
Abstract
Plasmodium knowlesi is a zoonotic parasite transmitted from macaques causing malaria in humans in Southeast Asia. Plasmodium parasites bind to red blood cell (RBC) surface receptors, many of which are sialylated. While macaques synthesize the sialic acid variant N-glycolylneuraminic acid (Neu5Gc), humans cannot because of a mutation in the enzyme CMAH that converts N-acetylneuraminic acid (Neu5Ac) to Neu5Gc. Here we reconstitute CMAH in human RBCs for the reintroduction of Neu5Gc, which results in enhancement of P. knowlesi invasion. We show that two P. knowlesi invasion ligands, PkDBPβ and PkDBPγ, bind specifically to Neu5Gc-containing receptors. A human-adapted P. knowlesi line invades human RBCs independently of Neu5Gc, with duplication of the sialic acid-independent invasion ligand, PkDBPα and loss of PkDBPγ. Our results suggest that absence of Neu5Gc on human RBCs limits P. knowlesi invasion, but that parasites may evolve to invade human RBCs through the use of sialic acid-independent pathways.
Similar content being viewed by others


Introduction
Diverse pathogens including bacteria1,2, parasites3,4 and many different viruses5,6 take advantage of the ubiquity of sialic acids on the surface of mammalian cells to attach to and infect host cells6. Sialic acids are a family of acidic sugars usually found at the termini of glycan chains on proteins and lipids7. The most common forms of sialic acid on mammalian cells are N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). Humans express Neu5Ac, but lack Neu5Gc because of an Alu-mediated exon deletion in the gene encoding cytidine monophosphate Neu5Ac hydroxylase (CMAH), which converts Neu5Ac to Neu5Gc8,9,10. This CMAH gene inactivation occurred ∼3.2 Myr ago11, around a time of active hominin evolution12, and is shared by Neanderthals13, extinct hominins closely related to humans.
In contrast to the human lineage, CMAH is functional in great apes. Variation between human (Neu5Ac) and great ape (Neu5Gc) sialic acid was posited to underlie the host specificity exhibited by the evolutionarily related Laverania parasites, P. falciparum and P. reichenowi, which infect humans and chimpanzees, respectively14. However, recent work has challenged the view that Neu5Gc is a key host determinant of the tropism exhibited by P. falciparum and other Laverania parasites15,16.
P. knowlesi is unique among the human malaria-causing parasites because it displays significant cross-species transmission. P. knowlesi chronically infects Neu5Gc-positive long-tailed and pig-tailed macaques, as well as Neu5Ac-positive humans17. Most human blood-stage infections are mild and associated with low parasitaemias18, but increasing numbers of severe infections accompanied by high parasitaemias are being reported19,20. There is growing concern that this simian parasite is adapting to infect humans more efficiently. It has been shown that P. knowlesi can expand its preferred host cell niche by invading older red blood cells (RBCs)21, and this is an important factor influencing adaptation of P. knowlesi to the human population.
In this study, we sought to test the hypothesis that Neu5Gc is a key determinant of P. knowlesi tropism. We modified human RBCs by introducing Neu5Gc on the cell surface through expression of chimpanzee CMAH in ex vivo-cultured RBCs (cRBCs)22,23. We show that while Neu5Gc expression has no effect on P. falciparum invasion, it significantly increases P. knowlesi invasion. Further, both PkDBPβ and PkDBPγ, which belong to the Duffy binding protein (DBP) ligand family, bind to Neu5Gc-sialylated receptors, potentially facilitating invasion of Neu5Gc-expressing human cRBCs. In demonstrating that a human-adapted P. knowlesi lab strain efficiently invades Neu5Gc-expressing human cRBCs in a sialic acid-independent manner, we identify a potential host factor that P. knowlesi must overcome for efficient adaptation to humans.
Results
CMAH expression introduces Neu5Gc on human RBCs
An analysis of the genomes of a large range of mammalian species indicates that CMAH loss is infrequent (Supplementary Fig. 1). The genomic sequence of a Denisovan24, an extinct species of the human lineage, has an exon deletion in CMAH like humans and Neanderthals, which would result in the loss of expression of Neu5Gc (Fig. 1a; Supplementary Fig. 2). Among primates, CMAH genetic inactivation has also occurred in New World monkeys25 (Fig. 1a, Supplementary Fig. 1), which serve as model organisms for the human P. falciparum and P. vivax parasites26.

(a) The phylogenetic tree for a range of primates indicates loss of CMAH (highlighted in red) and timing of this event in ancestral hominins and New World monkeys (marmoset and Saimiri monkey). Myr ago, million years ago. (b) Domain structures of pLVX-Puro and PtCMAH.pLVX-Puro. Ψ, packaging signal; CMV IE p, cytomegalovirus immediate early promoter; cPPT, central polypurine tract; PGK p, phosphoglycerate kinase promoter; PuroR, puromycin resistance gene; RRE, Rev-response element; WPRE, woodchuck hepatitis virus post-transcriptional regulatory element. (c) Normal morphology of day 21 pLVX and PtCMAH cRBCs. Scale bar, 10 μm. (d) Neu5Gc expression on RBCs as measured by flow cytometry using an α-Neu5Gc antibody. Shown are representative plots from at least six independent experiments. Normalized to mode—normalization to the modal fluorescence value. (e) Neu5Gc expression on RBCs as measured by western blot (left panel). The coomassie-stained protein gel (right panel) shows total protein from RBC samples analysed by western blot. Representative images are shown from two independent experiments. (f) Relative amounts of Neu5Ac and Neu5Gc on RBCs by HPLC analyses. The average of three independent experiments is shown. Error bars represent the s.e.m.
To determine whether host sialic acid variation impacts P. knowlesi tropism, we expressed the CMAH gene from chimpanzee (Pan troglodytes) in human CD34+ haematopoietic stem cells by transduction, with lentivirus harbouring the chimpanzee CMAH transgene (PtCMAH) or an empty vector (pLVX; Fig. 1b). Terminal differentiation to mature cRBCs22,23 resulted in transgenic human cRBCs expressing PtCMAH or wild-type control cRBCs with an inactive human CMAH. Both sets of cells displayed normal RBC morphology (Fig. 1c) and had similar expression of the major glycosylated receptors, glycophorin A and glycophorin C, as well as the Duffy antigen receptor for chemokines (DARC; Supplementary Fig. 3), a critical human receptor for P. knowlesi invasion27. PtCMAH cRBCs, but not pLVX cRBCs, expressed Neu5Gc as shown by flow cytometry (Fig. 1d), and western blot analysis where we observed that Neu5Gc is added to a range of proteins of varying molecular weights (Fig. 1e). Using high-performance liquid chromatography (HPLC), we found that on PtCMAH cRBCs, Neu5Gc constitutes ∼70% of total sialic acid, while Neu5Ac is reduced to ∼30% of the amount found on pLVX cells (Fig. 1f; Supplementary Table 1), consistent with the proportion of Neu5Gc and Neu5Ac observed in chimpanzees28.
