
Abstract
The end-Triassic mass extinction overlapped with the eruption of the Central Atlantic Magmatic Province (CAMP), and release of CO2 and other volcanic volatiles has been implicated in the extinction. However, the timing of marine biotic recovery versus CAMP eruptions remains uncertain. Here we use Hg concentrations and isotopes as indicators of CAMP volcanism in continental shelf sediments, the primary archive of faunal data. In Triassic–Jurassic strata, Muller Canyon, Nevada, Hg levels rise in the extinction interval, peak before the appearance of the first Jurassic ammonite, remain above background in association with a depauperate fauna, and fall to pre-extinction levels during significant pelagic and benthic faunal recovery. Hg isotopes display no significant mass independent fractionation within the extinction and depauperate intervals, consistent with a volcanic origin for the Hg. The Hg and palaeontological evidence from the same archive indicate that significant biotic recovery did not begin until CAMP eruptions ceased.
Similar content being viewed by others
Introduction
Various proxies reveal a dramatic rise in atmospheric pCO2 across the Triassic–Jurassic (T-J) boundary1,2 in association with the end-Triassic mass extinction ∼201.5 million years ago3,4 (for a summary of key biotic and geochemical events surrounding the T-J interval, see Supplementary Fig. 1). The extinction severely affected clades common to the modern ocean (the so-called Modern Fauna5) and reef-building scleractinian corals more significantly than any other extinction event and resulted in the lowest standing diversity in Phanerozoic time6. The robust coral reef ecosystem in the latest Triassic collapsed and reef/carbonate dwelling organisms were preferentially affected7, such that ocean acidification has been implicated in the extinction8,9,10. The extinction overlaps with the eruption of the Central Atlantic Magmatic Province (CAMP), a large igneous province emplaced as a result of the opening of the Atlantic during the rifting of Pangea11 (Fig. 1a) that was likely a significant source of CO2. High-resolution dating of CAMP basalts and sills from terrestrial successions in North America indicates that CAMP volcanism was geologically rapid and occurred in three or four pulses over ∼700 thousand years3 (Fig. 1b). Upper estimates of CO2 release to the atmosphere are ∼13 Gt CO2 per year (3 × 1017 mol CO2 released in discrete 1,000 year pulses)2, rivaling modern input rates (∼40 Gt CO2 per year). Although actual rates may have been somewhat lower, the T-J interval provides an opportunity to investigate the global consequences of a major carbon cycle perturbation.

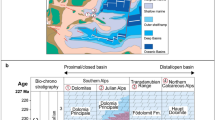
(a) Palaeogeographic map of the T-J interval showing the hypothesized extent of CAMP with the study site and other key T-J localities marked (with a red dot and black dots, respectively), modified from Greene et al.8 (b) Comparison of ammonoid biostratigraphy and U-Pb dates from interbedded volcanic ashes in Peru4,13,25 and U-Pb ages for CAMP magmatism largely from N. America and Morocco3 (top grey bar). The initiation of CAMP volcanism is coincident with the end-Triassic extinction (typically indicated by the last occurrence of C. crickmayi) and continues through the earliest Jurassic.
Dated volcanic ashes that are interbedded with biostratigraphically important ammonites from deeper water strata in northern Peru4,12,13 demonstrate that the extinction was essentially coincident with the first major eruption of CAMP basalts at 201.51±0.15 Ma (Fig. 1b). The last pulse of CAMP basalts in North America occurred ca 200.9±0.064 Ma (ref. 3) and postdates the appearance of the earliest Jurassic ammonite Psiloceras spelae ca 201.39±0.14 Ma (ref. 4), hinting that biotic recovery may have been underway while CAMP was still erupting3. However, the palaeoenvironmental settings of the currently well-dated successions—terrestrial for CAMP, deeper marine for Peru—makes quantitative evaluation of the marine biotic recovery problematic, as most of the fauna typically used to assess ecological recovery occur in relatively shallow marine settings.
Mercury chemostratigraphy has recently been used to investigate the role of large igneous provinces in driving climatic events and biotic crises during mass extinctions14,15,16,17,18,19. Large volcanic events may cause substantial short-term increases to the atmospheric Hg burden, and the long residence time of Hg0(g) in the atmosphere (∼1 year) makes it possible for large volcanic Hg fluxes to have global impacts20. Once released to the atmosphere, Hg may undergo complex cycling (for example, transformation, deposition, re-emission) before long-term burial, which occurs dominantly in marine sediments on million-year timescales21. Mercury primarily enters marine systems through direct atmospheric deposition or through riverine inputs, which transfer terrestrial Hg (derived from crustal and also atmospheric sources) to coastal sediments21. Terrestrial Hg of atmospheric origin may potentially be transported to coastal sediments bound to organics or clay particles22. In the ocean, organic carbon is a major scavenging pathway and sink for Hg, and marine sediments often preserve the strong association between Hg and organic matter23.
