly thermogenic origin is also consistent with the stable isotope composition of methane gas currently seeping from the seabed (δ13C-CH4 of −48‰ and δD-CH4 of −186‰ Vienna Standard Mean Ocean Water (VSMOW); Supplementary Fig. 2).

Isotope compositions of early generation cements and late generation cavity infills of samples hand-drilled from two representative carbonate crusts. The boxes deliniated by dashed lines indicate the δ18O range of carbonate (aragonite47 and Mg-calcite with 13–15% mol MgCO3 (refs 48, 49)) precipitating in equilibrium with present-day bottom water (temperature of 2–3 °C and seawater δ18O of 0‰ VSMOW).
The δ18O values of the authigenic carbonates provide information about the temperature and oxygen isotope composition of the ambient porewater in which carbonates precipitate. The oxygen isotope composition of carbonates precipitating in equilibrium with the bottom water after deglaciation, that is, at 15 ka (δ18O=1‰ VSMOW46 and 1 °C) would have a δ18O value of 4.7‰ VPDB for aragonite47 and 5.6‰ VPDB for Mg-calcite with 15% mol MgCO3 (refs 48, 49). The high δ18O values (that is, up to 6.4‰ VPDB; Fig. 3) measured in the early-generation Mg-calcite-cemented sediments cannot be explained by precipitation of Mg-calcite in equilibrium with the bottom waters during the early deglaciation period and probably indicate the influence of 18O-rich water potentially released during gas hydrate dissociation50.
U-Th geochronology
A total of 14 methane-derived authigenic carbonate crusts from the five sites were selected for U-Th dating; the sampling targeted both early-stage carbonate cements and late-stage cavity fills (Supplementary Data 1). The latter are characterized by low detrital content ([230Th/232Th]AR ranging between 5 and 427) and give dates with a precision on the order of ±0.5–1% (2σ). Early-stage cements prove to be more difficult to date effectively, owing to the presence of a high proportion of detrital material (Fig. 2c), with uncertainties magnified by the impact of the corrections required to account for the presence of initial detrital Th (Fig. 2b and Supplementary Data 2). We obtain 59 reliable dates (defined as [230Th/232Th]AR >2, see Fig. 4 and Supplementary Note), mostly from late-stage cavity fills, which show that 12 of the analysed crusts formed from 17 to 7 ka. Younger dates, between 5 and 2 ka, are obtained from two crusts, one each from the PR1 and PR5 sites (Fig. 4). Owing to the preponderance of late-stage aragonite cavity fills, the U-Th data set provides minimum age estimates for the onset of emissions, which is recorded by the less well-dated early-stage cements. Both the older (17–7 ka) and younger (5–2 ka) crusts span several 100–5,000 years (Fig. 4) and constrain the minimum duration and absolute timing of methane efflux at each site. Data from multiple subsamples collected from the same crust are consistent with the observed textural relationships (that is, cavity fills are younger than early-stage cements; Fig. 2b).

(a) U-Th ages (error bars are 2σ) on 14 studied carbonate crusts (up to 11 ages per crust) grouped by site (see Fig. 1 for locations). (b) Global sea-level curve (error bars are 1σ (ref. 61)) and seabed uplift curve (extracted from ref. 57). (c) Stable oxygen isotope records of ice core, North Greenland Ice Core Project (NGRIP)75 and of benthic foraminifera from a sediment core from the southwest Barents Sea76. The blue vertical shaded area depicts the period when grounded ice-sheet prevailed across the study area39.
Gas hydrate stability modelling
Gas hydrate stability was modelled at steady state based on the presence of an ∼1,100-m-thick grounded ice sheet51, the regional average geothermal gradient of 31 °C km−1 (ref. 52) and a gas composed of 96% methane, 3% ethane and 1% propane. Such a gas composition is consistent with the base of the GHSZ corresponding to the observed bottom-simulating seismic reflectors in the Barents Sea37,53. The model estimates the thickness of the GHSZ at up to 600 m below the seafloor during the Last Glacial Maximum (LGM), thinning gradually towards the shelf edge due to decreasing ice load (Fig. 5a). Following the retreat of grounded ice, which triggered isostatic rebound, sea-level rise and bottom water warming, the GHSZ thinned to ca. 100 m in most of the southwest Barents Sea, including the study area. A thicker GHSZ of up to 400 m persists in the deeper, central part of the southwest Barents Sea (Fig. 5b). Based on the volume change of gas hydrates between the LGM (Fig. 5a) and the present day (Fig. 5b), we estimate that 5–43 Gt of methane was released in the studied area (4 × 105 km2).

Thickness of GHSZ during the LGM (a) and at present-day conditions (b). Black contours denote (a) the ice-sheet thickness in metres during the LGM51 and (b) broad scale faulting system77. The average value of the geothermal gradient used is 31 °C km−1 (ref. 52). It is noteworthy that the abyssal plain has not been taken into account in our model.