Neu5Gc on human RBCs enhances P. knowlesi invasion
To determine the effect of human Neu5Gc expression on P. knowlesi, we tested invasion of the P. knowlesi H strain into RBCs expressing different sialic acid variants. As expected, we found that P. knowlesi invasion into Neu5Gc-positive rhesus macaque RBCs is more efficient than Neu5Ac-positive human RBCs (Fig. 2a). With the transgenic and isogenic cRBCs differing solely in the expressed sialic acid variant, we observed that P. knowlesi invades Neu5Gc-positive PtCMAH cRBCs at significantly increased levels (greater than fivefold, P=0.012; Student’s t-test (two-tailed)) compared with Neu5Ac-positive pLVX cRBCs (Fig. 2a), bringing the level of P. knowlesi invasion close to that found for macaque RBCs. This provides direct functional evidence that the absence of Neu5Gc is a major restriction factor for P. knowlesi invasion of human RBCs. Our in vitro culture system produces cRBCs of similar age, as indicated by CD71 expression (Supplementary Fig. 3), suggesting that Neu5Gc mediates invasion independently of RBC age21.

(a) P. knowlesi H invasion of PtCMAH cRBCs is significantly enhanced, compared with pLVX cRBCs. (b,c) Sialic acid-dependent and sialic acid-independent P. falciparum strains Dd2 (b) and 3D7 (c), respectively, invade PtCMAH cRBCs and pLVX cRBCs similarly. Human and macaque RBCs are included as controls for each strain. The parasitized erythrocyte multiplication rate for P. knowlesi H is the average of four independent experiments and for P. falciparum Dd2 and 3D7, three independent experiments. Error bars indicate the s.e.m. The Student’s t-test (two-tailed) was used to assess significant differences in invasion. *P=0.012. NS, not significant.
P. falciparum is not affected by Neu5Gc on human RBCs
To directly determine the impact of cell surface Neu5Gc on P. falciparum invasion, we assessed invasion of the P. falciparum sialic acid-dependent and sialic acid-independent laboratory-adapted strains, Dd2 and 3D7, respectively, into RBCs expressing different sialic acid variants. As expected, P. falciparum Dd2 and 3D7 invaded human RBCs, but not macaque RBCs29 (Fig. 2b,c). However, in contrast to P. knowlesi, both P. falciparum strains invaded PtCMAH cRBCs and pLVX cRBCs at similar levels (Fig. 2b,c), suggesting that P. falciparum can utilise both Neu5Ac and Neu5Gc. This supports the biochemical evidence that the major sialic acid-dependent invasion ligand P. falciparum erythrocyte-binding antigen-175 does not discriminate between Neu5Ac and Neu5Gc15,16. The PtCMAH cRBCs have ∼30% of the normal complement of Neu5Ac that might be sufficient for efficient invasion. However, this is not supported by the finding that an enzymatic reduction of Neu5Ac to ∼30% normal levels on human RBCs results in significantly decreased invasion by P. falciparum sialic acid-dependent lines (Supplementary Fig. 4).
PkDBPβ and PkDBPγ bind to sialylated receptors
To identify the molecular determinants of sialic acid-dependent invasion in P. knowlesi, we recombinantly expressed the binding domains of the P. knowlesi DBP ligands, PkDBPα, PkDBPβ and PkDBPγ30 (Supplementary Fig. 5a,b), as fusions with the cartilage oligomeric matrix protein (COMP) pentamerization domain31. PkDBPα binds to the DARC receptor32, while PkDBPβ and PkDBPγ have been shown to bind rhesus macaque RBCs33. We assessed binding of recombinant PkDBP domains to normal and enzyme-treated human and rhesus macaque RBC ghosts in protein overlay assays. As expected, PkDBPα did not bind to chymotrypsin-treated macaque RBC ghosts (chymotrypsin cleaves DARC). However, binding to untreated and trypsin-treated macaque ghosts, as well as macaque ghosts stripped of sialic acid by neuraminidase treatment was preserved (Fig. 3a), as expected32,33. In contrast, PkDBPβ and PkDBPγ did not bind to neuraminidase-treated macaque RBC ghosts, demonstrating that PkDBPβ and PkDBPγ are both sialic acid-binding ligands (Fig. 3b). The sialic acid dependence of PkDBPβ has been reported previously34, but the same study concludes that PkDBPγ binds to macaque RBCs in a sialic acid-independent manner. Our protein overlay and orthogonal flow cytometry-based-binding data (Fig. 3c; Supplementary Fig. 6) strongly suggest that PkDBPγ is also a sialic acid-dependent invasion ligand. The binding pattern for PkDBPβ and PkDBPγ to proteins within the RBC ghost overlays was different, however, implying a diversification for binding to different macaque receptors. As expected, PkDBPβ and PkDBPγ did not bind to human RBC ghosts, and glutathione s-transferase (GST) control protein did not bind to either cell type (Fig. 3b; Supplementary Fig. 5d).

(a) Left panel—PkDBPα-COMP binds to DARC on untreated (U), neuraminidase-treated (N) and trypsin-treated (T) macaque RBCs, but not chymotrypsin-treated (C) macaque RBCs as seen in a protein overlay. Right panel—α-DARC western blot confirms that the PkDBPα receptor on macaque RBCs is DARC (highlighted by the bracket). (b) Binding of PkDBPβ-COMP and PkDBPγ-COMP to untreated and protease-treated macaque RBCs, but not to neuraminidase-treated macaque RBCs. GST-COMP does not bind to macaque RBCs. Data are representative of at least four independent experiments. (c) PkDBPβ-COMP (red) and PkDBPγ-COMP (blue) bind to untreated (solid trace) PtCMAH cRBCs, as to macaque RBCs, but not to pLVX cRBCs, like human RBCs as determined by a flow cytometry-based assay. Binding to neuraminidase-treated (dashed trace) PtCMAH cRBCs and macaque RBCs is markedly decreased. COMP protein (black) does not bind to any cell type. The assay was performed twice, in triplicate; representative traces are shown.
PkDBPβ and PkDBPγ bind to Neu5Gc receptors
Since PkDBPβ and PkDBPγ bound macaque but not human RBCs in a sialic acid-dependent manner, we sought to determine if binding was a function of a preference for Neu5Gc over Neu5Ac. To this end, we assayed binding of increasing concentrations of PkDBPβ and PkDBPγ to PtCMAH cRBCs, using a flow cytometry-based binding assay (Supplementary Fig. 7a). We observed dose-dependent binding of both PkDBPβ and PkDBPγ to PtCMAH cRBCs, but not control pLVX cRBCs (Fig. 3c; Supplementary Fig. 7a), suggesting that Neu5Gc is strictly required for binding. Furthermore, neuraminidase treatment of PtCMAH cRBCs abrogated binding (Fig. 3c; Supplementary Fig. 7b). Similarly, we observed Neu5Gc-dependent binding to protein receptors in cRBC overlay assays (Supplementary Fig. 7c). Together, our experiments indicate that PkDBPβ and PkDBPγ are Neu5Gc-binding ligands that can enhance invasion into Neu5Gc-expressing RBCs.