An exemplary T-J shallow shelf succession deposited between the Sierran arc and the North American continent in eastern Panthalassa allows detailed analysis of local palaeoecological recovery that can be tightly correlated to worldwide biotic trends. The section is well-exposed in Muller Canyon of the Gabbs Valley Range, Nevada24 (Fig. 1), but has lacked detailed correlation with CAMP volcanism. Here, we investigate Hg concentrations and Hg isotopes in continental shelf sediments from Muller Canyon as tracers of volcanism and combine Hg measurements with palaeoecological data from this succession to assess the timing of marine biotic recovery relative to the eruption of the large igneous province. We also measure the amount of total organic carbon (TOC) to determine if variation in Hg concentrations are driven by lithological changes controlling organic matter content and provide organic carbon isotopic measurements in order to directly tie trends in Hg to globally recognized carbon isotope excursions across the T-J interval. In Muller Canyon, Nevada, mercury anomalies (as indicated by both Hg and Hg/TOC levels) appear in the extinction interval and persist in association with a depauperate (low diversity) early Jurassic fauna. They are not observed in pre-extinction strata, and Hg levels fall before significant pelagic and benthic faunal recovery begins. Furthermore, no significant mass independent fractionation (MIF) of Hg isotopes is present within the extinction and depauperate intervals, consistent with a volcanic origin for the Hg anomalies, versus small MIF in adjacent strata. Based on Hg concentrations and Hg isotope chemostratigraphy, we conclude that eruptions from CAMP ceased before significant biotic recovery was underway.
Results
Organic carbon isotopes
Our δ13Corg measurements (Fig. 2 and Table 1) are consistent with previous studies25,26,27,28 (Supplementary Fig. 2) and reveal a negative δ13Corg excursion coincident with the extinction interval (termed the initial carbon isotope excursion or I-CIE (ref. 29)), followed by a positive excursion. We display our carbon isotope data in Fig. 2 for direct comparison to our new Hg chemostratigraphy and provide the fault-corrected data from Ward et al.27 in Supplementary Fig. 2 for comparison. Although minor differences exist between the various δ13Corg profiles (see Guex et al.26), all δ13Corg profiles broadly record a negative δ13Corg excursion in association with the extinction interval, followed by a positive excursion in association with P. spelae.

Plots show (a) δ13Corg, (b) Hg, (c) Hg/TOC and (d) Δ199Hg for Muller Canyon, Nevada, along with lithology and key ammonites. Marker beds N3, N9 and N11 are after Guex et al.25 In panel b, error bars on Hg are 2 s.d. In panel c, error bars represent the combined error on Hg (2 s.d.) and TOC (1 s.d.) concentration measurements. In panel d, errors on individual Δ199Hg measurements are as reported in Supplementary Table 1 and represent either 2 s.e.m. of sample replicates or 2 s.d. of the JT Baker standard, whichever is higher (see the Methods for further explanation). The vertical grey bar in panel d is centred on Δ199Hg=0.00‰ and extends from −0.05‰ to +0.05‰. Samples for which the Δ199Hg values and associated error bars fall within the shaded grey region can be considered to have Δ199Hg values within experimental error of zero, and thus no measureable mass independent fractionation (MIF).
Palaeoecology
The uppermost Triassic strata of the Mount Hyatt Member of the Gabbs Formation represent a prolific, bivalve-dominated Late Triassic carbonate ramp assemblage30 (Figs 2 and 3). The shift to siliciclastic-dominated sedimentation in the overlying Muller Canyon Member signifies a collapse of the vibrant carbonate system in association with the mass extinction24, which is marked by the last occurrence of the Triassic ammonite Choristoceras crickmayi and is coincident with the onset of the negative δ13Corg excursion25,26,27,28. The first occurrence of Psiloceras spelae (official marker of the basal Jurassic) above the last occurrence of C. crickmayi brackets a 7-m-thick extinction interval within the Muller Canyon Member, which records benthic fossils limited to rare microscopic gastropods and sponge spicules24. Depauperate benthic macrofauna in the upper 10 m of the Muller Canyon Member comprise minor bioturbation31 and rare bivalves (for example, Agerchlamys27 and Modiolus32, also found in the immediate extinction aftermath in England33 and Austria34), which occur in tandem with a minor increase in ammonoid diversity25 (cosmopolitan genera ubiquitous across Panthalassa12,25,35; Figs 2 and 3). The first phase of ecological recovery following the depauperate interval is indicated by a substantial increase in ammonoid diversity12,25 in the pelagic realm and the appearance of a pervasive demosponge-dominated ecosystem in the benthic realm36,37 (recovery state 1 in Fig. 3). The early recovery phase represents an ecological state shift (sensu Hull et al.38), which is also recorded in Peru37 and Austria39. Trophic complexity matching or surpassing pre-extinction conditions40 and a return to carbonate-dominated benthic biota36, including the first North American Jurassic corals41, did not occur until ∼2 million years after the extinction (recovery state 2 in Fig. 3).