Discussion
The clustering of U-Th dates indicates that the main crust-forming methane flux episode took place between 17 and 7 ka (Fig. 4), which is consistent with the estimated timing of pockmark development in the Barents Sea31 and North Sea54, suggesting that fluid release activity was controlled by a regional process. Although the onset of crust growth is less well constrained, the oldest U-Th age (17.5±0.7 ka) coincides with the deglaciation of the southwest Barents Sea (∼18–16 ka) (Fig. 4), which is constrained by 14C dating of foraminifera associated with glaciomarine sediments39. The U-Th ages and the presence of isotopically heavy oxygen (>5‰ VPDB) support a model in which methane is discharged from gas hydrate dissociation, triggered by pressure changes on the continental shelf due to collapse and retreat of the SIS3,31. The U-Th data further indicate that substantial methane efflux continued along the ice-free northern Norwegian margin for another ca. 10 kyr. Although the carbonate crust U-Th data are limited in coverage, the clustering of U-Th dates (Fig. 4) does suggest that methane efflux may have been pulsed. U-Th ages of distinct layers within cavity infills provide evidence for sustained methane fluid flow through individual conduits over 900 years (Fig. 2b). Such focused fluid flow events may be analogous to the observed active water column acoustic gas flares and as such could provide a means to extrapolate observed fluxes.
The integrated geochemistry and geochronology of methane-derived authigenic carbonates, coupled with gas hydrate modelling, provides a conceptual and temporal framework for methane seepage in the southwest Barents Sea. During the LGM, the SIS covered most of Northern Europe, including the Norwegian continental shelf and the entire Barents Sea55,56. Glacial loading by about 1,100 m of grounded ice51 created a 150- to 200-m isostatic depression of the lithosphere57 and the resulting increase of hydrostatic pressure in the underlying sediments extended the GHSZ to up to 600 m below the seafloor throughout the Barents Sea and the Norwegian shelf (Figs 5a and 6). Ice loading probably reactivated widespread basement-penetrating fault systems enhancing the migration of gas originating in Triassic and Jurassic source rocks and hydrocarbon reservoirs58. The accumulation of thermogenic gas in the upper part of the sediment column, corresponding to the GHSZ, would have enabled widespread gas hydrate formation beneath the ice cap during the LGM (Fig. 6a).

(a) During the LGM, gas hydrate stability shown with the red area in the top-left corner was extending up to 600 m below the seabed. (b) Methane migrates through fractures and porous media as a result of gas hydrate dissociation triggered by grounded ice sheet retreat 18–16 ka. (c) Gas hydrate dissociation continues during the isostatic rebound and bottom water warming from ∼16 to ∼9 ka. (d) After ∼9 ka to present, gas plumes occur locally connected to open deep-seated faults. The average geothermal gradient and associated 2σ uncertainties (31±6 °C km−1 (ref. 52)) are shown by solid and dashed lines, respectively, at the base of gas hydrate stability fields (red areas). The red arrow depicts relative change of the base of the GHSZ (red dashed line). Temperature and pressure constraints used for assessing change in GHSZ are in Supplementary Table 3.
The retreat of the SIS from the shelf margin began at 19 ka and comprised several episodes of ice retreat and advance with grounding zone wedge formation35,39. The western Barents Sea, including our study area, was deglaciated over a 2-kyr period, with ice-free conditions reached at ∼16 ka39. The pressure drop associated with ice sheet unloading led to thinning of the GHSZ by as much as 400 m, resulting in gas hydrate dissociation and thereby increased pore pressure, triggering methane advection (Fig. 6b). Assuming a Darcy linear flow model, the dissipation of pore pressure from the seafloor to the base of the LGM GHSZ (600 m below seafloor) during glacial retreat is expected to have taken between 0.005 and 4.6 kyr, to reach the steady state (permeability of 0.04 mD to 1.51 D59 and water viscosity of 2.04 × 10−3 Pa˙s). In addition, the rate of gas hydrate dissociation, position within the sedimentary column and the fluid flow velocity would potentially affect the time over which the fluids reach the seafloor. As carbonate crusts are forming at fluid flow velocities between 20 and 60 cm per year60, the methane migration at such velocities from the base of the LGM GHSZ (600 m) to the seafloor would take between 1 and 3 kyr.
Following the SIS collapse, isostatic rebound caused a ca. 90 m uplift of the seafloor between 16 and 6 ka (Fig. 4), and at the same time sea-level rose ca. 120 m (ref. 61), resulting in a minor net increase in hydrostatic pressure. Changes in stress over this time interval associated with glacial unloading and isostatic rebound resulted in reactivation of pre-existing faults in certain orientations relative to the evolving stress field62 and would have facilitated fluid migration from shallow gas reservoirs generated by gas hydrate dissociation and/or from deeper petroleum systems. Gas hydrate dissociation was further promoted by the inflow of warm Atlantic water into the Barents Sea shelf after deglaciation, as bottom water temperatures rose from 4 °C at ∼15 ka to 6 °C at ∼10 ka with 1–2 °C fluctuations on 100-year timescales (Fig. 4), remaining relatively stable during the Holocene. The heat transfer would take several hundred years to propagate through the sediments and reach the base of the GHSZ24. Consequently, post-glacial gas hydrate destabilization possibly continued until ∼9–7 ka under the combined influence of seabed uplift and temperature increase (Fig. 6c). At present, gas seepage as evidenced by flares in the water column in the Barents Sea is mainly observed above deep-seated faults (Fig. 6d) with methane sourced from petroleum reservoirs.