We observed stronger binding of PkDBPγ to rhesus RBCs relative to PtCMAH cRBCs potentially because of a differing linkage between the terminal sialic acid and the proximal sugar or significant differences in the structure of the protein backbone of the orthologous receptor on PtCMAH cRBCs. Both sialic acid–sugar linkage and protein backbone have been shown to be important for sialic acid-dependent ligand–receptor interactions in P. falciparum35,36,37.
Human-adapted P. knowlesi invasion is Neu5Gc-independent
Increased invasion into human RBCs could be a major factor in P. knowlesi pathogenesis21. We have adapted P. knowlesi strain H (Pk H) to grow efficiently in human RBCs, generating a strain named Pk YH1 hereafter21. To determine how Neu5Gc impacts the human-adapted P. knowlesi strain, we assessed invasion of Pk YH1 into PtCMAH cRBCs and pLVX cRBCs. In contrast to the parental Pk H strain, Pk YH1 invasion into PtCMAH cRBCs was not enhanced relative to pLVX cRBCs (Figs 2a and 4a). As expected, Pk YH1 invaded human and macaque RBCs equally well (Fig. 4a). These results suggest that Pk YH1 has adapted to use an altered set of functional invasion ligands for human infection, circumventing the dependence on Neu5Gc for optimal invasion.

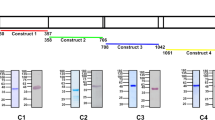
(a) A human-adapted laboratory strain, Pk YH1, invades Neu5Gc-positive and Neu5Gc-negative RBCs similarly. Shown is the average PEMR of three biological replicates performed in triplicate. Error bars represent the s.e.m. NS, not significant (Student’s t-test (two-tailed)). (b) Read coverage normalized to the average genome-wide coverage for regions containing the DBP loci in Pk YH1 and the parental Pk H strain. Pk YH1 has a PkDBPα duplication (left panel) and a PkDBPγ deletion (bottom panel). PkDBPβ in Pk YH1 is unchanged (right panel). The intron–exon structure of each gene is shown above the corresponding chromosomal region. (c) The IC50 of MGSA against Pk YH1 invasion of human RBCs is increased >10-fold compared with Pk H. Shown is the average invasion efficiency of three (Pk H) or four (Pk YH1) biological replicates. Error bars indicate the s.e.m.
Whole-genome sequencing of the Pk YH1 and parental Pk H strains was carried out to identify changes in invasion ligands of the DBP family and the recently described orthologues of the reticulocyte binding-like (RBL) family38. Sequencing revealed a duplication of the region containing the sialic acid-independent PkDBPα gene, as well as a complete deletion of the sialic acid-dependent PkDBPγ gene in Pk YH1 (Fig. 4b; Supplementary Fig. 8; Supplementary Table 2). These alterations suggest that the mechanism for the observed increase in Pk YH1 invasion into human RBCs is a shift towards greater usage of the sialic acid-independent PkDBPα ligand, and away from the PkDBPβ and PkDBPγ ligands that are unable to bind human Neu5Ac.
To test the hypothesis that Pk YH1 has increased usage of the PkDBPα sialic acid-independent ligand, we assessed the invasion of Pk YH1 and Pk H into human RBCs in the presence of increasing concentration of the chemokine melanoma growth stimulatory activity (MGSA; Fig. 4c). MGSA binds specifically to DARC and inhibits its interaction with PkDBPα39. We observed a greater than 10-fold increase in the half-maximal inhibitory concentration (IC50) for Pk YH1 (6 nM) compared with Pk H (0.5 nM) (Fig. 4c). The requirement for more MGSA for the inhibition of Pk YH1 is consistent with an increased number of PkDBPα–DARC interactions, resulting from PkDBPα amplification and loss of PkDBPγ.
Discussion
In this study, we used an ex vivo RBC culture system coupled with lentiviral transgene expression of CMAH to specifically and functionally study Neu5Gc, as a determinant of the tropism displayed by the zoonotic macaque parasite P. knowlesi. We provide evidence that P. knowlesi is restricted in its invasion of human RBCs due to the absence of surface Neu5Gc. Evolutionarily, this modified sialic acid was once present in ancestral humans Myr ago. It is plausible that selective pressure due to a Plasmodium parasite utilizing this invasion pathway could have resulted in loss of a functional CMAH in ancestral humans, given the considerable evidence that malaria has shaped the human genome4,40. However, fossil records of ancestral macaques in Africa do not place them in East Africa at the site of CMAH inactivation, but rather in North Africa, from where they migrated to Eurasia 5.5 Myr ago41. Thus, an ancestral zoonotic P. knowlesi species is less likely to have been the selective force for CMAH loss ∼3.2 Myr ago. We speculate that a Laverania parasite, such as P. reichenowi, that infects chimpanzees (which have Neu5Gc-positive RBCs) is a more likely candidate for the selective pressure leading to CMAH loss. Regardless of the origins, we suggest that CMAH gene inactivation in humans is currently a restriction factor limiting the emergence of P. knowlesi infections in the human population.
However, the human-adapted Pk YH1 strain used in this study suggests that P. knowlesi can adapt to overcome this restriction through duplication of the sialic acid-independent PkDBPα invasion ligand. Our hypothesis that Pk YH1 has greater and more efficient usage of the PkDBPα–DARC invasion pathway is supported by the increased IC50 for Pk YH1 in the MGSA invasion inhibition assay. We also observed deletion of the Neu5Gc-dependent PkDBPγ, although whether this was a necessary change concomitant with the duplication of PkDBPα, remains to be determined.
The space-filling model for the display of invasion ligands at the apical end of the invading parasite42,43 can be one way to rationalize how increased expression of PkDBPα and loss of PkDBPγ may enhance invasion of human RBCs. With limited space at the apex of the parasite for the deployment of invasion ligands before invasion, increased expression of PkDBPα and absence of PkDBPγ would result in greater release of PkDBPα to the apex of the parasite, resulting in an increased number of productive PkDBPα–DARC interactions. PkDBPβ and PkDBPγ are expressed together with PkDBPα in wild-type P. knowlesi44,45, making plausible the increased apical presentation of PkDBPα in the absence of PkDBPγ in Pk YH1.
The duplication of PkDBPα in Pk YH1 is reminiscent of the duplication of the closely related P. vivax DBP invasion ligand observed in P. vivax isolates in Madagascar46. This P. vivax DBP duplication, likely a recent event, may have been selected for the efficient invasion of Malagasy individuals, where RBCs that are either homozygous for DARC-negative alleles or heterozygous for DARC-negative/DARC-positive alleles are common.