Panels compare (a) Hg chemostratigraphy (this study), (b) ammonite species diversity from Guex et al.25; (c) benthic palaeoecology and microfacies modified from Ritterbush et al.36; and (d) ecosystem state for Muller Canyon, Nevada, in association with lithology and key dates. Nevada ash date is from Schoene et al.13 and approximate ages of key ammonites are from Wotzlaw et al.,4 and Schoene et al.13 (see Fig. 1b). These comparisons show that significant biotic recovery follows the Hg anomalies and provide evidence that biotic recovery began after the cessation of CAMP magmatism.
Mercury and organic carbon concentrations
Sediments of the Gabbs and Sunrise Formations have low organic carbon contents (<0.5% TOC) that do not correlate with Hg concentrations (Fig. 2 and Supplementary Fig. 3). First-order structure in Hg and Hg/TOC reveals a rapid rise and peak within the extinction interval, with a few smaller peaks in the overlying depauperate interval (Figs 2 and 3). Significant biotic recovery occurs as Hg concentrations return to pre-extinction levels36,37 (see recovery state 1 in Fig. 3). The decoupling of Hg and organic carbon within these strata suggests that variations in Hg content primarily result from changes in Hg loading to coastal waters rather than changes in the size of the organic carbon sink. It also implies that sources of Hg and organic carbon to these strata were, at least in part, different. Strata with the highest Hg/TOC in the Muller Canyon section have ∼600 p.p.b. of Hg per %Corg, far more Hg per unit Corg than found in many contaminated sediments today (for example, average p.p.b. of Hg per %TOC in contaminated sediments measured in San Francisco Bay is ∼200 (ref. 42)).
Mercury isotopes
We use Hg isotopic analyses to further explore the sources of Hg in the Muller Canyon succession. Mercury isotopes undergo large mass-dependent and mass-independent fractionations in nature and can be used to trace Hg sources and cycling (see Blum et al.43 and references therein). We report mass dependent fractionation (MDF) using δ202Hg values (see Methods for Hg isotope nomenclature). δ202Hg values of the Muller Canyon strata are primarily negative and range from −1.78 to 0.12‰ (see Table 1, Supplementary Fig 2 and Supplementary Table 1 for details). Because MDF can result from many physical, chemical and biological reactions, we do not interpret MDF signatures here. However, negative δ202Hg values are typical of both marine sediments and volcanic emissions. To investigate whether or not the Muller Canyon Hg anomalies were derived from CAMP volcanism, we focus on Hg-MIF signatures of these strata. Because Hg-MIF is primarily associated with photochemistry in natural samples and occurs during far fewer pathways than MDF (see Blum et al.43 for a summary of MIF and MDF pathways), it is unlikely that Hg-MIF signatures are altered by post-depositional processes.
Modern Earth surface environments, including marine sediments, often carry measurable amounts of odd isotope Hg-MIF (for example, see Yin et al.44), which is thought to result from the aqueous photochemical cycling of Hg (ref. 45). In comparison, direct isotopic measurements of volcanic Hg emissions display no measureable MIF46, and measurements of igneous rocks, ores and most hydrothermal precipitates also provide no evidence for Hg-MIF in solid Earth materials, except in contexts where sedimentary/surface Hg may be leached or recycled by geologic processes43.
Odd-isotope MIF (as indicated by Δ199Hg values in Fig. 2; see the Methods for Hg isotope nomenclature) is present in two of the lowermost samples analysed (both part of prolific Triassic carbonate ramp) and re-appears at the top of the depauperate interval where Hg is at background levels (Fig. 2 and Supplementary Fig. 2). Out of the seven samples analysed for isotopes below and above the extinction/depauperate intervals, five display measureable MIF, with Δ199Hg values ranging from 0.11 to −0.30‰ (Fig. 2, Table 1 and Supplementary Fig. 2). The odd-isotope MIF signatures present in the lower and upper-most intervals are consistent with those observed in modern coastal and oceanic sediments (for example, see Yin et al.44 and references therein) and suggest some similarity between modern and ancient Hg cycling. In contrast, no significant Hg-MIF is recorded throughout the extinction and depauperate intervals, when increases in Hg and Hg/TOC are observed (Fig. 2). The total range of measured Δ199Hg values within these intervals is −0.05 to 0.07‰ (Table 1), and thus most values fall within experimental error of zero. We interpret the paucity of MIF as evidence for a significant influx of volcanic Hg from CAMP during these periods.