We estimate that 10 × 103 – 88 × 103 Tg of methane was stored as gas hydrate in the southwest Barents Sea shelf during the LGM, which is equivalent to 0.6–4.9% of the current total oceanic gas hydrate reservoir63. As our study area only represents ca. 30% of the glaciated Barents Sea shelf and ca. 5% of all glaciated shelves with grounded ice sheets (that is, Norwegian margin, Antarctica and Greenland), it is likely that significant amounts of methane were stored in such shelf settings during the LGM. However, the impact of oceanic gas hydrate dissociation on the climate system is limited by the atmospheric transfer rate of methane, which is a function of timescales of dissociation and release rates at the seafloor, the efficiency of methane oxidizers and bubble dissolution in the water column, and the thickness of the water column. Modelling estimates show that under normal oceanographic conditions, methane released at water depths >200 m would be almost entirely (>80%) consumed during transit towards the sea surface20, and that a catastrophic bubble release is required, to drive transport of methane from deeper waters (>100 m) to the atmosphere20. As water depth in the investigated areas of the Norwegian and Barents Sea was on the order of 200–400 m after ice sheet collapse, the resulting impact on atmospheric methane concentrations was probably muted. Furthermore, methane release at the sediment–water interface was also probably modulated by the availability of excess methane and fluid pathways to the seafloor, with slower transit through the sediment column increasing the magnitude of microbial methane consumption within the sedimentary sulphate methane transition zone. Assuming that 90% of the methane released by gas hydrate dissociation is consumed by AOM11, and that 1–5% of it would pass through the water column and reach the atmosphere, the mean integrated flux to the atmosphere in the southwest Barents Sea over 10 kyr would be about 0.0005–0.0172 Tg·per year (minimum and maximum estimates) of methane, which is relatively small when compared with the ∼200 Tg·per year methane flux from all natural sources64. Such a scenario of protracted, relatively low-intensity methane efflux in response to abrupt environmental changes has rarely been evaluated by studies that postulate linkages between gas hydrate dissociation and transfer of methane to the marine realm/atmosphere. However, on shallow continental shelf regions (that is, <150–200 m water depth) that have also experienced glacial–interglacial cycles, it is plausible that a higher proportion of the methane released by gas hydrate dissociation may have passed through the water column and reached the atmosphere.
The main episode of carbonate crust formation in the Barents Sea after the collapse of the SIS broadly overlaps with elevated atmospheric methane concentrations as recorded by ice cores from Greenland and Antarctica65. The potential importance of gas hydrate destabilization due to warming of upper oceanic water masses as a cause for elevated atmospheric methane has been debated in several publications4,10. The hydrogen isotope and 14C characteristics of methane trapped in ice cores suggest insignificant emissions from marine gas hydrates during times of high atmospheric methane after deglaciation and stability of gas hydrates14,66. However, the general inference of gas hydrate stability within continental margin sediments through glacial–interglacial cycles is not globally applicable, as it is dependent on local changes in temperature and hydrostatic pressure. This is particularly important in case of glaciated continental margins where gas hydrate destabilization was triggered by local reduction in pressure effects of collapsing grounded ice sheets. Reliable assessment of the influence of hydrate-released methane from glaciated margins on the climate system after the LGM requires global quantification of methane storage, release and consumption budgets, constraints of timescales of hydrate dissociation, and temporal and volume estimates of the dynamics of ice sheets. Such global data are not currently available. Abrupt, globally synchronous methane release over a timescale of 102 years from deglaciated shelf areas with grounded ice appears unlikely given the protracted nature of hydrate-derived methane efflux after the LGM, as our findings from the southwest Barents Sea indicate, as well as the asynchronous deglaciation of different shelf areas.
The analysis of methane-derived authigenic carbonate through the integration of U-Th geochronology and geochemical proxies, combined with gas hydrate modelling provides a means for evaluating past methane release from glaciated continental margins where gas hydrate dynamics are governed by glacial–interglacial cycles. Modelling results demonstrate that gas hydrate accumulation beneath grounded ice-sheets on the Norwegian margin, in a setting analogous to the present day Antarctic shelf16, generates potentially significant methane reservoirs the stability of which is sensitive to environmental changes affecting local pressure and temperature regimes. Although significant amounts of methane could have been released at the seafloor and transferred to the water column, U-Th geochronology suggests methane release over a ca. 10 kyr interval implying that flux rates were modulated by the second-order processes controlling both the rate of dissociation (isostatic rebound and bottom water warming), fluid transport (changes in stress fields leading to fault reactivation under the control of isostatic rebound) and consumption (via efficiency of microbial oxidation). Overall, the protracted nature of methane release on the Norwegian margin and its minimal impact on atmospheric methane concentrations highlight the complexity of the gas hydrate system and the importance of mechanisms mediating gas hydrate dissociation and fluid advection in response to abrupt climatic change.