The rise in reported malaria cases due to P. knowlesi infection in Southeast Asia19,20 raises concern about the possible occurrence of human-to-human transmission. There is already evidence suggesting the clustering of P. knowlesi infections in households, where individuals had not been in proximity to macaques47,48. In the 1950s, the use of P. knowlesi as a treatment for tertiary neurosyphilis had to be stopped because of increased pathogenicity of the parasite, with continuous passaging in humans49,50. This suggests that increased parasite adaptation to humans may be associated with severe disease. We suggest that genetic changes in the DBP invasion ligands might be a sentinel indicator of increased adaptation to humans.
Methods
Investigation of CMAH gene loss in mammalian evolution
We examined hominid and mammalian genomes with genome alignments and gene loss analysis. The mammalian gene loss events were analysed with the program CAFE (Computational Analysis of gene Family Evolution51), a statistical tool for computing the gene family size evolution that is implemented in Ensembl 72. The gene loss event in hominins was further verified with independent genomic analysis; the sequence reads of the Neanderthal52 and Denisovan genomes24 were aligned to chimpanzee genome panTro3 at the CMAH gene locus with the University of California, Santa Cruz (UCSC) genome browser53.
The ferret54 and New World monkey25 CMAH gene loss events are published. Dog CMAH does not appear to be functional in certain breeds of dogs55,56.
Ex vivo culture of RBCs
Bone-marrow-derived CD34+ haematopoietic stem cells (2–3 × 105; Lonza) were cultured in media with 5% solvent/detergent virus-inactivated human plasma (Octaplas, Octapharma) as described previously57 with the following modifications. On day 6 or 7 of culture, cells were transduced with lentivirus harbouring either the chimpanzee (Pan troglodytes) CMAH complementary DNA (cDNA) sequence in pLVX-Puro (Clontech) or the empty vector, pLVX-Puro. The CMAH cDNA sequence with a C-terminal tobacco etch virus (TEV) cleavage site and FLAG-c-myc tags was codon-optimized for human expression and synthesized (GeneArt). CMAH cDNA was amplified using primers listed in Supplementary Table 3 and cloned into pLVX-Puro. Transduction with lentiviral vectors and subsequent selection on 2 μg ml−1 puromycin dihydrochloride (Sigma-Aldrich) were done as described23, but puromycin was maintained until day 12 or 13 when cells were co-cultured on a murine MS-5 stromal cell layer at a cell density of 3–6 × 105 cells per ml until day 18 as described58. On day 18, cells were replated on a fresh MS-5 stromal cell layer and then collected on day 20 or 21 at which time 70–90% of cells were enucleated. Cells were stored at 4°C in incomplete RPMI; (consisting of RPMI-1640 (Sigma-Aldrich) supplemented with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 50 mg l−1 hypoxanthine), until use in downstream experiments.
Neu5Gc or RBC receptor expression by flow cytometry
RBCs and cRBCs were washed three times in PBS containing 0.5% Neu5Gc-free blocking agent (Siamab, formerly Sialix) and pelleted at 500 g for 4 min in a 96-well plate at 5 × 105 cells per well. Cells were resuspended in either 50 μl of blocking buffer or 50 μl of the appropriate antibody solution. The following antibodies were used at the indicated dilutions: anti-Neu5Gc (1:5,000, Siamab), phycoerythrin(PE)-conjugated anti-DARC (1:10, Miltenyi Biotec), anti-CD71-PE (1:10, Miltenyi Biotec), fluorescein isothiocyanate-conjugated anti-glycophorin A (GPA); 1:50, Clone 2B7, STEMCELL Technologies) and anti-glycophorin C (GPC)-fluorescein isothiocyanate (1:500, BRIC 10, Santa Cruz). Cells were incubated for 1 h at room temperature and washed three times in blocking buffer. Unstained cells and cells stained with DARC, CD71, GPA and GPC antibodies were resuspended in 100 μl PBS for analysis on the MACSQuant flow cytometer (Miltenyi Biotec). Neu5Gc-stained cells were incubated in anti-chicken IgY-Alexa Fluor 488 secondary antibody (Life Technologies) at 1:1,000 for 30 min at room temperature. Control samples were similarly incubated in anti-mouse IgG2a-PE (1:10, Miltenyi Biotec), anti-chicken IgY-Alexa Fluor 488 antibody or anti-mouse IgG-Alexa Fluor 488 (1:1,000, Life Technologies). Cells were washed twice and subjected to flow cytometric analysis. The data were analysed in FlowJo 4 version 10.0.7 (Tree Star).
Measurement of sialic acid by HPLC
For measurement of cell surface sialic acid, RBC and cRBC ghosts were prepared for sialic acid release as described59, with some modifications. In brief, 1 × 107 cells were washed once in incomplete RPMI and twice in PBS pH 7.4. Cells were stored at −80 °C until RBC ghost preparation, where thawed RBCs were resuspended in cold 5 mM sodium phosphate pH 8.0 containing protease inhibitor cocktail. RBC ghosts were pelleted at 21,130 g for 10 min, washed three times in PBS containing protease inhibitor cocktail and stored at −80 °C until sialic acid preparation. Sialic acid release from RBC ghosts was achieved by mild acid hydrolysis as described60, with some modifications. In brief, RBC ghosts were resuspended in 75 μl 2 M acetic acid and heated at 80 °C for 3 h. For formation of fluorescent derivatives, 75 μl of derivatization solution was added to sialic acid samples and heated at 50 °C in the dark for 2.5-3 h. The derivatization solution was made up of 7 mM 1,2-diamino-4,5-methylenedioxybenzene dihydrochloride, acetic acid (1.7 M for experimental samples or 3.7 M for sialic acid standards), 0.75 M β-mercaptoethanol and 18 mM sodium hydrosulfite. Samples were run on an HPLC with a C18 column at a flow rate of 0.9 ml min−1, using a mobile phase of acetonitrile–methanol–water (9:7:84 v/v). Sialic acid elution was monitored by absorbance at 373 nm. Data were acquired on Chemstation Rev A. 10.01 (Agilent Technologies). Eluted sialic acids were compared with a sialic acid reference panel, and Neu5Ac and Neu5Gc solutions of known amounts for quantitation of sialic acids.
For measurement of sialic acid in invasion assays, neuraminidase treatment of RBCs was performed in 40 μl total volume. Supernatants were collected and sialic acid was quantified as described previously61. In brief, cell supernatants were treated with 20 μl of a freshly prepared solution of 0.2 M sodium periodate in 48% phosphoric acid for 20 min at room temperature. To terminate the reaction, 100 μl of freshly prepared 10% sodium arsenite in 0.1 N sulphuric acid was added slowly, then vortexed until clear and incubated 5 min at room temperature. Finally, 600 μl of thiobarbituric acid (6 mg ml−1) was added, and the reaction was incubated at 100 °C for 15 min, then chilled on ice. Samples were analysed on the HPLC as described above, but elution was performed using a running buffer consisting of 115 mM sodium perchlorate, 30% methanol and 1% phosphoric acid, and absorbance was monitored at 549 nm.
