
Abstract
Chronic lymphocytic leukemia (CLL) is a common lymphoid malignancy with strong heritability. To further understand the genetic susceptibility for CLL and identify common loci associated with risk, we conducted a meta-analysis of four genome-wide association studies (GWAS) composed of 3,100 cases and 7,667 controls with follow-up replication in 1,958 cases and 5,530 controls. Here we report three new loci at 3p24.1 (rs9880772, EOMES, P=2.55 × 10−11), 6p25.2 (rs73718779, SERPINB6, P=1.97 × 10−8) and 3q28 (rs9815073, LPP, P=3.62 × 10−8), as well as a new independent SNP at the known 2q13 locus (rs9308731, BCL2L11, P=1.00 × 10−11) in the combined analysis. We find suggestive evidence (P<5 × 10−7) for two additional new loci at 4q24 (rs10028805, BANK1, P=7.19 × 10−8) and 3p22.2 (rs1274963, CSRNP1, P=2.12 × 10−7). Pathway analyses of new and known CLL loci consistently show a strong role for apoptosis, providing further evidence for the importance of this biological pathway in CLL susceptibility.
Similar content being viewed by others

Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia among adults in western countries1. Although advances in treatment options have been made, CLL remains an incurable malignancy. Genome-wide association studies (GWAS) have identified multiple susceptibility loci for CLL2,3,4,5,6,7 with at least three loci having more than one independent signal5,8. However, these discovered loci only account for about a third of the estimated heritability attributed to common variants5. In a combined analysis of four GWAS and follow-up replication, including 3,888 cases and 12,539 controls of European ancestry, we recently discovered 11 independent single-nucleotide polymorphisms (SNPs) in nine novel loci associated with CLL risk5. To discover additional loci associated with susceptibility to CLL, we more than doubled our replication sample size in the present study, slightly increasing our statistical power, and investigated the association with 14 other promising SNPs identified from our GWAS meta-analysis.
Here, we identify four new independent SNPs in three novel loci as well as two promising new loci associated with the risk of CLL. Pathway analyses with these new loci as well as the previously identified loci suggest a strong role for the apoptosis in susceptibility to CLL, further enhancing our understanding.
Results
Discovery meta-analysis
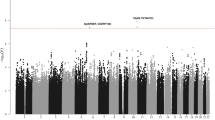
We conducted a meta-analysis of four genome-wide association studies4,5,9 comprising 3,100 unrelated cases and 7,667 controls of European ancestry (see ‘Methods’ section, Supplementary Tables 1–3). As these studies used different commercial SNP microarrays, we imputed the ∼8.5 million common SNPs present in the 1000 Genomes Phase 1 integrated data (version 3)10 for each study using IMPUTE2 (ref. 11; Supplementary Table 2) and tested for associations with CLL risk assuming a log-additive genetic model. After quality control exclusions, ∼8.5 million SNPs with minor allele frequency >1% were meta-analysed in the discovery stage using a fixed effects model.
A quantile–quantile plot of the meta-analysis results in the discovery stage showed an enrichment of small P values from the fixed-effects model compared with the null distribution, which persisted even after removal of the known loci (Supplementary Fig. 1). There was little evidence for inflation due to population stratification (lambda=1.028). Under a log-additive genetic model, a total of 16 unique loci (defined as separated by at least 1 Mb) reached genome-wide significance (P<5 × 10−8; Supplementary Fig. 2), all of which had been previously reported2,3,5,8. For each previously reported locus, we identified the SNP with the strongest P value within 1 Mb of the published index SNP. Of the 29 published loci, 21 were at least suggestively associated with CLL under a log-additive model in our discovery meta-analysis with P<5 × 10−7 (Supplementary Table 4). As the original reported SNPs at two loci (4q26 and 6q25.2) failed to show nominal significance (P<0.05) in our study, we meta-analysed our results with the published results for known loci from two other GWAS6,7. In this larger meta-analysis, 25 of the published loci were at least suggestively associated with CLL risk (P<5 × 10−7) based on a fixed-effects model; however, both rs6858698 at 4q26 and rs11631963 at 15q25.2 showed attenuated odds ratios and weak P values even with this increased sample size (P=0.002 and P=0.0003, respectively; Supplementary Table 5), questioning the certainty of these loci.
Joint meta-analysis of the discovery and replication
To identify additional loci associated with CLL risk, four SNPs in known regions that appeared to be possible secondary signals (r2<0.1 with the reported SNPs and P<5 × 10−7 in the discovery meta-analysis) and 10 SNPs in novel regions that reached a significance threshold of P<5 × 10−6 in the discovery meta-analysis were taken forward for replication in 1,958 cases and 5,530 controls. In the joint meta-analysis of the discovery and replication, four SNPs were identified as genome-wide significant under a fixed-effects model, three in novel regions and one as a new independent SNP in the previously reported 2q13 region: 3p24.1 (rs9880772, EOMES, P=2.55 × 10−11), 6p25.2 (rs73718779, SERPINB6, P=1.97 × 10−8), 3q28 (rs9815073, LPP, P=3.62 × 10−8) and 2q13 (rs9308731, BCL2L11, P=1.00 × 10−11; Table 1, Fig. 1, Supplementary Table 6). The new 2q13 SNP, rs9308731, was weakly correlated with the two previously identified2,5 independent SNPs at 2q13, rs17483466 (r2=0.008) and rs13401811 (r2=0.0005); when the three 2q13 SNPs were included in the same logistic regression model, all three remained genome-wide significant (Supplementary Table 7). Genome-wide suggestive evidence (P<5 × 10−7) was also found in the joint discovery/replication fixed-effects meta-analysis for two promising novel loci at 4q24 (rs10028805, BANK1, P=7.19 × 10−8) and 3p22.2 (rs1274963, CSRNP1, P=2.12 × 10−7; Table 1, Supplementary Fig. 3).