Discussion
If the mercury anomalies within the extinction and depauperate intervals are from volcanism as isotopes suggest, then significant biotic recovery did not begin until eruptions associated with CAMP ceased. The preservation of a volcanic Hg isotopic fingerprint in sediments distant in location from the CAMP eruptions (Fig. 1) implies that Hg emissions from CAMP dominated the Hg pools at Earth’s surface and altered the Hg cycle such that an insufficient amount of Hg released by volcanism underwent the aqueous photochemical transformations necessary to impart significant MIF. We have also considered the possibility that the input of volcanic Hg could be derived from arc volcanism proximal to the Muller Canyon succession, as indicated by the presence of an ash in the section. However, Hg concentrations in the strata closest to the ash layer (9.5 m) are relatively low (18.5–22.5 p.p.b.) when compared with other strata within the extinction and depauperate intervals (Fig. 2). It thus seems unlikely that Hg signals from arc volcanism are resolvable within the resolution of our sampling or could explain the elevated Hg levels on the timescale represented by the Muller Canyon succession. This interpretation is also supported by evidence that aquatic sediments do not reliably archive short-term Hg releases associated with sporadic large explosive eruptions21.
In summary, we show for the first time that Hg concentrations and isotopic compositions record the timing of massive volcanism in a marine section that spans the T-J interval, strengthening the case for CAMP’s potential role in the mass extinction. Robust biotic recovery, which initially occurred in the form of bio-siliceous deposition36, did not begin until Hg concentrations returned to pre-extinction levels and Hg-MIF re-appeared, indicating the cessation of major CAMP volcanism. This inferred timing of recovery contrasts with previous suggestions3 that the recovery was underway as CAMP was still erupting.
Ocean acidification via CO2 input from CAMP has been suggested as a potential kill mechanism for the end-Triassic extinction9. An initial lowering of carbonate saturation may have contributed to the extinction of carbonate biota, but the Nevada section reveals that the carbonate-dominated ecosystems did not recover for nearly 2 million years after the extinction and ∼1 million years after the cessation of CAMP volcanism. Ocean acidification models typically predict much shorter recovery time scales (typically 10–100 ka)10, suggesting that ocean acidification alone cannot explain the prolonged disruption of metazoan carbonate-dominated ecosystems in the aftermath of the end-Triassic mass extinction. Other factors (for example, the initial shift in ecological state dominated by siliceous sponges, among others) may have played a role in the pattern of carbonate recovery37. Whatever the case, our new data from Nevada suggest that the long process of biotic recovery began in earnest once CAMP volcanism drew to a close.
Methods
Carbon measurements
Samples were collected from the field following the stratigraphy of Guex et al.25,26 (see Supplementary Fig. 4 for an image of the collection site). Samples were inspected and those with veins and weathered surfaces were removed. Samples were crushed in a jaw crusher and then pulverized in an agate ball mill at the University of Southern California. An aliquot of powder from each sample (∼0.5 g) was dissolved in 40 ml of 1 M hydrochloric acid and heated to 70 °C for 4 h to remove all carbonate mineral phases. This method is similar to that described by Ward et al.27. Samples were washed with deionized water three times and dried at 50 °C.
Weight percent organic carbon was determined on decarbonated powder using a Picarro cavity ring down spectrometer (G2131-i) coupled via a Picarro Liason (A0301) to a Costech Elemental Combustion System (EA 4010). This determination of organic carbon content was converted to a value of % TOC taking into account the amount of carbonate loss during acid treatment. Errors were calculated by replicate analyses of samples and standards. The 1 s.d. uncertainty was assigned as 10% of the reported value, which takes into account uncertainties associated with decarbonation. Standards included both internal CaCO3 standards and the USGS-40 reference material (L-glutamic acid).
The isotopic composition of organic carbon was also determined using the Picarro cavity ring down spectrometer and is reported in delta notation (δ13Corg) relative to the Vienna Pee Dee Belemnite standard. The uncertainty on the δ13Corg values was assessed from replicate runs of standards (including NBS-18 calcite, USGS-40 and internal carbonate standards) and samples. Replicate analyses were run on 33% of the samples. Standard deviation on replicate analyses was on average <0.1‰. Uncertainties and blanks associated with this methodology are further discussed in Subhas et al.47
Mercury concentration measurements
Samples were inspected, crushed and pulverized at the University of Southern California, as described above. Total Hg was measured using a Hydra IIc Direct Mercury Analyzer (Teledyne Leeman Labs) at the University of Toronto. Within the Hydra IIc, samples were combusted in two stages under an oxygen flow of 350 ml min−1. First they were heated to 300 °C for 30–60 s, and then decomposed at 800 °C for 300–500 s. After combustion, the evolved gases were carried through a heated catalyst tube to remove possible interferences (for example, halogen compounds, sulfur oxides, nitrous oxides) and Hg was captured on a gold amalgamation trap while combustion gases were removed from the detection cell. The gold trap was then heated for 30 s at 600 °C to release Hg. Hg was carried to the detection cell where absorbance from a mercury lamp was measured at 253.7 nm.
Calibration was performed using a fresh, gravimetrically prepared NIST 3133 Hg standard in a 0.25% L-cysteine solution. Blank absorbance was <2% of typical sample signals and always <4%. Sample boats were periodically re-combusted to check that all available Hg had been released during the initial analysis. To determine measurement precision, the NIST 3133 L-cysteine solution was periodically combusted and analysed alongside samples. The measured concentrations of the NIST 3133 standard are within 5% of nominal values.