Plasmodium parasite cultures
P. falciparum Dd2attB/BSD-GFP (BSD: blasticidin S-deaminase) and 3D7attB/BSD-GFP62 used in cRBC/RBC invasion assays were kind gifts from David Fidock (Columbia University). Pk H SSUDattB/BSD-GFP (SSUD: small subunit of the D-type ribosomal RNA gene) was generated in our lab as follows. The targeting vector, pDEF-DSSU-hDHFR63, was integrated into Pk H (provided by A.W. Thomas; Biomedical Primate Research Center, Rijswijk) to form Pk H SSUD. The P. knowlesi d-ssu gene was then amplified using Pk H SSUD genomic DNA and primers listed in Supplementary Table 3, and then cloned into pCG6-attB62. The resulting vector, pSSUD-CG6-attB, was integrated into Pk H SSUD to form Pk H SSUDattB. In the final step, pBSD-GFP-INT-attP62 was integrated into Pk H SSUDattB. Pk YH1 was generated in our laboratory from the parental Pk H strain by growth for 3 months in human O+ RBCs supplemented with 16% reticulocytes purified from haemochromatosis patient blood21. The percentage of reticulocytes was decreased to 2–4% for an additional 2 months, after which parasites proliferated stably in the absence of reticulocyte-enriched blood. P. falciparum Dd2 and W2mef strains used in invasion assays with neuraminidase-treated RBCs were obtained from the Malaria Research and Reference Reagent Resource.
P. falciparum cultures and the human-adapted Pk YH1 line were maintained in human RBCs (Research Blood Components), while P. knowlesi H SSUDattB/BSD-GFP was grown in rhesus macaque RBCs (New England Primate Research Center). Parasites were grown at 2% haematocrit in complete RPMI (RPMI supplemented with 25 mM HEPES, 50 mg l−1 hypoxanthine, 2.57 mM sodium bicarbonate and 0.5% AlbuMAX II (Life Technologies)).
Enzyme treatments
For RBC binding assays, enzyme treatments were performed as described42. In brief, RBCs or cRBCs were washed three times in incomplete RPMI and treated with 1 mg ml−1 trypsin (Sigma-Aldrich) and/or 1 mg ml−1 chymotrypsin (Worthington) and/or 66.7 mU ml−1 Vibrio cholerae neuraminidase (Sigma-Aldrich) at 37 °C with gentle mixing for 1 h. RBCs were then washed twice in incomplete RPMI and once in PBS pH 7.4.
For neuraminidase treatment for invasion assays, human RBCs were washed in wash media (RPMI supplemented with 25 mM HEPES-KOH, pH 7.4) and treated with decreasing amounts of neuraminidase from Clostridium perfringens (100, 50, 25, 12.5, 6.25 and 3.125 U; New England Biolabs) at 37 °C with gentle mixing for 30 min. The neuraminidase-treated RBCs were then washed extensively in complete RPMI before use. The neuraminidase-treated RBCs were subjected to a second neuraminidase treatment to remove and quantify the remaining sialic acid. This second treatment was performed at 250 U neuraminidase at 50% haematocrit for 30 min at 37 °C in PBS pH 6.0.
Invasion assays
For invasion assays using RBCs and cRBCs, cells were washed in complete RPMI and counted on a haemocytometer. Cells (1.5 × 106) were added to respective wells of a half-area 96-well plate at 0.5% haematocrit in triplicate. Synchronised schizont-stage parasites were purified by magnetic separation on a MACS cell separator (Miltenyi Biotec) and added to RBCs at ∼2% parasitaemia in a final volume of 30 μl. Cells (2.5 × 105; 5 μl) were taken from each well for cytospin preparations to assess initial parasitaemia by light microscopy. Assay plates were kept in a modulator incubator chamber gassed with 1% O2, 5% CO2 and balance of N2, and reinvasion and growth were monitored over time by thin smear of mock wells. At trophozoite/schizont stage, final parasitaemia was estimated by light microscopy from counts of at least 1,000 cells on May-Grünwald- and Giemsa-stained cytospins. The parasitized erythrocyte multiplication rate (final parasitaemia (after single round of invasion)/initial parasitaemia) was determined for each strain in each cell type. The Student’s unpaired t-test (two-tailed) was used to assess significant differences in invasion.
For invasion assays with neuraminidase-treated RBCs, parasite invasion into neuraminidase-treated RBCs was measured by SYBR Green I (Life Technologies) staining and flow cytometry as described previously64, with several modifications. Synchronised schizont-stage parasites at ∼10% parasitaemia that had been treated with 250 U C. perfringens neuraminidase at ring stage to reduce reinvasion, were washed, and then mixed with untreated and neuraminidase-treated target cells. Invasion assays were seeded in triplicate 200 μl samples at ∼1% parasitaemia at 0.5% haematocrit in a 96-well plate. To quantify inoculum parasitaemia, a subset of samples was fixed immediately with formaldehyde and stored at 4 °C. The remaining samples were incubated for 48 h, then fixed for 1 h. Inoculum and post-invasion samples were then stained with SYBR Green I and analysed by flow cytometry. Expansion rates were expressed as parasitized erythrocyte multiplication rate.
For MGSA invasion inhibition assays, human RBCs were plated in a 96-well plate at 4% haematocrit in 2 × complete RPMI (complete RPMI with twice the amount of AlbuMAX II and sodium bicarbonate). MGSA (PeproTech) of varying concentrations made up in incomplete RPMI were added to respective wells. Schizont-stage parasites purified by magnetic separation, as described above, were added to each well at ∼1% parasitaemia. After allowing for a single round of invasion, parasitaemia was measured the following day by either SYBR green I or Vybrant DyeCycle Violet (Life Technologies) staining and flow cytometry. The parasitaemia of MGSA-treated samples was normalized to a mock-treated control for each strain. The data were analysed in GraphPad Prism 5.
Protein expression plasmids
Region II of PkDBPα (gene ID: PkH_062300) and Region II of PkDBPβ (gene ID: PkH_000490) were amplified from P. knowlesi H genomic DNA, while Region II of PkDBPγ (gene ID: PkH_134580) was amplified from a P. knowlesi fosmid obtained from a P. knowlesi fosmid library65, using primers listed in Supplementary Table 3. PCR products were cloned into the mammalian expression vector, pSDP32, a version of pTT66 with a modified multiple cloning site and with the rat COMP domain downstream of the multiple cloning site. For the GST-COMP-expressing plasmid, GST was amplified from pET-42a(+) (Supplementary Table 3) and cloned into pSDP32. For the COMP-negative control plasmid, NheI- and NotI-digested pSDP32 was modified with T4 DNA polymerase (New England Biolabs) to generate blunt ends and then ligated. Plasmid constructs were verified by sequencing.