(a) Chromosome 3p24.1 (rs9880772), (b) chromosome 6p25.2 (rs73718779), (c) chromosome 3q28 (rs9815073) and (d) chromosome 2q13 (rs9308731). Shown are the −log10 association P values from the discovery fixed effects meta-analysis (dots) and combined discovery and replication fixed effects meta-analysis (diamonds). The lead SNPs are shown in purple. Estimated recombination rates (from 1000 Genomes) are plotted in blue. The SNPs surrounding the most significant SNP are colour-coded to reflect their correlation with this SNP. Pairwise r2 values are from 1000 Genomes European data (March 2012 release). Genes, position of exons and direction of transcription from UCSC genome browser (genome.ucsc.edu) are noted. Plots were generated using LocusZoom (http://csg.sph.umich.edu/locuszoom).
Discussion
All the three novel loci are located in or near genes implicated in apoptosis and/or immune function. The novel 3p24.1 SNP (rs9880772) resides 13 kb 5′ of eomesodermin (EOMES), a member of the T-box gene family and a key regulator in cell-mediated immunity and CD8+ T-cell differentiation12. EOMES is critical for lymphoproliferation due to Fas-deficiency13, which has been observed in inherited lymphoproliferative disorders associated with autoimmunity14,15. Overexpression of EOMES has been observed among extranodal natural killer/T (NK/T)-cell and peripheral T-cell lymphomas16. Interestingly, highly correlated SNPs within the same 15 kb region 5′ of EOMES have also been associated with two autoimmune diseases, rheumatoid arthritis17 (rs3806624, r2=0.96) and multiple sclerosis18 (rs11129295, r2=0.72), as well as Hodgkin’s lymphoma19 (rs3806624, r2=0.96), underscoring the importance of this genetic region for susceptibility to both lymphoma and autoimmune disease. Regions locally centromeric and telomeric of rs9880772 show strong regulation and promoter signatures by histone marks, DNaseI hypersensitivity and transcription factor binding sites, and the correlated SNP, rs3806624, is located within a poised promoter in the lymphoblastoid cell line, GM12878 (Supplementary Table 8).
The novel 6p25.2 SNP (rs73718779) is located within an intron of SERPINB6, which encodes a member of the serine protease inhibitor (serpin) superfamily. Although the physiological role of SERPINB6 is not well understood, it inhibits cathepsin G20, which activates the pro-apoptotic proteinase caspase 7 (ref. 21). In eQTL and methylation QTL analyses, we found that the T allele for rs6939693, an SNP completely correlated with rs73718779 (r2=1), was associated with significantly reduced SERPINB6 expression in blood in a weighted z-score meta-analysis (P=1.40 × 10−52, Supplementary Table 9) and increased DNA methylation levels based on a linear mixed model (P=1.70 × 10−11, Supplementary Table 10), suggesting strong potential functional relevance.
The 3q28 SNP (rs9815073) is an intronic variant within the LIM domain containing preferred translocation partner in lipoma gene (LPP). The SNP is located within a strong enhancer in the lymphoblastoid cell line, GM12878 (Supplementary Fig. 4). Moderately correlated SNPs in LPP have previously been associated with diseases related to autoimmunity and/or immune dysregulation, including celiac disease22 (rs1464510, r2=0.51), allergy23 (rs9860547, r2=0.68) and vitiligo24 (rs1464510, r2=0.51). SNPs within this region have also been associated with follicular lymphoma25 (rs6444305, r2=0.001) and B-cell lymphoma in Asians (rs6773854, r2=0.002); however, the association with rs9815073 appears to be independent of both of these SNPs in the fixed-effects meta-analysis (Prs9815073=9.11 × 10−7 after conditioning on rs6444305 and Prs9815073=5.11 × 10−7 after conditioning on rs6773854 compared with Prs9815073=5.35 × 10−7 without adjustment).
The suggestive 4q24 SNP (rs10028805) is located within an intron of B-cell scaffold protein with ankyrin repeats 1 (BANK1), which encodes a protein adaptor that is predominantly expressed in B-cells. BANK1 is a putative tumour suppressor gene in B-cell lymphomagenesis26, and BANK1-deficient cells show enhanced CD40-mediated proliferation and survival with Akt activation27. Rs10028805 is moderately correlated with rs10516487 (r2=0.70), a non-synonymous SNP in exon 2 that has been associated with systemic lupus erythematosus28 and shown to alter mRNA splicing and the quantity of the BANK1 protein29. Consistent with this, we observed rs10028805 to be associated with BANK1 expression in lymphoblastoid cells (P=6.89 × 10−13, Supplementary Table 11).
