
Abstract
Cellular transformation and cancer progression is accompanied by changes in the metabolic landscape. Master co-regulators of metabolism orchestrate the modulation of multiple metabolic pathways through transcriptional programs, and hence constitute a probabilistically parsimonious mechanism for general metabolic rewiring. Here we show that the transcriptional co-activator peroxisome proliferator-activated receptor gamma co-activator 1α (PGC1α) suppresses prostate cancer progression and metastasis. A metabolic co-regulator data mining analysis unveiled that PGC1α is downregulated in prostate cancer and associated with disease progression. Using genetically engineered mouse models and xenografts, we demonstrated that PGC1α opposes prostate cancer progression and metastasis. Mechanistically, the use of integrative metabolomics and transcriptomics revealed that PGC1α activates an oestrogen-related receptor alpha (ERRα)-dependent transcriptional program to elicit a catabolic state and metastasis suppression. Importantly, a signature based on the PGC1α–ERRα pathway exhibited prognostic potential in prostate cancer, thus uncovering the relevance of monitoring and manipulating this pathway for prostate cancer stratification and treatment.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

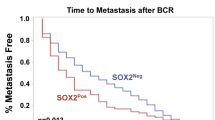
Change history
22 May 2017
In the original version of this Article, the name of author James David Sutherland was coded wrongly, resulting in it being incorrect when exported to citation databases. This has now been corrected, though no visible changes will be apparent.
References
Loo, J. M. et al. Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406 (2015).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Vander Heiden, M. G. et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329, 1492–1499 (2010).
Carracedo, A., Cantley, L. C. & Pandolfi, P. P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer 13, 227–232 (2013).
Yang, M., Soga, T. & Pollard, P. J. Oncometabolites: linking altered metabolism with cancer. J. Clin. Invest. 123, 3652–3658 (2013).
Ortega-Molina, A. et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 15, 382–394 (2012).
Garcia-Cao, I. et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149, 49–62 (2012).
Salmena, L., Carracedo, A. & Pandolfi, P. P. Tenets of PTEN tumor suppression. Cell 133, 403–414 (2008).
Song, M. S., Salmena, L. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 (2012).
Di Cristofano, A., Pesce, B., Cordon-Cardo, C. & Pandolfi, P. P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355 (1998).
Chen, Z. et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005).
Majumder, P. K. et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc. Natl Acad. Sci. USA 100, 7841–7846 (2003).
Magnon, C. et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013).
Ding, Z. et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273 (2011).
Nandana, S. & Chung, L. W. Prostate cancer progression and metastasis: potential regulatory pathways for therapeutic targeting. Am. J. Clin. Exp. Urol. 2, 92–101 (2014).
Cho, H. et al. RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov. 4, 318–333 (2014).
Mouchiroud, L., Eichner, L. J., Shaw, R. J. & Auwerx, J. Transcriptional coregulators: fine-tuning metabolism. Cell Metab. 20, 26–40 (2014).
Lapointe, J. et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl Acad. Sci. USA 101, 811–816 (2004).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
Grasso, C. S. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012).
Taylor, B. S. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010).
Tomlins, S. A. et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 39, 41–51 (2007).
Varambally, S. et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8, 393–406 (2005).
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015).
Lin, J., Handschin, C. & Spiegelman, B. M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 (2005).
Haq, R. et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 23, 302–315 (2013).
Vazquez, F. et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 23, 287–301 (2013).
LeBleu, V. S. et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003 (2014).
LaGory, E. L. et al. Suppression of PGC-1α is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep. 12, 116–127 (2015).
D’Errico, I. et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-α (PGC1α) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl Acad. Sci. USA 108, 6603–6608 (2011).
Sancho, P. et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 22, 590–605 (2015).
Audet-Walsh, E. et al. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep. 14, 920–931 (2016).
Lin, J. et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135 (2004).
Eisele, P. S. & Handschin, C. Functional crosstalk of PGC-1 coactivators and inflammation in skeletal muscle pathophysiology. Semin. Immunopathol. 36, 27–53 (2014).
Saint-Geniez, M. et al. PGC-1α regulates normal and pathological angiogenesis in the retina. Am. J. Pathol. 182, 255–265 (2013).