HEK293-6E culture and protein expression and purification
HEK293-6E cells, a kind gift from Yves Durocher (National Research Council, Canada), were maintained in serum-free Freestyle 293 expression media (Life Technologies) supplemented with 0.1% Pluronic F-68 (Life Technologies) and 25 μg ml−1 Geneticin (Life Technologies). Cells were grown in suspension in Erlenmeyer flasks at 110 r.p.m., at 37 °C in 8% CO2. Recombinant protein expression by transient transfection was achieved as described66 with some modifications. In brief, cells were transfected at a cell density of 1 × 106 cells per ml using 1 μg of plasmid DNA per 1 × 106 cells and a DNA:polyethylenimine (PEI) ratio of 1:3 (w/w). DNA and the transfection reagent, linear PEI (Polysciences, Inc.), were diluted separately in complete Freestyle 293 media. The DNA was vortexed for 10 min, and DNA and PEI were combined, vortexed for 30 s, allowed to incubate at room temperature for 12 min and finally added to the cell culture. Between 24 and 48 h post transfection, a solution of Tryptone-N1 (Organotechnie SAS) was added to the transfected cell culture at 0.5% (w/v). Cultures were processed on day 5 or 6 post transfection by pelleting and collecting the culture supernatant containing the expressed protein. Protein expression was confirmed by western blot using Strep-Tactin-HRP (IBA). Culture supernatant containing recombinant protein was used in protein overlay assays. For flow cytometry-based-binding assays, PkDBPβ, PkDBPγ and COMP protein were affinity-purified from culture supernatant on a StrepTactin sepharose column (StrepTrap HP; GE Healthcare) using an ÄKTAFPLC Fast Protein Liquid Chromatography system (GE Healthcare). Bound protein was eluted by 2.5 mM desthiobiotin (Sigma-Aldrich). Buffer exchange into PBS pH 7.4 was achieved using centrifugal filter units. Protein concentration was determined from absorbance measurements at 280 nm and the protein-specific extinction coefficient.
Western blot analyses
HEK293-6E culture supernatant, RBCs or RBC ghosts were electrophoresed on 4–15% gradient gels and transferred to nitrocellulose membrane. Membranes were blocked in PBS–0.05% Tween (PBST)–10% milk (for probing with Strep-Tactin-HRP), tris-buffered saline–0.1% Tween–0.5% Neu5Gc-free blocking agent (for probing with anti-Neu5Gc) or PBST–5% milk (for probing with anti-DARC; Sigma-Aldrich). Membranes were probed with the respective antibodies at the indicated dilutions—anti-DARC: 1:1,000 in blocking buffer, anti-Neu5Gc: 1:10,000 in blocking buffer and Strep-Tactin-HRP: 1:2,500 in PBST–20 μg ml−1 avidin (PBST-A) after pre-incubation in PBST-A for 10 min. The DARC western blot was probed with anti-rabbit-HRP (GE Healthcare) at 1:10,000, while the Neu5Gc western blot was probed with anti-chicken/turkey-HRP (Life Technologies) at 1:5,000 in blocking buffer. All membranes were developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and imaged by autoradiography.
Protein overlay assay
Untreated and enzyme-treated rhesus macaque, and human RBC ghosts and cRBC ghosts were prepared by cell lysis, and multiple washes in cold 5 mM sodium phosphate pH 8.0. Protein content of macaque and human RBC ghosts was quantified by Bradford assay. RBC/cRBC ghosts were stored at −80 °C until use. Before use, cRBC ghosts were treated with DNase I (New England Biolabs). Samples of enzyme-treated or untreated macaque and human RBC ghosts or cRBC ghosts, not treated with reducing agent or heat-denatured, were electrophoresed; 10 μg of macaque or human RBC ghosts (corresponding to ∼3 × 107 cells) were loaded per lane, while cRBC ghosts made from ∼1.7 × 106 cells were loaded per lane. Electrophoresed protein was transferred to nitrocellulose membrane. Membranes were blocked in PBST-10% milk for 2 h at room temperature and then washed in PBST. HEK293-6E culture supernatant containing recombinant protein with Tween 20 added to a final concentration of 0.1% was added to membranes and incubated overnight at 4 °C with gentle rotation. Membranes were washed, blocked in PBST-A for 10 min, incubated in Strep-Tactin-HRP at a dilution of 1:2,500 in PBST-A for 1 h at room temperature and finally developed with Pico substrate. Bound protein was detected by autoradiography.
Flow cytometry-based binding assay
RBCs or cRBCs were washed three times in PBS–0.5% Neu5Gc-free blocking buffer. Cells were pelleted in a 96-well plate at 3 × 105 RBCs per well and resuspended in either 50 μl blocking buffer or PkDBPβ-COMP, PkDBPγ-COMP or COMP protein diluted in blocking buffer at the appropriate concentrations. Samples were incubated for 1 h at room temperature, washed three times in blocking buffer and incubated in a monoclonal anti-strepII-tag antibody (IBA) at 2 μg ml−1 for 1 h at room temperature. Samples were washed three times in blocking buffer then incubated in anti-mouse IgG-Alexa Fluor 488 antibody for 30 min at room temperature. Samples were washed twice, then resuspended in 100 μl PBS for analysis on the MACSQuant flow cytometer. Data were analysed by FlowJo 4 version 10.0.7.
Whole-genome sequencing
Genomic DNA was extracted from schizont-stage Pk H and Pk YH1 parasites using the QIAamp DNA blood kit (Qiagen). Genomic DNA was used to construct Illumina sequencing libraries for both P. knowlesi strains, with a library insert size of 200 bp. Each library was sequenced to a depth of at least 50-fold coverage using 101-bp paired-end reads on an Illumina HiSeq 2000 sequencer. Reads were aligned to the reference genome (P. knowlesi H, PlasmoDB release 11) by applying the bwa mem algorithm from the Burrows–Wheeler Aligner67 to the left and right read files using default settings. Alignments were converted from SAM to BAM format, sorted and indexed using SAMtools68. Coverage was calculated using the BEDtools genomeCoverageBed function69.
Whole-genome sequencing resulted in 23,386,393 reads for Pk H and 21,200,464 reads for Pk YH1. 95.05% of Pk H reads and 88.77% of Pk YH1 reads mapped to the P. knowlesi reference genome. The average coverage depth for Pk H was 89.43% and for Pk YH1 74.59%. These results represent a genome coverage of 99.86% for Pk H and 99.87 for Pk YH1.
Additional information
Accession codes: Whole-genome sequence data for P. knowlesi H and P. knowlesi YH1 have been deposited in the NCBI Sequence Read Archive (SRA) under accession codes SRX1175910 and SRX1553878, respectively.
How to cite this article: Dankwa, S. et al. Ancient human sialic acid variant restricts an emerging zoonotic malaria parasite. Nat. Commun. 7:11187 doi: 10.1038/ncomms11187 (2016).
Accession codes
References
Deng, L. et al. Host adaptation of a bacterial toxin from the human pathogen Salmonella Typhi. Cell 159, 1290–1299 (2014).