The 3p22.2 SNP (rs1274963) is an intronic variant in the gene CSRNP1 (cysteine-serine-rich nuclear protein 1), which is induced by AXIN1, a scaffold protein that is a negative regulator of the Wnt/signalling pathway30. A putative tumour suppressor with potential apoptosis activity31, CSRNP1 plays an important role in the development of haematopoiesis progenitors in zebrafish32 and has been shown to be expressed in many tissues, with leukocytes being among those with the highest abundance30. The SNP resides in an area with strong regulatory potential based on histone marks, DNaseI hypersensitivity and transcription factor binding sites (Supplementary Table 8) and is located within a strong enhancer in the lymphoblastoid cell line, GM12878 (Supplementary Fig. 4). Of potential functional relevance, in lymphocytes and blood, the rs1274963A risk allele was associated with reduced WDR48 expression (Supplementary Tables 9 and 11), a gene shown to induce apoptosis and suppress tumour cell proliferation33.
To explore potential biological pathways associated with the newly discovered loci as well as the previously established loci for CLL, we conducted pathway analyses using GRAIL34, Webgestalt and GeneMania (see ‘Methods’ section). All the three pathway analyses identified apoptosis or apoptosis-related pathways as either the top key words (GRAIL, Supplementary Table 12, Fig. 2a) or their most significantly enriched pathway: regulation of apoptotic signalling (GeneMania, P=2.06 × 10−17, false discovery rate-corrected hypergeometric test, Supplementary Table 13, Fig. 2b) and activation of pro-apoptotic gene products (Webgestalt, P=5.49 × 10−11, false discovery rate-corrected hypergeometric test, Supplementary Table 14). Other enriched pathways included related apoptotic functions and pathways, such as cytochrome c release from mitochondria (Webgestalt, P=2.16 × 10−6; GeneMania, P=7.50 × 10−13) and mitochondrial outer membrane (Webgestalt, P=3.89 × 10−6; GeneMania, P=7.18 × 10−17; Supplementary Tables 13 and 14, Supplementary Fig. 5). Lymphocyte-related pathways, such as lymphocyte homeostasis (Webgestalt, P=2.16 × 10−6), haematopoietic or lymphoid organ development (GeneMania, P=0.009), and lymphoid (GRAIL) were also observed in all the three analyses.

(a) The GRAIL results are depicted in a circle plot with the connections between the SNPs and corresponding gene for the established CLL loci. The width of the line corresponds to the strength of the literature-based connectivity with thicker lines representing stronger connections. (b) Depiction of GeneMania results. Query genes are shown in large circles with hatch marks and tightly connected neighbouring genes were shown in small solid circles. The genes belonging to the top function, ‘regulation of apoptotic signalling pathway’, are highlighted with red colour. The colour of the line indicates the network: co-expression (lavender), co-localization (purple), genetic interactions (grey), pathway (blue), physical interactions (pink), predicted (orange) and shared protein domains (beige). The figure was created with GeneMania Application version: 3.1.2.8.
We constructed a polygenetic risk score that included the four new SNPs from this study as well as 30 previously identified SNPs at known loci (Supplementary Table 5) to evaluate the possibility of risk stratification for CLL (see ‘Methods’ section). Those in the top 20% of the risk distribution had a 1.9-fold increased risk (95% confidence interval: 1.70–2.21) compared with those in the middle quintile of the distribution. The newly discovered SNPs explain ∼1% of the familial risk. Together with the previously identified loci, we estimate that the identified loci for CLL thus far explain ∼16.5% of the familial risk, which is similar to previous estimates5,6.
In conclusion, our meta-analysis of GWAS identified four new independent SNPs and two additional promising loci for CLL, furthering our knowledge of the underpinnings of genetic susceptibility to CLL. Pathway analyses of known and new CLL loci point to regulation of apoptosis as one of the key biological processes underlying the genetic loci to date and suggest new avenues for disease prevention and treatment.
Methods
Discovery meta-analysis
Our discovery meta-analysis included four CLL GWAS of European ancestry: National Cancer Institute NHL GWAS (NCI GWAS)5, Utah Chronic Lymphocytic Leukemia GWAS (UTAH), Genetic Epidemiology of CLL Consortium GWAS (GEC)4, and Molecular Epidemiology of Non-Hodgkin Lymphoma GWAS (UCSF)9. Details of the case and control ascertainment and study design of the four GWAS, including the 22 studies that comprise the NCI GWAS, are described in Supplementary Table 1. In brief, CLL cases were ascertained from cancer registries, clinics or hospitals, or through self-report verified by medical and pathology reports. For the NCI GWAS, phenotype information for the cases was reviewed centrally at the International Lymphoma Epidemiology Consortium (InterLymph) Data Coordinating Center and harmonized according to the hierarchical classification proposed by the Interlymph Pathology Working Group based on the World Health Organization classification (2008)35,36. All the studies obtained informed consent from their participants and approval from their respective Institutional Review Boards for this study5.
To maximize our statistic power, all cases with sufficient DNA and a subset of available controls were genotyped for this study. Subjects in these studies were genotyped using the Illumina OmniExpress, Omni2.5, HumanHap610K, HumanCNV360-Duo or Affymetrix 6.0. For the NCI GWAS, the majority of subjects were genotyped with the Illumina OmniExpress; however, a subset of controls (N=3,536) and one case were genotyped using the Omni2.5, so to prevent potential platform artifacts, extensive quality control metrics were used, including the removal of assays with low completion rates or monomorphic calls from either platform, before combining the data5. For all four GWAS, rigorous quality control metrics were applied to each study to ensure high quality results. Samples with poor call rates, gender discordance, abnormal heterozygosity or of non-European ancestry were excluded, and SNPs with a call rate <95% or Hardy–Weinberg equilibrium P value <1 × 10−6 were removed from the analysis (Supplementary Table 2).