Nardella, C., Carracedo, A., Salmena, L. & Pandolfi, P. P. Faithfull modeling of PTEN loss driven diseases in the mouse. Curr. Top. Microbiol. Immunol. 347, 135–168 (2011).
Nardella, C. et al. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 22, 2172–2177 (2008).
Li, S. et al. Genome-wide coactivation analysis of PGC-1α identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 8, 105–117 (2008).
Tennakoon, J. B. et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 33, 5251–5261 (2014).
Shiota, M. et al. Peroxisome proliferator-activated receptor gamma coactivator-1α interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol. Endocrinol. 24, 114–127 (2010).
Buijs, J. T. & van der Pluijm, G. Osteotropic cancers: from primary tumor to bone. Cancer Lett. 273, 177–193 (2009).
Garcia, M. et al. Cyclooxygenase-2 inhibitor suppresses tumour progression of prostate cancer bone metastases in nude mice. BJU Int. 113, E164–E177 (2014).
Feige, J. N. & Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 17, 292–301 (2007).
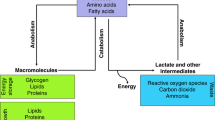
Finley, L. W., Zhang, J., Ye, J., Ward, P. S. & Thompson, C. B. SnapShot: cancer metabolism pathways. Cell Metab. 17, e462–466 (2013).
Stein, R. A. et al. Estrogen-related receptor alpha is critical for the growth of estrogen receptor-negative breast cancer. Cancer Res. 68, 8805–8812 (2008).
Gaillard, S. et al. Receptor-selective coactivators as tools to define the biology of specific receptor-coactivator pairs. Mol. Cell 24, 797–803 (2006).
Chang, C. Y. et al. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 20, 500–510 (2011).
Piskounova, E. et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015).
Lunt, S. Y. & Vander Heiden, M. G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 (2011).
Carracedo, A. et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Invest. 122, 3088–3100 (2012).
Schafer, Z. T. et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 (2009).
Arroyo-Berdugo, Y. et al. Involvement of ANXA5 and ILKAP in susceptibility to malignant melanoma. PLoS ONE 9, e95522 (2014).
Song, M. S. et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199 (2011).
Chen, Z. et al. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci. Signal 2, ra44 (2009).
Nardella, C. et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci. Signal 2, ra2 (2009).
Guiu, M., Arenas, E. J., Gawrzak, S., Pavlovic, M. & Gomis, R. R. Mammary cancer stem cells reinitiation assessment at the metastatic niche: the lung and bone. Methods Mol. Biol. 1293, 221–229 (2015).
Carracedo, A. et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 118, 3065–3074 (2008).
Ugalde-Olano, A. et al. Methodological aspects of the molecular and histological study of prostate cancer: focus on PTEN. Methods 77–78, 25–30 (2015).
Finley, L. W. et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428 (2011).
Caro-Maldonado, A. et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 192, 3626–3636 (2014).
Wojtala, A. et al. Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol. 542, 243–262 (2014).
Ittmann, M. et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 73, 2718–2736 (2013).
Liu, X. et al. High resolution metabolomics with acyl-CoA profiling reveals widespread remodeling in response to diet. Mol. Cell. Proteomics 14, 1489–1500 (2015).
Liu, X., Ser, Z. & Locasale, J. W. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal. Chem. 86, 2175–2184 (2014).
Shestov, A. A. et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife 3, e03342 (2014).
Rodriguez, R. M. et al. Regulation of the transcriptional program by DNA methylation during human alphabeta T-cell development. Nucleic Acids Res. 43, 760–774 (2015).
Bergmann, R., Ludbrook, J. & Spooren, W. P. J. M. Statistical computing and graphics: different outcomes of the Wilcoxon—Mann—Whitney test from different statistics packages. Am. Statistician 54, 72–77 (2000).
Quinn, G. & Keough, M. Experimental Design and Data Analysis for Biologists (Cambridge Univ. Press, 2002).