Samples measured more than once are reported as the mean of duplicate measurements (Supplementary Table 1). Reproducibility of sample concentrations was better than 10%. To check measurement accuracy, powders of NIST SRM 1944 (New York/New Jersey Waterway Sediment) and NIST SRM 1646a (Estuarine Sediment) were repeatedly combusted over the period of sample analysis. The average value for NIST 1944 was 3,496±334 p.p.b. (2 s.d., n=2), which is within the certified value of 3,400±500 p.p.b., and the average value for NIST 1646a was 27.7±2.8 p.p.b. (2 s.d., n=9). Although NIST 1646a is not certified for Hg, we used it as in-house external standard because our batch had a similar Hg content to the samples. The measured concentrations of NIST 1646a are consistent with the long-term values obtained on this standard in our laboratory. Based on the reproducibility of samples and external standards, errors on Hg concentration measurements are estimated to be 10% (2 s.d.).
Mercury isotope nomenclature
Mercury isotope compositions are reported using nomenclature suggested by Blum and Bergquist48. Isotopic compositions are reported using δ-notation relative to the NIST SRM 3133 standard according to equation (1):

where x is the mass number of each Hg isotope from 199Hg to 204Hg. We use δ202Hg to report MDF. MIF is reported as ΔxHg, which is defined using equation (2):

where x is the mass number of each Hg isotope (199, 200, 201 and 204) and β is the scaling constant used to estimate theoretical MDF based on kinetic mass fractionation49. β is 0.2520, 0.5024, 0.7520 and 1.493 for 199Hg, 200Hg, 201Hg and 204Hg, respectively.
Mercury isotope measurements
Before isotope analysis, Hg was extracted and purified from samples by combustion separation using the furnace module of the Hydra IIc with the gold trap removed. The decomposition procedure was the same as described for the Hg concentration measurements. To trap Hg, the gas outflow containing elemental Hg was sparged directly into a freshly prepared solution of ∼10% trace metal grade H2SO4 (v/v) and ∼1% KMnO4 (w/w), where the Hg0 gas was oxidized to Hg(II). After the combustion of each sample, 50 μl of Milli-Q water was loaded into a nickel boat and combusted according to the same procedures as samples to ensure removal of any residual Hg in the furnace. During this step, the line linking the gas outflow to the sparger was also heated with a heat gun to ensure full recovery of Hg.
Aqueous solutions of NIST 3133, powders of NIST 1646a and blanks were combusted and trapped alongside samples as procedural standards and blanks. Procedural blanks were <0.02 ng g−1, which is <1–2% of the sample Hg. Recovery of Hg from samples and process standards was checked by neutralizing an aliquot of each solution with NH2OH-HCl immediately after trapping and measuring its concentration using a Tekran 2600 cold vapour atomic fluorescence spectrometer. The recoveries of samples were 99.3±10.6%, (2 s.d., n=35) and of procedural standards were 99.6±4.8% (2 s.d., n=8). The ∼10% variation in sample recoveries reflects both the uncertainty in concentration method and sample heterogeneity.
Isotopic analysis was conducted using a cold vapour multi-collector inductively coupled plasma mass spectrometer (Neptune Plus, Thermo-Finnigan) at the University of Toronto. Sample solutions were first neutralized with NH2OH-HCl in order to reduce KMnO4 and then diluted to 1–2 ng g−1 using a pre-neutralized 1% KMnO4 solution (the same matrix as samples). Hg was introduced into the plasma as Hg(0) using online SnCl2 reduction and Hg(0) vapour separation. To correct for instrumental mass bias, we used an internal Tl standard (NIST 997; introduced as a desolvated aerosol) and strict standard-sample bracketing with the NIST 3133 Hg standard. In addition, an in-house secondary aqueous Hg standard (J.T.Baker Chemicals) was measured at least seven times in each analytical session to determine the external reproducibility of the method. Both the NIST 3133 bracketing standards and the J.T.Baker Hg standards were prepared in the same matrix solution as samples and procedural standards. Signal concentrations and intensities of all standards and samples were matched within 10%. Isobaric interference from 204Pb was monitored using 206Pb, but was always negligible (correction never altered the calculated δ204Hg). On-peak blank corrections were made on all Hg and Pb masses and the Hg intensities of the blank measurements were monitored to ensure negligible carry-over and build up of Hg.