Each GWAS was imputed separately using IMPUTE2 (ref. 11). In contrast to the previous study5 where a hybrid reference panel was used for imputation, all the studies in this analysis were imputed using the 1000 Genomes Project version 3 (March 2012 release) as the reference panel. Poorly imputed SNPs (INFO score <0.3) and SNPs with minor allele frequency <1% were excluded from each study, leaving roughly ∼8.5 million SNPs for analysis. After quality control filters, a total of 3,100 cases and 7,667 controls across the four studies remained for analysis (Supplementary Table 3). For each study, principal component analyses were conducted separately. Association testing was conducted for each study separately using SNPTEST version 2, adjusting for age, sex and significant principal components (P<0.05 in null model with age and sex). Meta-analyses were performed using the fixed-effects inverse variance method based on the beta estimates and standard errors from each study.
Replication and technical validation
Replication of potential novel SNPs was undertaken in 1,958 additional cases and 5,530 controls from six different studies (Supplementary Tables 1 and 3). Fourteen promising SNPs that reached a significance threshold of P<5 × 10−6 in the discovery meta-analysis were taken forward for replication, including 10 SNPs in novel regions (defined as at least 1 Mb from a known CLL locus) and four SNPs in known regions that appeared to be possible secondary signals (r2<0.1 with the reported SNPs and P<5 × 10−7 in the discovery meta-analysis). To conduct conditional analyses with the potential secondary signals, the previously reported index SNP(s) in each of these four regions were also genotyped. TaqMan custom genotyping assays (Applied Biosystems) were designed and optimized for the 14 promising SNPs as well as five previously reported index SNPs. Taqman or Sequenom genotyping was conducted separately for each replication study at their own centre. Each study included duplicates for quality control, and HapMap samples genotyped across the centres yielded excellent concordance (100%). Association testing was conducted separately for each study, adjusting for age, sex and for MSKCC, Ashkenazi ancestry. The replication studies were then meta-analysed together and with the discovery GWAS using an inverse variance fixed effects model. All the SNPs reaching genome-wide or suggestive significance in the joint meta-analysis were either directly genotyped or well imputed (INFO>0.78 for all SNPs with average INFO=0.95) in the GWAS. Technical validation comparing genotype calls or imputed data from the NCI GWAS with Taqman assays for 639 samples revealed moderate concordance for rs9815073 (r2=0.67), but high concordance (r2>0.97) for the other SNPs. Although the concordance was lower than expected and further confirmation is needed, an analysis of the Taqman validation data for rs9815073 showed an odds ratio=1.30, which is similar to the odds ratio observed in the full discovery data set.
Polygenic risk score analysis
To evaluate possible stratification for CLL risk based on the 34 independent SNPs from the 30 loci, we performed a polygenic risk score analysis using the discovery sample data. Polygenic risk scores were derived for each person by taking the weighted sum of the risk alleles (0, 1 or 2) for each of the 34 SNPs. The weights for each SNP were the per-allele log odds ratios estimated from our meta-analysis of the discovery data. We then computed the quintiles of the polygenic risk scores and used logistic regression models to estimate the odds ratio for CLL risk for each quintile with the middle quintile as the reference. Departures from a multiplicative model were assessed by testing for all pair-wise SNP interactions. No evidence of significant interactions was observed.
Heritability analysis
To estimate the familial risk explained by both the novel and previously established loci for CLL, we estimated the contribution of each independent SNP to the heritability using the equation h2SNP=β22f(1−f), where β is the log-odds ratio per copy of the risk allele from the replication stage analyses and f is the allele frequency, and summed the contributions of all novel and established SNPs37. We then estimated the total heritability from the sibling relative risk (relative risk=8.5 from Goldin et al.38), using the equation derived by Pharoah et al.39 We then calculated the proportion of familial risk explained by dividing the summed contributions of the novel and established SNPs by the total heritability.
Expression quantitative trait loci and other related analyses
To explore the potential functional relevance of the CLL-associated SNPs, we conducted expression quantitative trait loci (eQTL) and methylation quantitative trait loci (meQTL) analyses using three independent data sets: (1) a childhood asthma study of gene expression in lymphoblastoid cell lines40, (2) a meta-analysis of eQTL associations from whole blood41, and (3) meQTL in CD4+ lymphocytes from the GOLDN study42. In the childhood asthma study40, RNA was extracted from lymphoblastoid cell lines from 830 parents and offspring from 206 families of European ancestry. Gene expression was assessed with the Affymetrix HG-U133 Plus 2.0 chip, and subjects were genotyped using the Illumina Human-1 and HumanHap300K beadchips with subsequent imputation using data from the 1000 Genomes Project. The four new and two suggestive SNPs were tested for cis associations (defined as gene transcripts within 1 Mb), adjusting for non-genetic effects in the gene expression value and relatedness using MERLIN43. To gain insight into the relative importance of associations with our SNPs compared with other SNPs in the region, conditional analyses were also conducted, in which both the CLL SNP and the most significant SNP for the particular gene transcript (that is, the peak SNP) were included in the same model. The meta-analysis of eQTL associations from whole blood41 included eQTL data generated using Illumina gene expression arrays from seven studies consisting of a total of 5,311 unrelated Europeans. Gene expression arrays were harmonized by matching probe sequences, and all the studies were imputed using the HapMap European reference panel. SNPs that were strongly correlated (r2>0.8) with the newly discovered and suggestive CLL SNPs were examined for possible cis associations. In the GOLDN study42, over 450,000 CpG methylation sites were genotyped in CD4+ T-cells from 593 participants. Subjects were genotyped with the Affymetrix Human SNP Array 6.0, and the 2.5 million SNPs available in the HapMap2 release were imputed. We updated the analysis by including more participants (n=717) and expanded the scope of cis-meQTL to SNPs and CpG sites within 50 kb of each other. The association between the CLL-associated SNPs (as well as strongly correlated SNPs, r2>0.8) and methylation beta values was tested using the linear mixed models, adjusting for family structure and other covariates including age, sex, recruitment centres and principal components. Finally, we also utilized HaploReg44, a tool for exploring noncoding functional annotation using ENCODE data, to evaluate the genome surrounding our SNPs.