Acknowledgements
Apologies to those whose related publications were not cited owing to space limitations. We would like to thank the following researchers: B. Spiegelman (Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts, USA; Department of Cell Biology, Harvard Medical School, Boston, Massachusetts, USA) for providing the Pgc1aLoxP mice; D. Santamaría and M. Barbacid (Experimental Oncology, Molecular Oncology Programme, Centro Nacional de Investigaciones Oncológicas (CNIO), Madrid, Spain) for technical help and advice with doxycycline-enriched diets in xenograft experiments; P. Puigserver (Department of Cell Biology, Harvard Medical School, and in the Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, Massachusetts, USA) for providing Pgc1α-expressing constructs; B. Carver (Department of Surgery, Division of Urology, Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, USA) for help and advice with data set analysis, D. McDonnell (Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, North Carolina, USA) for providing mutant Pgc1αL2L3M-expressing constructs and M. D. Boyano (Department of Cell Biology and Histology, School of Medicine and Dentistry, University of the Basque Country (UPV/EHU), Leioa, Bizkaia, Spain) and A. Buqué (Medical Oncology Research Laboratory, Cruces Universtity Hospital, Bizkaia, Spain) for providing melanoma cell lines. The work of A.C. is supported by the Ramón y Cajal award, the Basque Department of Industry, Tourism and Trade (Etortek), health (2012111086) and education (PI2012-03), Marie Curie (277043), Movember, ISCIII (PI10/01484, PI13/00031), FERO VIII Fellowship and the European Research Council Starting Grant (336343). N.M.-M. is supported by the Spanish Association Against Cancer (AECC). A.C.-M. is supported by the MINECO postdoctoral program and the CIG program from the European commission (660191). A.A.-A. and L.V.-J. are supported by the Basque Government of Education. P.Pinton is grateful to C. degli Scrovegni for continuous support and the work in his laboratory was supported by the Italian Association for Cancer Research (AIRC: IG-14442), the Italian Ministry of Education, University and Research (COFIN no. 20129JLHSY_002, FIRB no. RBAP11FXBC_002, and Futuro in Ricerca no. RBFR10EGVP_001) and the Italian Ministry of Health. R.B. is supported by MINECO (BFU2014-52282-P, BFU2011-25986) and the Basque Government (PI2012/42). The work of V.S.-M. was supported by Cancer Research UK C33043/A12065; Royal Society RG110591. P.Pandya was supported by King’s Overseas Scholarship. Work by the group of G.V. was supported by grants from the Spanish Ministry of Economy and Competitiveness/Instituto de Salud Carlos III (MINECO/ISCIII) together with the European Regional Development Fund (ERDF/FEDER): PS09/01401; PI12/02248 and PI15/00339, Fundación Mutua Madrileña and Fundació la Marató de TV3. C.C.-C. and M.C.-M. were financially supported by NIH P01CA087497. J.W.L. is supported by R00CA168997, R01CA193256 and R21CA201963 from the National Institutes of Health. Work in the M.Graupera laboratory was supported by SAF2014-59950-P from MINECO (Spain), 2014-SGR-725 from the Catalan Government, from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/ (REA grant agreement 317250), and the Institute of Health Carlos III (ISC III) and the European Regional Development Fund (ERDF) under the integrated Project of Excellence no. PIE13/00022 (ONCOPROFILE). J.U. is a Juan de la Cierva Researcher (MINECO). A.Bellmunt is a FPI-Severo Ochoa fellowship grantee (MINECO). R.R.G. research support was provided by the Spanish Government (MINECO) and FEDER grant SAF2013-46196, as well as the Generalitat de Catalunya AGAUR 2014-SGR grant 535.