The average value of the JT Baker Hg standard over all analytical sessions was −0.60±0.09‰ for δ202Hg and 0.02±0.03‰ for Δ199Hg (2 s.d., n=31; Supplementary Table 2), which is consistent with previous values on this standard50,51. All samples and procedural standards were measured at least twice. Sample isotope values are reported as the mean of duplicate or triplicate measurements (Supplementary Table 1). Isotopic values obtained on the NIST 3133 procedural standards are within error of our bracketing standard with an average δ202Hg of 0.02±0.03‰ and Δ199Hg of −0.01±0.01‰ (2 s.e.m., n=4; Supplementary Table 2). Isotopic values for the NIST 1646a procedural standards are consistent with previously measured values for this standard in our laboratory with an average δ202Hg of −0.90±0.05‰ and Δ199Hg of 0.08±0.01‰ (2 s.e.m., n=4; Supplementary Table 2)50. We chose NIST 1646a as a procedural isotope standard for this study because it has Hg concentrations and slight MIF similar to our samples. Over the same time period in the lab, NIST 1944 was also measured by the above combustion procedure and had an average δ202Hg of −0.43±0.03‰ and Δ199Hg of 0.01±0.02‰ (2 s.e.m., n=5), which is within error of published values50. Sample errors are reported as 2 s.e.m. of sample replicates unless that value is smaller than 2 s.d. of the in-house JT Baker Hg standard. If the 2 s.e.m. of sample replicates is smaller than the 2 s.d. of the JT Baker standard, then the 2 s.d. of the JT Baker standard is used as the error for the sample.
Additional information
How to cite this article: Thibodeau, A. M. et al. Mercury anomalies and the timing of biotic recovery following the end-Triassic mass extinction. Nat. Commun. 7:11147 doi: 10.1038/ncomms11147 (2016).
References
Steinthorsdottir, M., Jeram, A. J. & McElwain, J. C. Extremely elevated CO2 concentrations at the Triassic/Jurassic boundary. Palaeogeogr. Palaeoclimatol. Palaeoecol. 308, 418–432 (2011).
Schaller, M. F., Wright, J. D., Kent, D. V. & Olsen, P. E. Rapid emplacement of the Central Atlantic Magmatic Province as a net sink for CO2 . Earth Planet. Sci. Lett. 323-324, 27–39 (2012).
Blackburn, T. J. et al. Zircon U-Pb geochronology links the end-Triassic extinction with the Central Atlantic Magmatic Province. Science 340, 941–945 (2013).
Wotzlaw, J. F. et al. Towards accurate numerical calibration of the Late Triassic: High-precision U-Pb geochronology constraints on the duration of the Rhaetian. Geology 42, 571–574 (2014).
Sepkoski, J. J. Jr. A factor analytic description of the Phanerozoic marine fossil record. Paleobiology 7, 36–53 (1981).
Alroy, J. The shifting balance of diversity among major marine animal groups. Science 329, 1191–1194 (2010).
Kiessling, W., Aberhan, M., Brenneis, B. & Wagner, P. J. Extinction trajectories of benthic organisms across the Triassic–Jurassic boundary. Palaeogeogr. Palaeoclimatol. Palaeoecol. 244, 201–222 (2007).
Greene, S. E. et al. Recognising ocean acidification in deep time: An evaluation of the evidence for acidification across the Triassic-Jurassic boundary. Earth Sci. Rev. 113, 72–93 (2012).
Hautmann, M., Benton, M. J. & Tomašových, A. Catastrophic ocean acidification at the Triassic-Jurassic boundary. N. Jb. Geol. Palaont. Abh 249, 119–127 (2008).
Berner, R. A. & Beerling, D. J. Volcanic degassing necessary to produce a CaCO3 undersaturated ocean at the Triassic–Jurassic boundary. Palaeogeogr. Palaeoclimatol. Palaeoecol. 244, 368–373 (2007).
Marzoli, A. et al. Extensive 200-Million-Year-Old Continental Flood Basalts of the Central Atlantic Magmatic Province. Science 284, 616–618 (1999).
Guex, J. et al. Geochronological constraints on post-extinction recovery of the ammonoids and carbon cycle perturbations during the Early Jurassic. Palaeogeogr. Palaeoclimatol. Palaeoecol. 346-347, 1–11 (2012).
Schoene, B., Guex, J., Bartolini, A., Schaltegger, U. & Blackburn, T. J. Correlating the end-Triassic mass extinction and flood basalt volcanism at the 100 ka level. Geology 38, 387–390 (2010).
Sial, A. N. et al. High-resolution Hg chemostratigraphy: A contribution to the distinction of chemical fingerprints of the Deccan volcanism and Cretaceous–Paleogene Boundary impact event. Palaeogeogr. Palaeoclimatol., Palaeoecol. 414, 98–115 (2014).
Sial, A. N. et al. Mercury as a proxy for volcanic activity during extreme environmental turnover: The Cretaceous–Paleogene transition. Palaeogeogr. Palaeoclimatol. Palaeoecol. 387, 153–164 (2013).
Percival, L. M. E. et al. Globally enhanced mercury deposition during the end-Pliensbachian extinction and Toarcian OAE: A link to the Karoo–Ferrar Large Igneous Province. Earth Planet. Sci. Lett. 428, 267–280 (2015).
Sanei, H., Grasby, S. E. & Beauchamp, B. Latest Permian mercury anomalies. Geology 40, 63–66 (2011).