Pathway analyses
To explore potential biological pathways underlying known CLL loci to date, we conducted analyses using GRAIL34, Webgestalt45 and GeneMania46. GRAIL34 is a text-based mining tool that is used to evaluate the relationship between genes at different disease loci. Genes within 250 kb of known loci were included, and the 2006 text database was used to avoid overweighting the previously published loci. Webgestalt45 is a web-based pathway analysis server offering hypergeometric tests for Gene Ontology (GO) term enrichments and visualization of enriched GO terms in a graph depicting the GO hierarchy. GeneMania46 is a network-based analysis server that finds an expanded set of genes including the query genes and additional genes closely linked with the query genes via protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. For both Webgestalt and GeneMania, the nearest gene for each locus was included. For all pathways analyses, only newly discovered loci and the previously identified loci that reached at least P<1 × 10−5 in the combined meta-analysis with the published results from two other GWAS6,7 (Supplementary Table 5) were included.
Chromatin state dynamics analysis
To assess chromatin state dynamics, we used Chromos47, which utilizes Chip-Seq data from ENCODE48 on nine cell types: B-lymphoblastoid cells (GM12878), hepatocellular carcinoma cells (HepG2), embryonic stem cells (hESC), erythrocytic leukemia cells (hK562), umbilical vein endothelial cells (hUVEC), skeletal muscle myoblasts (hSMM), normal lung fibroblasts (hNHLF), normal epidermal keratinocytes (hNHEK) and mammary epithelial cells (hMEC). This programme uses pre-computed data with genome-segmentation performed using a multivariate hidden Markov-model to reduce the combinatorial space to a set of interpretable chromatin states. The output from Chromos lists data into 15 chromatin states corresponding to repressed, poised and active promoters, strong and weak enhancers, putative insulators, transcribed regions and large-scale repressed and inactive domains. For this study, we focused on the results observed for the lymphoblastoid cell line (GM12878).
Additional information
How to cite this article: Berndt, S. I. et al. Meta-analysis of genome-wide association studies discovers multiple loci for chronic lymphocytic leukemia. Nat. Commun. 7:10933 doi: 10.1038/ncomms10933 (2016).
References
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30 (2013).
Di Bernardo, M. C. et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 40, 1204–1210 (2008).
Crowther-Swanepoel, D. et al. Common variants at 2q37.3, 8q24.21, 15q21.3 and 16q24.1 influence chronic lymphocytic leukemia risk. Nat. Genet. 42, 132–136 (2010).
Slager, S. L. et al. Common variation at 6p21.31 (BAK1) influences the risk of chronic lymphocytic leukemia. Blood 120, 843–846 (2012).
Berndt, S. I. et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat. Genet. 45, 868–876 (2013).
Speedy, H. E. et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 46, 56–60 (2014).
Sava, G. P. et al. Common variation at 12q24.13 (OAS3) influences chronic lymphocytic leukemia risk. Leukemia 29, 748–751 (2014).
Slager, S. L. et al. Genome-wide association study identifies a novel susceptibility locus at 6p21.3 among familial CLL. Blood 117, 1911–1916 (2011).
Conde, L. et al. Genome-wide association study of follicular lymphoma identifies a risk locus at 6p21.32. Nat. Genet. 42, 661–664 (2010).
1000 Genomes Project Consortium. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Pearce, E. L. et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043 (2003).
Kinjyo, I. et al. Cutting edge: lymphoproliferation caused by Fas deficiency is dependent on the transcription factor eomesodermin. J. Immunol. 185, 7151–7155 (2010).
Fisher, G. H. et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81, 935–946 (1995).
Drappa, J., Vaishnaw, A. K., Sullivan, K. E., Chu, J. L. & Elkon, K. B. Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N. Engl. J. Med. 335, 1643–1649 (1996).
Zhang, S., Li, T., Zhang, B., Nong, L. & Aozasa, K. Transcription factors engaged in development of NK cells are commonly expressed in nasal NK/T-cell lymphomas. Hum. Pathol. 42, 1319–1328 (2011).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Sawcer, S. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Frampton, M. et al. Variation at 3p24.1 and 6q23.3 influences the risk of Hodgkin's lymphoma. Nat. Commun. 4, 2549 (2013).
Scott, F. L. et al. The intracellular serpin proteinase inhibitor 6 is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, cathepsin G. Blood 93, 2089–2097 (1999).
Zhou, Q. & Salvesen, G. S. Activation of pro-caspase-7 by serine proteases includes a non-canonical specificity. Biochem. J. 324, (Pt 2): 361–364 (1997).
Hunt, K. A. et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 40, 395–402 (2008).
Hinds, D. A. et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat. Genet. 45, 907–911 (2013).
Jin, Y. et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N. Engl. J. Med. 362, 1686–1697 (2010).