Byres, E. et al. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature 456, 648–652 (2008).
Iyer, J., Gruner, A. C., Renia, L., Snounou, G. & Preiser, P. R. Invasion of host cells by malaria parasites: a tale of two protein families. Mol. Microbiol. 65, 231–249 (2007).
Varki, A. & Gagneux, P. Human-specific evolution of sialic acid targets: explaining the malignant malaria mystery? Proc. Natl Acad. Sci. USA 106, 14739–14740 (2009).
Suzuki, Y. et al. Sialic acid species as a determinant of the host range of influenza A viruses. J. Virol. 74, 11825–11831 (2000).
Lehmann, F., Tiralongo, E. & Tiralongo, J. Sialic acid-specific lectins: occurrence, specificity and function. Cell. Mol. Life Sci. 63, 1331–1354 (2006).
Angata, T. & Varki, A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem. Rev. 102, 439–469 (2002).
Chou, H. H. et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl Acad. Sci. USA 95, 11751–11756 (1998).
Irie, A., Koyama, S., Kozutsumi, Y., Kawasaki, T. & Suzuki, A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 273, 15866–15871 (1998).
Hayakawa, T., Satta, Y., Gagneux, P., Varki, A. & Takahata, N. Alu-mediated inactivation of the human CMP- N-acetylneuraminic acid hydroxylase gene. Proc. Natl Acad. Sci. USA 98, 11399–11404 (2001).
Hayakawa, T., Aki, I., Varki, A., Satta, Y. & Takahata, N. Fixation of the human-specific CMP-N-acetylneuraminic acid hydroxylase pseudogene and implications of haplotype diversity for human evolution. Genetics 172, 1139–1146 (2006).
Wood, B. Colloquium paper: reconstructing human evolution: achievements, challenges and opportunities. Proc. Natl Acad. Sci. USA 107, (suppl 2): 8902–8909 (2010).
Chou, H. H. et al. Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc. Natl Acad. Sci. USA 99, 11736–11741 (2002).
Martin, M. J., Rayner, J. C., Gagneux, P., Barnwell, J. W. & Varki, A. Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc. Natl Acad. Sci. USA 102, 12819–12824 (2005).
Wanaguru, M., Liu, W., Hahn, B. H., Rayner, J. C. & Wright, G. J. RH5-Basigin interaction plays a major role in the host tropism of Plasmodium falciparum. Proc. Natl Acad. Sci. USA 110, 20735–20740 (2013).
Wanaguru, M., Crosnier, C., Johnson, S., Rayner, J. C. & Wright, G. J. Biochemical analysis of the Plasmodium falciparum erythrocyte-binding antigen-175 (EBA175)-glycophorin-A interaction: implications for vaccine design. J. Biol. Chem. 288, 32106–32117 (2013).
Singh, B. & Daneshvar, C. Human infections and detection of Plasmodium knowlesi. Clin. Microbiol. Rev. 26, 165–184 (2013).
Cox-Singh, J. et al. Anti-inflammatory cytokines predominate in acute human Plasmodium knowlesi infections. PLoS ONE 6, e20541 (2011).
William, T. et al. Changing epidemiology of malaria in Sabah, Malaysia: increasing incidence of Plasmodium knowlesi. Malar. J. 13, 390 (2014).
Barber, B. E. et al. A prospective comparative study of knowlesi, falciparum, and vivax malaria in Sabah, Malaysia: high proportion with severe disease from Plasmodium knowlesi and Plasmodium vivax but no mortality with early referral and artesunate therapy. Clin. Infect. Dis. 56, 383–397 (2013).
Lim, C. et al. Expansion of host cellular niche can drive adaptation of a zoonotic malaria parasite to humans. Nat. Commun. 4, 1638 (2013).
Bei, A. K., Brugnara, C. & Duraisingh, M. T. In vitro genetic analysis of an erythrocyte determinant of malaria infection. J. Infect. Dis. 202, 1722–1727 (2010).
Egan, E. S. et al. Malaria. A forward genetic screen identifies erythrocyte CD55 as essential for Plasmodium falciparum invasion. Science 348, 711–714 (2015).
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
Springer, S. A., Diaz, S. L. & Gagneux, P. Parallel evolution of a self-signal: humans and new world monkeys independently lost the cell surface sugar Neu5Gc. Immunogenetics 66, 671–674 (2014).
Herrera, S., Perlaza, B. L., Bonelo, A. & Arevalo-Herrera, M. Aotus monkeys: their great value for anti-malaria vaccines and drug testing. Int. J. Parasitol. 32, 1625–1635 (2002).
Miller, L. H., Mason, S. J., Dvorak, J. A., McGinniss, M. H. & Rothman, I. K. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science 189, 561–563 (1975).
Muchmore, E. A., Diaz, S. & Varki, A. A structural difference between the cell surfaces of humans and the great apes. Am. J. Phys. Anthropol. 107, 187–198 (1998).
Miller, L. H. et al. Evidence for differences in erythrocyte surface receptors for the malarial parasites, Plasmodium falciparum and Plasmodium knowlesi. J. Exp. Med. 146, 277–281 (1977).
Adams, J. H. et al. A family of erythrocyte binding proteins of malaria parasites. Proc. Natl Acad. Sci. USA 89, 7085–7089 (1992).
Bushell, K. M., Sollner, C., Schuster-Boeckler, B., Bateman, A. & Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res. 18, 622–630 (2008).
Haynes, J. D. et al. Receptor-like specificity of a Plasmodium knowlesi malarial protein that binds to Duffy antigen ligands on erythrocytes. J. Exp. Med. 167, 1873–1881 (1988).
Chitnis, C. E. & Miller, L. H. Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506 (1994).
Ranjan, A. & Chitnis, C. E. Mapping regions containing binding residues within functional domains of Plasmodium vivax and Plasmodium knowlesi erythrocyte-binding proteins. Proc. Natl Acad. Sci. USA 96, 14067–14072 (1999).
Orlandi, P. A., Klotz, F. W. & Haynes, J. D. A malaria invasion receptor, the 175-kilodalton erythrocyte binding antigen of Plasmodium falciparum recognizes the terminal Neu5Ac(alpha 2-3)Gal- sequences of glycophorin A. J. Cell. Biol. 116, 901–909 (1992).
Mayer, D. C., Mu, J. B., Feng, X., Su, X. Z. & Miller, L. H. Polymorphism in a Plasmodium falciparum erythrocyte-binding ligand changes its receptor specificity. J. Exp. Med. 196, 1523–1528 (2002).
Maier, A. G., Baum, J., Smith, B., Conway, D. J. & Cowman, A. F. Polymorphisms in erythrocyte binding antigens 140 and 181 affect function and binding but not receptor specificity in Plasmodium falciparum. Infect. Immun. 77, 1689–1699 (2009).
Meyer, E. V. et al. The reticulocyte binding-like proteins of P. knowlesi locate to the micronemes of merozoites and define two new members of this invasion ligand family. Mol. Biochem. Parasitol. 165, 111–121 (2009).