Font, E. et al. Mercury anomaly, Deccan volcanism, and the end-Cretaceous mass extinction. Geology 44, 171–174 (2016).
Grasby, S. E., Beauchamp, B., Bond, D. P. G., Wignall, P. B. & Sanei, H. Mercury anomalies associated with three extinction events (Capitanian Crisis, Latest Permian Extinction and the Smithian/Spathian Extinction) in NW Pangea. Geol. Mag. 153, 285–297 (2016).
Pyle, D. M. & Mather, T. A. The importance of volcanic emissions for the global atmospheric mercury cycle. Atmos. Environ. 37, 5115–5124 (2003).
Fitzgerald, W. F. & Lamborg, C. H. in Treatise on Geochemistry 2nd edn (eds Holland H., Turekian K. 91–129Elsevier (2014).
Kongchum, M., Hudnall, W. H. & Delaune, R. D. Relationship between sediment clay minerals and total mercury. J. Environ. Sci. Health A 45, 534–539 (2011).
Fitzgerald, W. F., Lamborg, C. H. & Hammerschmidt, C. R. Marine biogeochemical cycling of mercury. Chem. Rev. 107, 641–662 (2007).
Lucas, S. G., Taylor, D. G., Guex, J., Tanner, L. H. & Krainer, K. The proposed global stratotype section and point for the base of the Jurassic System in the New York Canyon area, Nevada, USA. N. M. Mus. Nat. Hist. Sci. Bull. 40, 139–168 (2007).
Guex, J., Bartolini, A., Atudorei, V. & Taylor, D. High-resolution ammonite and carbon isotope stratigraphy across the Triassic–Jurassic boundary at New York Canyon (Nevada). Earth Planet. Sci. Lett. 225, 29–41 (2004).
Guex, J. et al. Comment on: ‘The organic carbon isotopic and paleontological record across the Triassic-Jurassic boundary at the candidate GSSP section at Ferguson Hill, Muller Canyon, Nevada, USA’ by Ward et al. (2007). Palaeogeogr. Palaeoclimatol. Palaeoecol. 273, 200–204 (2009).
Ward, P. D. et al. The organic carbon isotopic and paleontological record across the Triassic–Jurassic boundary at the candidate GSSP section at Ferguson Hill, Muller Canyon, Nevada, USA. Palaeogeogr. Palaeoclimatol. Palaeoecol. 244, 281–289 (2007).
Ward, P., McRoberts, C. & Williford, K. Reply to comment on: ‘The organic carbon isotopic and paleontological record across the Triassic–Jurassic boundary at the candidate GSSP section at Ferguson Hill, Muller Canyon, Nevada, USA’ by Ward et al. (2007). Palaeogeogr. Palaeoclimatol. Palaeoecol. 273, 205–206 (2009).
Hesselbo, S. P., Robinson, S. A. & Surlyk, F. Sea-level change and facies development across potential Triassic-Jurassic boundary horizons, SW Britain. J. Geol. Soc. 161, 365–379 (2004).
Laws, R. A. Late Triassic depositional environments and molluscan associations from west-central Nevada. Palaeogeogr. Palaeoclimatol. Palaeoecol. 37, 131–148 (1982).
Twitchett, R. J. & Barras, C. G.. in The Application of Ichnology to Paleoenvironmental and Stratigraphic Analysis ed. Mcllroy D. Special Publication 228, 397–418Geological Society (2004).
Hallam, A. & Wignall, P. B. Facies changes across the Triassic–Jurassic boundary in Nevada, USA. J. Geol. Soc. 157, 49–54 (2000).
Mander, L. & Twitchett, R. J. Quality of the Triassic-Jurassic bivalve fossil record in Northwest Europe. Palaeontology 51, 1213–1223 (2008).
Hillebrandt, von, A., Krystyn, L. & Kuerschner, W. M. A candidate GSSP for the base of the Jurassic in the Northern Calcareous Alps (Kuhjoch section, Karwendel Mountains, Tyrol, Austria). Int. Subcommission Jurassic Stratigr. Newslett. 34, 2–20 (2007).
Hillebrandt, von, A. & Krystyn, L. On the oldest Jurassic ammonites of Europe (Northern Calcareous Alps, Austria) and their global significance. N. Jb. Geol. Palaont. Abh 253, 163–195 (2009).
Ritterbush, K. A., Bottjer, D. J., Corsetti, F. A. & Rosas, S. New evidence on the role of siliceous sponges in ecology and sedimentary facies development in eastern Panthalassa following the Triassic-Jurassic mass extinction. Palaios 29, 652–668 (2014).
Ritterbush, K. A., Rosas, S., Corsetti, F. A., Bottjer, D. J. & West, A. J. Andean sponges reveal long-term benthic ecosystem shifts following the end-Triassic mass extinction. Palaeogeogr. Palaeoclimatol. Palaeoecol. 420, 193–209 (2015).