Skibola, C. F. et al. Genome-wide association study identifies five susceptibility loci for follicular lymphoma outside the HLA region. Am. J. Hum. Genet. 95, 462–471 (2014).
Yan, J. et al. Inactivation of BANK1 in a novel IGH-associated translocation t(4;14)(q24;q32) suggests a tumor suppressor role in B-cell lymphoma. Blood Cancer J. 4, e215 (2014).
Aiba, Y. et al. BANK negatively regulates Akt activation and subsequent B cell responses. Immunity 24, 259–268 (2006).
Kozyrev, S. V. et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 40, 211–216 (2008).
Kozyrev, S. V., Bernal-Quiros, M., Alarcon-Riquelme, M. E. & Castillejo-Lopez, C. The dual effect of the lupus-associated polymorphism rs10516487 on BANK1 gene expression and protein localization. Genes Immun. 13, 129–138 (2012).
Ishiguro, H. et al. Identification of AXUD1, a novel human gene induced by AXIN1 and its reduced expression in human carcinomas of the lung, liver, colon and kidney. Oncogene 20, 5062–5066 (2001).
Glavic, A., Molnar, C., Cotoras, D. & de Celis, J. F. Drosophila Axud1 is involved in the control of proliferation and displays pro-apoptotic activity. Mech. Dev. 126, 184–197 (2009).
Espina, J., Feijoo, C. G., Solis, C. & Glavic, A. csrnp1a is necessary for the development of primitive hematopoiesis progenitors in zebrafish. PLoS ONE 8, e53858 (2013).
Gangula, N. R. & Maddika, S. WD repeat protein WDR48 in complex with deubiquitinase USP12 suppresses Akt-dependent cell survival signaling by stabilizing PH domain leucine-rich repeat protein phosphatase 1 (PHLPP1). J. Biol. Chem. 288, 34545–34554 (2013).
Raychaudhuri, S. et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 5, e1000534 (2009).
Morton, L. M. et al. Proposed classification of lymphoid neoplasms for epidemiologic research from the Pathology Working Group of the International Lymphoma Epidemiology Consortium (InterLymph). Blood 110, 695–708 (2007).
Turner, J. J. et al. InterLymph hierarchical classification of lymphoid neoplasms for epidemiologic research based on the WHO classification (2008): update and future directions. Blood 116, e90–e98 (2010).
Park, J. H. et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 42, 570–575 (2010).
Goldin, L. R., Bjorkholm, M., Kristinsson, S. Y., Turesson, I & Landgren, O. Elevated risk of chronic lymphocytic leukemia and other indolent non-Hodgkin's lymphomas among relatives of patients with chronic lymphocytic leukemia. Haematologica 94, 647–653 (2009).
Pharoah, P. D. et al. Polygenic susceptibility to breast cancer and implications for prevention. Nat. Genet. 31, 33–36 (2002).
Dixon, A. L. et al. A genome-wide association study of global gene expression. Nat. Genet. 39, 1202–1207 (2007).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Zhi, D. et al. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 8, 802–806 (2013).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Wang, J., Duncan, D., Shi, Z. & Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41, W77–W83 (2013).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220 (2010).
Barenboim, M. & Manke, T. ChroMoS: an integrated web tool for SNP classification, prioritization and functional interpretation. Bioinformatics 29, 2197–2198 (2013).
Ernst, J. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011).
Acknowledgements
We thank I. Brock, K. Butterbach, A. Chabrier, D. Chan-Lam, D. Connley, H. Cramp, R. Cutting, C. Dalley, H. Dykes, A. Gabbas, P. Gaddam, P. Hui, L. Irish, L. Jacobus, S. Kaul, L. Klareskog, A. Lai, J. Lunde, M. McAdams, L. Padyukov, D. Parisi, V. Rajamanickam, T. Rattle, L. Rigacci, R. Sargent, G. Specchia, M. Stagner, P. Taylor, C. Tornow, J. WiIliams and G. Wood. The overall GWAS project was supported by the intramural programme of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, US National Institutes of Health.
ATBC - This research was supported in part by the Intramural Research Program of the NIH and the National Cancer Institute. Additionally, this research was supported by U.S. Public Health Service contracts N01-CN-45165, N01-RC-45035, N01-RC-37004 and HHSN261201000006C from the National Cancer Institute, Department of Health and Human Services.
BC – Canadian Institutes for Health Research (CIHR); Canadian Cancer Society; Michael Smith Foundation for Health Research.
CPS-II - The Cancer Prevention Study-II (CPS-II) Nutrition Cohort is supported by the American Cancer Society. Genotyping for all CPS-II samples were supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program.
ELCCS - Leukaemia & Lymphoma Research.
ENGELA - Association pour la Recherche contre le Cancer (ARC), Institut National du Cancer (INCa), Fondation de France, Fondation contre la Leucémie, Agence nationale de sécurité sanitaire de l'alimentation, de l'environnement et du travail (ANSES)
EPIC - Coordinated Action (Contract #006438, SP23-CT-2005-006438); HuGeF (Human Genetics Foundation), Torino, Italy; Cancer Research UK.