Methods
Mineralogy
Petrographical characterization of carbonates was achieved by a combination of optical and scanning electron microscopy techniques. Mineralogy was determined at the Geological Survey of Norway by X-ray diffraction (Philips X'pert apparatus, Cu Kα radiation in 2–70° 2θ range) on bulk carbonate powder. Identification of the main carbonate phases was done using EVA software (Supplementary Table 1). The d104 displacement was used to estimate the MgCO3 mol% in calcite67.
Stable isotope compositions
Stable carbon and oxygen isotopes of hand-drilled carbonate were analysed with a GasBench II preparation line connected to a Thermo Scientific Delta V Advantage IRMS (Thermo Fisher Scientific). Reproducibility is better than ±0.2‰ for both δ13C and δ18O. The stable carbon and hydrogen isotopes of methane (Supplementary Fig. 2) were analysed by a Delta plus XP IRMS (Thermo Fisher Scientific) with analytical reproducibility better than ±1‰ for δ13C values and better than ±10‰ for δD values, respectively. Stable isotope compositions are reported in conventional delta (δ) units relative to the VPDB reference for carbon and oxygen, and relative to the VSMOW reference for hydrogen isotopes.
U-Th dating
U-Th dating was carried out on 76 microdrilled carbonate samples weighing between 2–80 mg (Supplementary Data 1 and Supplementary Fig. 3). A further seven unconsolidated sediment samples (three to six replicates from each, weighing 30–130 mg) were also analysed, to constrain the impact of incorporated detrital material on the U-Th systematics of the carbonate crusts. Analytical protocols for the separation and purification of U and Th from carbonate crust and detritus samples are similar to those of Douarin et al.68, with modifications aimed at ensuring the following: (i) complete dissolution of the detrital material incorporated in the crusts and (ii) oxidation of organic material liable to produce isobaric interferences during measurements of Th isotope ratios69. All evaporation steps took place in a closed EvapoClean device, to minimize cross-contamination and reduce fall-in blanks. Samples were leached in ∼8 M HNO3 for 1 h and centrifuged to separate soluble carbonate-rich fractions from less-soluble detrital material. Detritus fractions were dissolved over 1–7 days in a mixture of HClO4:HF:HNO3 (1:2:2.5), using ∼50 μl HF per estimated mg of material, dried down, re-dissolved in 8 M HCl and dried down again, to ensure the conversion of residual fluorides to chlorides. Detritus fractions were then dissolved in 8 M HNO3, recombined with their respective carbonate fractions, spiked with a mixed 229Th-236U tracer, left to equilibrate overnight and dried down. All samples went through two overnight oxidation steps in a mixture of 2 ml 16 M HNO3 and 0.2 ml 30% H2O2 followed by evaporation to dryness. Pre-concentration of U and Th through Fe co-precipitation and initial separation on 0.6 ml columns using AG-1 × 8 anion exchange resin were done following the procedure of Edwards et al.70. Th fractions were further purified using a second pass through AG-1 × 8 resin and were filtered using 0.22-μm pore-size syringe filters to remove resin particles. Both U and Th fractions were oxidized twice in 2 ml 16 M HNO3 and 0.2 ml 30% H2O2, and dissolved in 1 ml 0.1 M HCl and 0.035 M HF. Before mass spectrometry analyses, all samples were filtered to remove particles originating from the perfluoroalkoxy alkane (FEP) beakers used for sample preparation.
Isotope ratio measurements were made on a Thermo Neptune Plus multi-collector ICP-MS, with samples introduced via an Aridus II desolvating nebulizer using an ESI PFA nebulizer tip with an ∼50 μl min−1 uptake rate. Typical operating conditions included the addition of 4–8 ml min−1 high-purity N2 to the sample-carrying Ar stream, to minimize U and Th oxide formation in the plasma.
U measurements were made using a normal sample cone and X-skimmer cone, and a static multicollector data collection protocol with 234U measured on an axial secondary electron multiplier, and 233U, 235U, 236U and 238U measured on Faraday cups equipped with 1011 Ω resistors. Blocks of five samples were bracketed by analyses of CRM 112a U and CRM 112a U+IRMM 3636 spike. Exponential mass bias corrections were based on the measured values of the 233U/236U ratio of the IRMM 3636 spike normalized to a value of 1.01906. Secondary electron multiplier/Faraday gain corrections were based on the 234U/235U ratio of bracketing unspiked CRM 112a analyses. Hydride formation and tailing were monitored at the beginning of each analytical session, with measurements made at mass 237 and 239, while aspirating an unspiked CRM 112a solution, and were corrected during offline data reduction.
Owing to the young age of the samples, Th measurements required better sensitivity and were predominantly made using a Jet sample cone and X-skimmer cone. 230Th was measured on the axial secondary electron multiplier, with 229Th and 232Th measured on Faraday cups equipped with 1011 Ω resistors. Blocks of ten samples were bracketed by blocks of five analyses of an in-house 229Th-230Th-232Th reference solution calibrated against CRM 112a, which was used to monitor mass bias and secondary electron multiplier/Faraday gain.