Horuk, R. et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science 261, 1182–1184 (1993).
Kwiatkowski, D. P. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 77, 171–192 (2005).
Fooden, J. Systematic review of the Barbary macaque (Linnaeus 1758). Fieldiana Zool. 113, 1–58 (2007).
Duraisingh, M. T. et al. Phenotypic variation of Plasmodium falciparum merozoite proteins directs receptor targeting for invasion of human erythrocytes. EMBO J. 22, 1047–1057 (2003).
Duraisingh, M. T., DeSimone, T., Jennings, C., Refour, P. & Wu, C. Erythrocyte invasion by Plasmodium falciparum: multiple ligand-receptor interactions and phenotypic switching. Subcell. Biochem. 47, 46–57 (2008).
Adams, J. H. et al. The Duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell 63, 141–153 (1990).
Singh, A. P. et al. Targeted deletion of Plasmodium knowlesi Duffy binding protein confirms its role in junction formation during invasion. Mol. Microbiol. 55, 1925–1934 (2005).
Menard, D. et al. Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl. Trop. Dis. 7, e2489 (2013).
Barber, B. E. et al. Epidemiology of Plasmodium knowlesi malaria in north-east Sabah, Malaysia: family clusters and wide age distribution. Malar. J. 11, 401 (2012).
Fornace, K. M. et al. Asymptomatic and Submicroscopic Carriage of Plasmodium knowlesi Malaria in Household and Community Members of Clinical Cases in Sabah, Malaysia. J. Infect. Dis. 213, 784–787 (2015).
Ciuca, M., Badenski, T. P., Badenski, G. & Terintianu PIM, A. Contribution à l’étude de la virulence du Pl. knowlesi chez l’homme. Caractères de la maladie et biologie du parasitie. Arch. Roum. Pathol. Exp. Microbiol. 10, 5–28 (1937).
Ciuca, M. & Romine, A. R. P. Contribution experimentale a l’etude de l’immunite dans le paludisme L’Academie, Bucharest, Romania (1955).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Ng, P. S. et al. Ferrets exclusively synthesize Neu5Ac and express naturally humanized influenza A virus receptors. Nat. Commun. 5, 5750 (2014).
Yasue, S. et al. Difference in form of sialic acid in red blood cell glycolipids of different breeds of dogs. J. Biochem. 83, 1101–1107 (1978).
Hashimoto, Y., Yamakawa, T. & Tanabe, Y. Further studies on the red cell glycolipids of various breeds of dogs. A possible assumption about the origin of Japanese dogs. J. Biochem. 96, 1777–1782 (1984).
Giarratana, M. C. et al. Proof of principle for transfusion of in vitro-generated red blood cells. Blood 118, 5071–5079 (2011).
Giarratana, M. C. et al. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat. Biotechnol. 23, 69–74 (2005).
Hayton, K. et al. Various PfRH5 polymorphisms can support Plasmodium falciparum invasion into the erythrocytes of owl monkeys and rats. Mol. Biochem. Parasitol. 187, 103–110 (2013).
Hara, S. et al. Determination of mono-O-acetylated N-acetylneuraminic acids in human and rat sera by fluorometric high-performance liquid chromatography. Anal. Biochem. 179, 162–166 (1989).
Powell, L. D. & Hart, G. W. Quantitation of picomole levels of N-acetyl- and N-glycolylneuraminic acids by a HPLC-adaptation of the thiobarbituric acid assay. Anal. Biochem. 157, 179–185 (1986).
Nkrumah, L. J. et al. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat. Methods 3, 615–621 (2006).
Wel, A., Kocken, C. H., Pronk, T. C., Franke-Fayard, B. & Thomas, A. W. New selectable markers and single crossover integration for the highly versatile Plasmodium knowlesi transfection system. Mol. Biochem. Parasitol. 134, 97–104 (2004).
Bei, A. K. et al. A flow cytometry-based assay for measuring invasion of red blood cells by Plasmodium falciparum. Am. J. Hematol. 85, 234–237 (2010).
Pain, A. et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455, 799–803 (2008).
Durocher, Y., Perret, S. & Kamen, A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, E9 (2002).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34 (2014).
Acknowledgements
This work was supported by a National Institutes of Health grant AI091787 to M.T.D., a Center for Disease Control grant (R36-CK000119-01) and an Epidemiology of Infectious Disease and Biodefense Training Grant (2-T32-AI007535-12) to A.K.B., a National Institute of General Medical Sciences (NIGMS) Biotechnology Training Grant 5-T32-GM08334 to J.R.A. and National Institutes of Health Director’s New Innovator Award 1DP2OD007124, NIGMS Center for Integrative Synthetic Biology Grant P50-GM098792, Ferry Award and Massachusetts Institute of Technology Startup funds to J.C.N. S.D.P. was supported by a National Institutes of Health Pediatric Scientist Development Program grant 5-K12-HD000850. Y.M. is a recipient of the Louis-Berlinguet Program Postdoctoral Research Scholarship in Genomics-Fonds de recherche du Quebec, Nature et technologies. Whole-genome sequencing was performed using the Illumina platform at the Broad Institute.
Author information
Authors and Affiliations
Contributions
M.T.D. conceived the experiments, and M.T.D. and J.C.N. supervised the experiments. S.D. performed most parasitological and red blood cell genetic and binding experiments, as well as data analyses. A.K.B. performed some parasitological and red blood cell genetic experiments. S.D.P. carried out some genetic and protein expression experiments. C.L. and J.M.G. performed whole-genome sequencing studies and C.L. performed MGSA inhibition assays. Y.M. generated the culture-adapted P. knowlesi YH1 line and M.K. generated the P. knowlesi H SSUDattB/BSD-GFP strain. J.R.A. carried out HPLC analyses and some invasion assays. R.H.Y.J. performed the bioinformatic analysis. M.T.D. and S.D. wrote the manuscript with contributions from co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8 and Supplementary Tables 1-3 (PDF 3052 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dankwa, S., Lim, C., Bei, A. et al. Ancient human sialic acid variant restricts an emerging zoonotic malaria parasite. Nat Commun 7, 11187 (2016). https://doi.org/10.1038/ncomms11187
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms11187
This article is cited by
-
Genetic polymorphism of the thrombospondin-related apical merozoite protein (TRAMP) of Plasmodium knowlesi in Malaysia
Parasitology Research (2023)
-
Plasmodium knowlesi: the game changer for malaria eradication
Malaria Journal (2022)
-
Metabolic fate of dietary sialic acid and its influence on gut and oral bacteria
Systems Microbiology and Biomanufacturing (2022)
-
Clinical recovery of Macaca fascicularis infected with Plasmodium knowlesi
Malaria Journal (2021)
-
Analysis of erythrocyte signalling pathways during Plasmodium falciparum infection identifies targets for host-directed antimalarial intervention
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