EpiLymph – European Commission (grant references QLK4-CT-2000-00422 and FOOD-CT-2006-023103); the Spanish Ministry of Health (grant references CIBERESP, PI11/01810, PI14/01219, RCESP C03/09, RTICESP C03/10 and RTIC RD06/0020/0095), the Marató de TV3 Foundation (grant reference 051210), the Agència de Gestiód'AjutsUniversitarisi de Recerca – Generalitat de Catalunya (grant reference 2014SRG756) who had no role in the data collection, analysis or interpretation of the results; the NIH (contract NO1-CO-12400); the Compagnia di San Paolo—Programma Oncologia; the Federal Office for Radiation Protection grants StSch4261 and StSch4420, the José Carreras Leukemia Foundation grant DJCLS-R12/23, the German Federal Ministry for Education and Research (BMBF-01-EO-1303); the Health Research Board, Ireland and Cancer Research Ireland; Czech Republic supported by MH CZ – DRO (MMCI, 00209805) and RECAMO, CZ.1.05/2.1.00/03.0101; Fondation de France and Association de Recherche Contre le Cancer.
GEC/Mayo GWAS - National Institutes of Health (CA118444, CA148690, CA92153). Intramural Research Program of the NIH, National Cancer Institute. Veterans Affairs Research Service. Data collection for Duke University was supported by a Leukemia & Lymphoma Society Career Development Award, the Bernstein Family Fund for Leukemia and Lymphoma Research, and the National Institutes of Health (K08CA134919), National Center for Advancing Translational Science (UL1 TR000135).
HPFS - The HPFS was supported in part by National Institutes of Health grants CA167552, CA149445, CA098122, CA098566, and K07 CA115687. We would like to thank the participants and staff of the Health Professionals Follow-up Study for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
Iowa-Mayo SPORE – NCI Specialized Programs of Research Excellence (SPORE) in Human Cancer (P50 CA97274); National Cancer Institute (P30 CA086862, P30 CA15083); Henry J. Predolin Foundation.
Italian GxE - Italian Association for Cancer Research (AIRC, Investigator Grant 11855) (PC); Fondazione Banco di Sardegna 2010-2012, and Regione Autonoma della Sardegna (LR7 CRP-59812/2012) (MGE).
Mayo Clinic Case-Control – National Institutes of Health (R01 CA92153); National Cancer Institute (P30 CA015083).
MCCS – The Melbourne Collaborative Cohort Study recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR).
MCC-Spain - The MCC-Spain study is funded by The Instituto de Salud Carlos III (ISCIII – Spanish Government) (PI11/01810, PI14/01219, RCESP C03/09, and CIBERESP); the Agencia de Gestio d'Ajuts Universitaris i de Recerca (AGAUR) – Generalitat de Catalunya (Catalonian Government) (2014SGR756). Nadia García and Marleny Vergara (ICO-IDIBELL) provided technical support for this study.
MD Anderson – Institutional support to the Center for Translational and Public Health Genomics.
MSKCC – Geoffrey Beene Cancer Research Grant, Lymphoma Foundation (LF5541); Barbara K. Lipman Lymphoma Research Fund (74419); Robert and Kate Niehaus Clinical Cancer Genetics Research Initiative (57470); U01 HG007033; ENCODE; U01 HG007033.
NCI-SEER – Intramural Research Program of the National Cancer Institute, National Institutes of Health, and Public Health Service (N01-PC-65064,N01-PC-67008, N01-PC-67009, N01-PC-67010, N02-PC-71105).
NHS –The NHS was supported in part by National Institutes of Health grants CA186107, CA87969, CA49449, CA149445, CA098122, CA098566, and K07 CA115687. We would like to thank the participants and staff of the Nurses' Health Study for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
NSW - NSW was supported by grants from the Australian National Health and Medical Research Council (ID990920), the Cancer Council NSW, and the University of Sydney Faculty of Medicine.
NYU-WHS - National Cancer Institute (R01 CA098661, P30 CA016087); National Institute of Environmental Health Sciences (ES000260).
PLCO - This research was supported by the Intramural Research Program of the National Cancer Institute and by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS.
SCALE – Swedish Cancer Society (2009/659). Stockholm County Council (20110209) and the Strategic Research Program in Epidemiology at Karolinska Institute. Swedish Cancer Society grant (02 6661). National Institutes of Health (5R01 CA69669-02); Plan Denmark.
UCSF2 – The UCSF studies were supported by the NCI, National Institutes of Health, CA1046282 and CA154643. The collection of cancer incidence data used in this study was supported by the California Department of Health Services as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885; the National Cancer Institute's Surveillance, Epidemiology, and End Results Program under contract HHSN261201000140C awarded to the Cancer Prevention Institute of California, contract HHSN261201000035C awarded to the University of Southern California, and contract HHSN261201000034C awarded to the Public Health Institute; and the Centers for Disease Control and Prevention's National Program of Cancer Registries, under agreement #1U58 DP000807-01 awarded to the Public Health Institute. The ideas and opinions expressed herein are those of the authors, and endorsement by the State of California, the California Department of Health Services, the National Cancer Institute, or the Centers for Disease Control and Prevention or their contractors and subcontractors is not intended nor should be inferred.
UTAH/Sheffield - National Institutes of Health CA134674. Partial support for data collection at the Utah site was made possible by the Utah Population Database (UPDB) and the Utah Cancer Registry (UCR). Partial support for all datasets within the UPDB is provided by the Huntsman Cancer Institute (HCI) and the HCI Cancer Center Support grant, P30 CA42014. The UCR is supported in part by NIH contract HHSN261201000026C from the National Cancer Institute SEER Program with additional support from the Utah State Department of Health and the University of Utah. Partial support for data collection in Sheffield, UK was made possible by funds from Yorkshire Cancer Research and the Sheffield Experimental Cancer Medicine Centre. We thank the NCRI Haemato-oncology Clinical Studies Group, colleagues in the North Trent Cancer Network the North Trent Haemato-oncology Database.