U-Th age calculations were performed in Isoplot v. 3.75 (ref. 71). The impact of measured detritus 232Th/238U activity ratio variability on the geological interpretation of U-Th dates within the scope of this study was found to be negligible for samples with (230U/232Th) activity ratios above ∼2 (see Supplementary Note). Corrected U-Th dates from samples with (230Th/232Th)<2 (n=16) were either negative or statistically equivalent to zero (at the 2σ level) and were consequently excluded from the data set (Supplementary Data 1 and 2). The remaining analyses were corrected using a calculated average composition of carbonate-free detritus samples (Option 3, Supplementary Table 2).
Gas hydrate modelling
The gas hydrate stability modelling was carried out assuming steady state using CSMHYD software72. Consequently, this approach illustrates only changes in gas hydrate stability independent of the transient behaviour of the system. We used the bathymetry grids of the southwest Barents Sea shelf (https://www.ngdc.noaa.gov/mgg/global/global.html) and World Ocean Database 2005 bottom water temperature (http://www.nodc.noaa.gov/OC5/indprod.html) from NOAA and a feeding thermogenic gas composition of 96% methane, 3% ethane and 1% propane37. As the underlying overturned sediments have varying consolidation with a range of different thermal conductivity and hence heat flow values59, the geothermal gradient in Barents Sea can vary from 22.8 to 69.3 °C km−1, but is mostly confined around 31±6 °C km−1 (2σ)52. For the Last Glacial Maximum (LGM), ice thickness models predicted for the Barents Sea varies in coverage and thicknesses, ranging from 750 to 2,000 m in the study area51,56,73. For predicting the gas hydrate stability during the LGM model, we used the data from ref. 56, which predicts an ice-sheet thickness of ∼1,100 m for the southwest Barents Sea falling within the most accepted range and we applied a bottom ice temperature of −1.5 °C.
The model of gas hydrate stability was used to estimate the volume of the GHSZ (12.7 × 104 km3) in the southwest Barents Sea shelf during the LGM. Considering a sediment porosity58 of 0.1–0.3 conservatively filled by 1–2% of gas hydrate in which methane occupies 70–100% of the cages74, the weight of methane stored in gas hydrate would range between 10 and 88 Gt, calculated with a volumetric ratio between methane and gas hydrate of 164 and a methane density of 0.70805, kg m−3 at standard conditions for temperature and pressure (0 °C and 1 bar).
Additional information
How to cite this article: Crémière, A. et al. Timescales of methane seepage on the Norwegian margin following collapse of the Scandinavian Ice Sheet. Nat. Commun. 7:11509 doi: 10.1038/ncomms11509 (2016).
References
Milkov, A. V. Global estimates of hydrate-bound gas in marine sediments: how much is really out there? Earth Sci. Rev. 66, 183–197 (2004).
Kvenvolden, K. A. Gas hydrates—geological perspective and global change. Rev. Geophys. 31, 173–187 (1993).
Chand, S. et al. Multiple episodes of fluid flow in the SW Barents Sea (Loppa High) evidenced by gas flares, pockmarks and gas hydrate accumulation. Earth Planet. Sci. Lett. 331–332, 305–314 (2012).
Kennett, J. P. Methane Hydrates in Quaternary Climate Change: The Clathrate Gun Hypothesis American Geophysical Union (2003).
Portnov, A., Vadakkepuliyambatta, S., Mienert, J. & Hubbard, A. Ice-sheet-driven methane storage and release in the Arctic. Nat. Commun. 7, 10314 (2016).
Dickens, G. R., O’Neil, J. R., Rea, D. K. & Owen, R. M. Dissociation of oceanic methane hydrate as a cause of the carbon isotope excursion at the end of the Paleocene. Paleoceanography 10, 965–971 (1995).
Hesselbo, S. P. et al. Massive dissociation of gas hydrate during a Jurassic oceanic anoxic event. Nature 406, 392–395 (2000).
Marín-Moreno, H., Minshull, T. A., Westbrook, G. K., Sinha, B. & Sarkar, S. The response of methane hydrate beneath the seabed offshore Svalbard to ocean warming during the next three centuries. Geophys. Res. Lett. 40, 5159–5163 (2013).
Reagan, M. T., Moridis, G. J., Elliott, S. M. & Maltrud, M. Contribution of oceanic gas hydrate dissociation to the formation of Arctic Ocean methane plumes. J. Geophys. Res. Oceans 116, C09014 (2011).
Nisbet, E. G. Have sudden large releases of methane from geological reservoirs occurred since the Last Glacial Maximum, and could such releases occur again? Philos. Trans. Math. Phys. Eng. Sci. 360, 581–607 (2002).
Reeburgh, W. S. Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513 (2007).
Valentine, D. L., Blanton, D. C., Reeburgh, W. S. & Kastner, M. Water column methane oxidation adjacent to an area of active hydrate dissociation, Eel river Basin. Geochim. Cosmochim. Acta 65, 2633–2640 (2001).