Hull, P. M., Norris, R. D., Bralower, T. J. & Schueth, J. D. A role for chance in marine recovery from the end-Cretaceous extinction. Nat. Geosci. 4, 856–860 (2011).
Delecat, S., Arp, G. & Reitner, J. in Advances in Stromatolite Geobiology 131, 355–390Springer Berlin Heidelberg, (2011).
Ritterbush, K. A., Ibarra, Y. & Tackett, L. S. Post-extinction ecosystem engineering on the first carbonate ramp of the Early Jurassic (Sinemurian) in NE Panthalassa (New York Canyon, Nevada, USA). Palaios (in the press).
Hodges, M. S. & Stanley, G. D. Jr. North American coral recovery after the End-Triassic mass extinction, New York Canyon, Nevada, USA. GSA Today 25, 4–7 (2015).
Conaway, C. H., Squire, S., Mason, R. P. & Flegal, A. R. Mercury speciation in the San Francisco Bay estuary. Mar. Chem. 80, 199–225 (2003).
Blum, J. D., Sherman, L. S. & Johnson, M. W. Mercury isotopes in earth and environmental sciences. Annu. Rev. Earth Planet. Sci. 42, 249–269 (2014).
Yin, R. et al. Identifying the sources and processes of mercury in subtropical estuarine and ocean sediments using Hg isotopic composition. Environ. Sci. Technol. 49, 1347–1355 (2015).
Bergquist, B. A. & Blum, J. D. Mass-dependent and -independent fractionation of Hg isotopes by photoreduction in aquatic systems. Science 318, 417–420 (2007).
Zambardi, T., Sonke, J. E., Toutain, J. P., Sortino, F. & Shinohara, H. Mercury emissions and stable isotopic compositions at Vulcano Island (Italy). Earth Planet. Sci. Lett. 277, 236–243 (2009).
Subhas, A. V. et al. A novel determination of calcite dissolution kinetics in seawater. Geochim. Cosmochim. Acta 170, 51–68 (2015).
Blum, J. D. & Bergquist, B. A. Reporting of variations in the natural isotopic composition of mercury. Anal. Bioanal. Chem. 388, 353–359 (2007).
Young, E. D., Galy, A. & Nagahara, H. Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance. Geochimica et Cosmochimica Acta 66, 1095–1104 (2002).
Zheng, W., Xie, Z. & Bergquist, B. A. Mercury stable isotopes in ornithogenic deposits as tracers of historical cycling of mercury in Ross Sea, Antarctica. Environ. Sci. Technol. 49, 7623–7632 (2015).
Chandan, P., Ghosh, S. & Bergquist, B. A. Mercury isotope fractionation during aqueous photoreduction of monomethylmercury in the presence of dissolved organic matter. Environ. Sci. Technol. 49, 259–267 (2015).
Acknowledgements
This research was supported by a NSF Earth Life Transitions grant (EAR-1338329) to F.C., the Roger E. Deane Postdoctoral Fellowship to A.T. and Geological Society of America, Society for Sedimentary Geology and American Philosophical Society grants to K.R. Mercury concentration and isotope measurements were funded by the NSERC-Discovery and the Canadian Institute for Advanced Research (CIFAR). We thank Nick Rollins and Ellie Hara for their help with the carbon measurements.
Author information
Authors and Affiliations
Contributions
D.J.B., F.A.C. and A.J.W. conceived the study and designed it with A.M.T. and B.A.B. K.R., J.A.Y. and Y.I. collected the samples. A.M.T. and J.A.Y. collected, analysed and interpreted the mercury and carbon data, respectively, with B.A.B., A.J.W., W.M.B. and F.A.C. K.R. provided the stratigraphic and paleoenvironmental context as well as the paleoecological data. K.R., Y.I. and F.A.C. were responsible for paleoenvironmental interpretation. F.A.C., J.A.Y., A.J.W. and A.M.T. conceived and drafted the figures. F.A.C. and A.M.T. wrote the paper with contributions from all other co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-4, Supplementary Tables 1-2 and Supplementary References. (PDF 520 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Thibodeau, A., Ritterbush, K., Yager, J. et al. Mercury anomalies and the timing of biotic recovery following the end-Triassic mass extinction. Nat Commun 7, 11147 (2016). https://doi.org/10.1038/ncomms11147
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms11147
This article is cited by
-
Early Jurassic large igneous province carbon emissions constrained by sedimentary mercury
Nature Geoscience (2024)
-
Nuclear volume effects in kinetic isotope fractionation: A case study of mercury oxidation by chlorine species
Acta Geochimica (2024)
-
Mercury evidence for combustion of organic-rich sediments during the end-Triassic crisis
Nature Communications (2022)
-
Intensified continental chemical weathering and carbon-cycle perturbations linked to volcanism during the Triassic–Jurassic transition
Nature Communications (2022)
-
Global diversity dynamics in the fossil record are regionally heterogeneous
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