WHI – WHI investigators are: Program Office - (National Heart, Lung, and Blood Institute, Bethesda, Maryland) Jacques Rossouw, Shari Ludlam, Dale Burwen, Joan McGowan, Leslie Ford, and Nancy Geller; Clinical Coordinating Center - (Fred Hutchinson Cancer Research Center, Seattle, WA) Garnet Anderson, Ross Prentice, Andrea LaCroix, and Charles Kooperberg; Investigators and Academic Centers - (Brigham and Women's Hospital, Harvard Medical School, Boston, MA) JoAnn E. Manson; (MedStar Health Research Institute/Howard University, Washington, DC) Barbara V. Howard; (Stanford Prevention Research Center, Stanford, CA) Marcia L. Stefanick; (The Ohio State University, Columbus, OH) Rebecca Jackson; (University of Arizona, Tucson/Phoenix, AZ) Cynthia A. Thomson; (University at Buffalo, Buffalo, NY) Jean Wactawski-Wende; (University of Florida, Gainesville/Jacksonville, FL) Marian Limacher; (University of Iowa, Iowa City/Davenport, IA) Robert Wallace; (University of Pittsburgh, Pittsburgh, PA) Lewis Kuller; (Wake Forest University School of Medicine, Winston-Salem, NC) Sally Shumaker; Women's Health Initiative Memory Study - (Wake Forest University School of Medicine, Winston-Salem, NC) Sally Shumaker. The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C.
YALE – National Cancer Institute (CA62006); National Cancer Institute (CA165923).
Other support: NSFC - the National Natural Science Foundation of China (No. 61471078).
Author information
Authors and Affiliations
Contributions
S.I.B., N.J.C., C.F.S., A.N., K.E.S., W.C., S.S.W., L.R.T., A.R.B.-W., P.H., M.P.P., B.M.B., P.C., Y.Z., A.Z.-J., C.L., R.M., H.H., J.M., P.V., J.J.S., A.K., J.R.C., S.J.C., N.R. and S.L.S. organized and designed the study. N.J.C., C.F.S., A.C., L.B., A.H., J.M.Cu., L.C., P.M.B., E.A.H., J.R.C., S.J.C. and S.L.S. conducted and supervised the genotyping of samples. S.I.B., N.J.C., C.F.S., J. Vijai, Z.W., M. Machado, M.Y., D.K.A., D.Z., J.M.L., L.L., B.M., J.H., J.-H.P., N.C., J.R.C., S.J.C., N.R. and S.L.S. contributed to the design and execution of data analysis. S.I.B., N.J.C., C.F.S., J. Vijai, Z.W., A.N., N.C., J.R.C., S.J.C., N.R. and S.L.S. wrote the first draft of the manuscript. S.I.B., N.J.C., C.F.S., J. Vijai, J.G., A.N., R.S.K., K.E.S., A.Mo., W.C., A.C., S.S.W., Q.L., L.R.T., A.R.B.-W., P.H., M.P.P., B.M.B., C.M.Vajdic, P.C., Y.Z., G.G.G., A.Z.-J., Y.Y., T.G.C., T.D.S., A.J.N., N.E.K., M.L., J.M.Cu., C.A., H.H., H.-O.A., M. Melbye, B.G., E.T.C., M.G., K.C., L.A.C.-A., W.R.D., B.K.L., G.J.W., L.C., P.M.B., J.R., E.A.H., R.D.J., L.F.T., Y.B., N.S., N.B., P.Bo., P.Br., L.F., M. Maynadie, J.M., A.S., K.G.C., S.J.A., C.M. Vachon, L.R.G., S.S.S., J.F.L., J.B.W., N.E.C., A.D.N., A.J.D.R., L.M.M., R.K.S., E.R., P.V., R.K., G.M., E.W., M.-D.C., R.C.H.V., R.C.T., M.C.S., R.L.M., D.A., J. Virtamo, S.W., J.C., T.Z., T.R.H., D.J.V., A.Ma., J.J.S., R.D.G., J.M.Co., K.A.B., E.G., P.K., A.K., J.T., M.G.E., G.M.F., L.M., S.C., K.E.N., J.A.S., J.W., J.F.F., K.O., X.W., S.d.S., J.R.C., N.R. and S.L.S. conducted the epidemiological studies and contributed samples to the GWAS and/or follow-up genotyping. All the authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-5 and Supplementary Tables 1-14 (PDF 2554 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Berndt, S., Camp, N., Skibola, C. et al. Meta-analysis of genome-wide association studies discovers multiple loci for chronic lymphocytic leukemia. Nat Commun 7, 10933 (2016). https://doi.org/10.1038/ncomms10933
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms10933
This article is cited by
-
Predictors of response of rituximab in rheumatoid arthritis by weighted gene co-expression network analysis
Clinical Rheumatology (2023)
-
Meiotic drive in chronic lymphocytic leukemia compared with other malignant blood disorders
Scientific Reports (2022)
-
Distinct germline genetic susceptibility profiles identified for common non-Hodgkin lymphoma subtypes
Leukemia (2022)
-
Validation and functional characterization of GWAS-identified variants for chronic lymphocytic leukemia: a CRuCIAL study
Blood Cancer Journal (2022)
-
BIRC3 and BIRC5: multi‐faceted inhibitors in cancer
Cell & Bioscience (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