Rise, L., Bellec, V. K., Chand, S. & Bøe, R. Pockmarks in the southwestern Barents Sea and Finnmark fjords. Nor. J. Geol. 94, 263–282 (2014).
Petrenko, V. V. et al. 14CH4 measurements in Greenland Ice: investigating Last Glacial termination CH4 sources. Science 324, 506–508 (2009).
Bock, M. et al. Hydrogen isotopes preclude marine hydrate CH4 emissions at the onset of Dansgaard-Oeschger events. Science 328, 1686–1689 (2010).
Wadham, J. L. et al. Potential methane reservoirs beneath Antarctica. Nature 488, 633–637 (2012).
Hinrichs, K. & Boetius, A. The Anaerobic Oxidation of Methane: New Insights in Microbial Ecology and Biogeochemistry (2002).
Wallmann, K., Drews, M., Aloisi, G. & Bohrmann, G. Methane discharge into the Black Sea and the global ocean via fluid flow through submarine mud volcanoes. Earth Planet. Sci. Lett. 248, 545–560 (2006).
Boetius, A. & Wenzhöfer, F. Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 6, 725–734 (2013).
McGinnis, D. F., Greinert, J., Artemov, Y., Beaubien, S. E. & Wüest, A. Fate of rising methane bubbles in stratified waters: How much methane reaches the atmosphere? J. Geophys. Res. Oceans 111, C09007 (2006).
Berndt, C. et al. Temporal constraints on hydrate-controlled methane seepage off Svalbard. Science 343, 284–287 (2014).
Phrampus, B. J. & Hornbach, M. J. Recent changes to the Gulf Stream causing widespread gas hydrate destabilization. Nature 490, 527–530 (2012).
Skarke, A., Ruppel, C., Kodis, M., Brothers, D. & Lobecker, E. Widespread methane leakage from the sea floor on the northern US Atlantic margin. Nat. Geosci. 7, 657–661 (2014).
Darnell, K. N. & Flemings, P. B. Transient seafloor venting on continental slopes from warming-induced methane hydrate dissociation. Geophys. Res. Lett. 42, 10765–10772 (2015).
Bayon, G., Henderson, G. M. & Bohn, M. U–Th stratigraphy of a cold seep carbonate crust. Chem. Geol. 260, 47–56 (2009).
Teichert, B. M. A. et al. U/Th systematics and ages of authigenic carbonates from Hydrate Ridge, Cascadia Margin: recorders of fluid flow variations. Geochim. Cosmochim. Acta 67, 3845–3857 (2003).
Crémière, A., Bayon, G., Ponzevera, E. & Pierre, C. Paleo-environmental controls on cold seep carbonate authigenesis in the Sea of Marmara. Earth Planet. Sci. Lett. 376, 200–211 (2013).
Liebetrau, V., Eisenhauer, A. & Linke, P. Cold seep carbonates and associated cold-water corals at the Hikurangi Margin, New Zealand: new insights into fluid pathways, growth structures and geochronology. Mar. Geol. 272, 307–318 (2010).
Feng, D. et al. U/Th dating of cold-seep carbonates: an initial comparison. Deep Sea Res. Part II Top. Stud. Oceanogr. 57, 2055–2060 (2010).
Watanabe, Y., Nakai, S., Hiruta, A., Matsumoto, R. & Yoshida, K. U–Th dating of carbonate nodules from methane seeps off Joetsu, Eastern Margin of Japan Sea. Earth Planet. Sci. Lett. 272, 89–96 (2008).
Pau, M., Hammer, Ø. & Chand, S. Constraints on the dynamics of pockmarks in the SW Barents Sea: evidence from gravity coring and high-resolution, shallow seismic profiles. Mar. Geol. 355, 330–345 (2014).
Mienert, J. & Posewang, J. Evidence of shallow- and deep-water gas hydrate destabilizations in North Atlantic polar continental margin sediments. Geo-Mar. Lett. 19, 143–149 (1999).
Vogt, P. R., Gardner, J. & Crane, K. The Norwegian–Barents–Svalbard (NBS) continental margin: introducing a natural laboratory of mass wasting, hydrates, and ascent of sediment, pore water, and methane. Geo-Mar. Lett. 19, 2–21 (1999).
Laberg, J. S. & Vorren, T. O. Late Weichselian submarine debris flow deposits on the Bear Island Trough Mouth Fan. Mar. Geol. 127, 45–72 (1995).
Ottesen, D., Dowdeswell, J. A. & Rise, L. Submarine landforms and the reconstruction of fast-flowing ice streams within a large Quaternary ice sheet: the 2500-km-long Norwegian-Svalbard margin (57°–80°N). Geol. Soc. Am. Bull. 117, 1033–1050 (2005).
Sættem, J., Poole, D. A. R., Ellingsen, L. & Sejrup, H. P. Glacial geology of outer Bjørnøyrenna, southwestern Barents Sea. Mar. Geol. 103, 15–51 (1992).
Chand, S. et al. Gas hydrate stability zone modelling in areas of salt tectonics and pockmarks of the Barents Sea suggests an active hydrocarbon venting system. Mar. Pet. Geol. 25, 625–636 (2008).
Rüther, D. C., Mattingsdal, R., Andreassen, K., Forwick, M. & Husum, K. Seismic architecture and sedimentology of a major grounding zone system deposited by the Bjørnøyrenna Ice Stream during Late Weichselian deglaciation. Quat. Sci. Rev. 30, 2776–2792 (2011).
Winsborrow, M. C. M., Andreassen, K., Corner, G. D. & Laberg, J. S. Deglaciation of a marine-based ice sheet: Late Weichselian palaeo-ice dynamics and retreat in the southern Barents Sea reconstructed from onshore and offshore glacial geomorphology. Quat. Sci. Rev. 29, 424–442 (2010).
Solheim, A. & Elverhøi, A. Gas-related sea floor craters in the Barents Sea. Geo-Mar. Lett. 13, 235–243 (1993).
Jung, W.-Y. & Vogt, P. R. Effects of bottom water warming and sea level rise on Holocene hydrate dissociation and mass wasting along the Norwegian-Barents Continental Margin. J. Geophys. Res. Solid Earth 109, B06104 (2004).
Chand, S. et al. Active venting system offshore Northern Norway. Eos Trans. Am. Geophys. Union 89, 261–262 (2008).
Peckmann, J. et al. Methane-derived carbonates and authigenic pyrite from the northwestern Black Sea. Mar. Geol. 177, 129–150 (2001).
Aloisi, G. et al. CH4-consuming microorganisms and the formation of carbonate crusts at cold seeps. Earth Planet. Sci. Lett. 203, 195–203 (2002).
Schoell, M. The hydrogen and carbon isotopic composition of methane from natural gases of various origins. Geochim. Cosmochim. Acta 44, 649–661 (1980).
Fairbanks, R. G. A 17,000-year glacio-eustatic sea level record: influence of glacial melting rates on the Younger Dryas event and deep-ocean circulation. Nature 342, 637–642 (1989).
Kim, S.-T., O’Neil, J. R., Hillaire-Marcel, C. & Mucci, A. Oxygen isotope fractionation between synthetic aragonite and water: Influence of temperature and Mg2+ concentration. Geochim. Cosmochim. Acta 71, 4704–4715 (2007).
Tarutani, T., Clayton, R. & Mayeda, T. Effect of polymorphism and magnesium substitution on oxygen isotope fractionation between calcium carbonate and water. Geochim. Cosmochim. Acta 33, 987–98 (1969).
Friedman, I. & O’Neil, J. R. Compilation of Stable Isotope Fractionation Factors of Geochemical Interest U.S. G.P.O. (1977).
Bohrmann, G., Greinert, J., Suess, E. & Torres, M. Authigenic carbonates from the Cascadia subduction zone and their relation to gas hydrate stability. Geology 26, 647–650 (1998).
Siegert, M. J., Dowdeswell, J. A., Hald, M. & Svendsen, J.-I. Modelling the Eurasian Ice Sheet through a full (Weichselian) glacial cycle. Glob. Planet. Change 31, 367–385 (2001).
Vadakkepuliyambatta, S., Hornbach, M. J., Bünz, S. & Phrampus, B. J. Controls on gas hydrate system evolution in a region of active fluid flow in the SW Barents Sea. Mar. Pet. Geol. 66, (Part 4): 861–872 (2015).
Laberg, J. S. & Andreassen, K. Gas hydrate and free gas indications within the Cenozoic succession of the Bjørnøya Basin, western Barents Sea. Mar. Pet. Geol. 13, 921–940 (1996).
Forsberg, C. F. et al. Formation Of Pockmarks In The Norwegian Channel Society of Underwater Technology (2007).
Landvik, J. Y. et al. The last glacial maximum of Svalbard and the Barents Sea area: ice sheet extent and configuration. Quat. Sci. Rev. 17, 43–75 (1998).
Svendsen, J. I. et al. Late Quaternary ice sheet history of northern Eurasia. Quat. Sci. Rev. 23, 1229–1271 (2004).
Fjeldskaar, W. The amplitude and decay of the glacial forebulge in Fennoscandia. Nor. Geol. Tidsskr. 74, 2–8 (1994).
Henriksen, E. et al. Chapter 17 Uplift and erosion of the greater Barents Sea: impact on prospectivity and petroleum systems. Geol. Soc. Lond. Mem. 35, 271–281 (2011).
Bugge, T. et al. Shallow stratigraphic drilling applied in hydrocarbon exploration of the Nordkapp Basin, Barents Sea. Mar. Pet. Geol. 19, 13–37 (2002).
Luff, R., Wallmann, K. & Aloisi, G. Numerical modeling of carbonate crust formation at cold vent sites: significance for fluid and methane budgets and chemosynthetic biological communities. Earth Planet. Sci. Lett. 221, 337–353 (2004).
Lambeck, K., Rouby, H., Purcell, A., Sun, Y. & Sambridge, M. Sea level and global ice volumes from the Last Glacial Maximum to the Holocene. Proc. Natl. Acad. Sci. 111, 15296–15303 (2014).
Brandes, C., Steffen, H., Steffen, R. & Wu, P. Intraplate seismicity in northern Central Europe is induced by the last glaciation. Geology G36710, 1 (2015).
Boswell, R. & Collett, T. S. Current perspectives on gas hydrate resources. Energy Environ. Sci. 4, 1206–1215 (2011).
Ciais, P. et al. in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change 465–570Cambridge University Press (2013).
Bender, M. et al. Climate correlations between Greenland and Antarctica during the past 100,000 years. Nature 372, 663–666 (1994).
Sowers, T. Late quaternary atmospheric CH4 isotope record suggests marine clathrates are stable. Science 311, 838–840 (2006).
Goldsmith, J. & Graf, D. Relation between lattice constants and composition of the Ca-Mg carbonates. Am. Mineral. 43, 84–101 (1958).
Douarin, M. et al. Growth of north-east Atlantic cold-water coral reefs and mounds during the Holocene: a high resolution U-series and 14C chronology. Earth Planet. Sci. Lett. 375, 176–187 (2013).
Shen, C.-C. et al. Uranium and thorium isotopic and concentration measurements by magnetic sector inductively coupled plasma mass spectrometry. Chem. Geol. 185, 165–178 (2002).
Edwards, R. L., Chen, J. H. & Wasserburg, G. J. U-238 U-234-TH-230-TH-232 systematics and the precise measurement of time over the past 50 000 years. Earth Planet. Sci. Lett. 81, 175–192 (1987).
Ludwig, K. R. User’s Manual for Isoplot 3.75: A Geochronological Toolkit for Microsoft Excel 5, Kenneth R. Ludwig (2003).
Sloan, E. D. & Koh, C. A. Clathrate Hydrates of Natural Gases CRC Press (2008).
Siegert, M. J. & Dowdeswell, J. A. Numerical reconstructions of the Eurasian Ice Sheet and climate during the Late Weichselian. Quat. Sci. Rev. 23, 1273–1283 (2004).
Lorenson, T. D. & Collett, T. S. in Proceedings of the Ocean Drilling Program, Scientific Results 164, 37–46 (2000).
Andersen, K. K. et al. High-resolution record of Northern Hemisphere climate extending into the last interglacial period. Nature 431, 147–151 (2004).
Aagaard-Sørensen, S., Husum, K., Hald, M. & Knies, J. Paleoceanographic development in the SW Barents Sea during the Late Weichselian–Early Holocene transition. Quat. Sci. Rev. 29, 3442–3456 (2010).
Gabrielsen, R. H., Færseth, R. B., Jensen, L. N., Kalheim, J. E. & Riis, F. Structural Elements of the Norwegian Continental Shelf, Part I: The Barents Sea Region 47, NPD Bulletin (1990).
Acknowledgements
This study benefited from reviews from H. Marín Moreno and two anonymous reviewers. We are grateful to G. Bayon, C. Pierre, A. Prave and B. Schoene for helpful discussions. We acknowledge J. Schönenberger for her help with XRD analyses. We thank Lundin Norway AS and the Geological Survey of Norway for permission to publish the data. This research is part of the Centre of Excellence: Arctic Gas hydrate, Environment and Climate (CAGE) funded by the Norwegian Research Council (grant No. 223259). T.M. acknowledges the Estonian Research Council (Grant PUT611).
Author information
Authors and Affiliations
Contributions
A.C., A.L., D.J.C., S.C. and D.S. designed the study and wrote the manuscript. D.S. produced U-Th data with input from A.C., A.L., D.J.C. and S.R.N. T.M. performed stable isotope analyses. S.C. and A.C. carried out the gas hydrate stability modelling. A.L., T.T., S.C. and H.B. carried out site surveys and sample collection. S.S. helped with sample preparation. All authors contributed to the final editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-3, Supplementary Tables 1-3, Supplementary Note 1 and Supplementary References. (PDF 1099 kb)
Supplementary Data 1
U-Th isotopic data (DOC 199 kb)
Supplementary Data 2
U-Th detritus-corrected age (DOC 137 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Crémière, A., Lepland, A., Chand, S. et al. Timescales of methane seepage on the Norwegian margin following collapse of the Scandinavian Ice Sheet. Nat Commun 7, 11509 (2016). https://doi.org/10.1038/ncomms11509
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms11509
This article is cited by
-
Evidence of Arctic methane emissions across the mid-Pleistocene
Communications Earth & Environment (2023)
-
Visualization of shallow subseafloor fluid migration in a shallow gas hydrate field using high-resolution acoustic mapping and ground-truthing and their implications on the formation process: a case study of the Sakata Knoll on the eastern margin of the Sea of Japan
Marine Geophysical Research (2022)
-
Deglacial bottom water warming intensified Arctic methane seepage in the NW Barents Sea
Communications Earth & Environment (2021)
-
Pore-water geochemistry in methane-seep sediments of the Makran accretionary wedge off Pakistan: Possible link to subsurface methane hydrate
Acta Oceanologica Sinica (2021)
-
The benthic foraminiferal δ34S records flux and timing of paleo methane emissions
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