Author information
Authors and Affiliations
Contributions
V.T. and L.V.-J. performed all in vitro and in vivo experiments, unless specified otherwise. A.R.C. carried out the bioinformatic and biostatistical analysis. A.Berenguer and N.S. provided support and advice in data set retrieval and normalization. S.F.-R. performed the histochemical stainings. P.S.-M. and S.F.-R. performed genotyping analyses. X.L. and J.W.L. contributed to the experimental design and executed the metabolomic analyses. G.M. and P.Pinton performed the biochemical ATP measurement in vitro and mitochondria analysis. G.V., P.Z.-G. and M.L. performed or coordinated (G.V.) subcutaneous xenograft experiments. J.U., A.Bellmunt, M.Guiu and R.R.G. performed or coordinated (R.R.G.) the intra-cardiac and intra-tibial metastasis assays. R.R.G. contributed to the design of the patient gene signature analysis. M.Graupera carried out microvessel staining and quantifications. P.Pandya and V.S.-M. provided technical advice and contributed to in vitro analysis. N.M.-M., A.A.-A. and A.Z.-L. contributed to the experimental design and discussion. A.C.-M. and N.E. performed Seahorse assays. J.D.S. and R.B. performed or coordinated (R.B.) the cloning of Pgc1a in lentiviral vectors. C.C.-C. and M.C.-M. carried out the pathological analysis and scoring of the xenografts and GEMMs. A.U.-O., I.L.-V., A.L.-I. and M.U.-U. provided BPH and PCa samples for gene expression analysis from Basurto University Hospital. A.M.A. contributed to the discussion of the results. A.C. directed the project, contributed to data analysis and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1
(A) Expression of 23 metabolic co-regulators in Taylor1 dataset (N: normal; PCa: prostate cancer). (B) Expression of 7 metabolic co-regulators from figure 1a in four additional prostate cancer datasets (N: normal; PCa: prostate cancer). In Varambally2 dataset gene expression levels are presented in Log2. In Tomlins3, Grasso4 and Lapointe5 datasets gene expression levels are presented in median centred Log2. (C,D) Association of PGC1A expression with Gleason score in TCGA provisional data6,7 (C) and Taylor1 datasets (D). (E) Analysis of PGC1A expression in benign prostatic hyperplasia (BPH) and PCa specimens from Basurto University Hospital cohort (qRTPCR, BPH n = 14 patient specimens and Cancer n = 16 patient specimens). (F) PGC1A expression in normal prostate (N), primary tumour (PT) and metastatic (Met) specimens in Grasso dataset4. Points outlined by circles indicate statistical outliers (A–D and F). Error bars represent minimum and maximum values (A,C,D and F) or median with interquartile range (E). Statistic test: two-tailed Student T test (A,B), two-tailed Mann Whitney U test (E) and ANOVA (C,D and F).
Supplementary Figure 2
(A) Analysis of Pten and Pgc1a gene expression in GEMMs of the indicated genotype (Ptenwt Pgc1awtn = 3 mice; Pten pc−/−Pgc1apc+/+n = 7 mice; Ptenpc+/+ Pgc1apc−/−n = 6 mice; Ptenpc−/−Pgc1apc−/−n = 12 mice; data is normalized to Gapdh expression). (B) Age comparison between experimental cohorts (n as in Fig. 2d). C Quantification of prostate tissue with histological vascular invasion signs in Pten KO (2 mice) and DKO mice (9 mice) (limited to mice with invasive signs). (D) Histological analysis of inflammatory signs (stromal and glandular infiltration) in Pten KO and DKO mice (Pten KO, n = 7 mice; DKO n = 12 mice). (E) Quantification of Vefga mRNA expression in Pten KO and DKO mice (Pten KO, n = 7 mice; DKO, n = 6 mice; data is normalized to Gapdh expression). (F) Quantification of microvessel density (MVD) (dot plot, left panels) and representative images of CD31 immunodetection (right panels) in Pten KO and DKO mice (Pten KO, n = 3 mice; DKO, n = 5 mice). (G) Representative haematoxylin and eosin staining depicting liver metastasis in DKO (bar = 500 μm). (H) Incidence of small groups of Pan-cytokeratin (Pan-CK) positive cells in the lymph nodes of Pten KO mice (6 mice). I-J, representative immunohistochemical detection (200X) of Pan-CK positive cells in the bone marrow (BM) of Pten KO and DKO (I) and androgen receptor (AR) in the bone marrow of DKO (J) (bar = 100 μm). Pink arrows indicate immunoreactive cells. (K) Quantification of BM dissemination frequency (Pten KO, 6 mice; DKO, 8 mice). (L,M) Histopathological characterization of the prostate tissue (L) and frequency of vascular invasion signs (M, only in mice with invasion signs) in Ptenpc−/−, Pgc1apc+/− mice (3 mice). (N) Frequency of metastatic lesions in lymph nodes (LN), liver and lung of Ptenpc−/−, Pgc1apc+/− mice (3 mice). (O) Correlation between PGC1A and PTEN gene expression in prostate cancer specimens (left panel) and the association of PTEN genomic loss to PGC1A gene expression (right panel), in TCGA provisional dataset. pc, prostate-specific allelic changes; +, Wildtype allele; –, deleted allele; wt: any given genotype resulting in the lack of deletion of Pgc1a and Pten alleles. Pten KO = Ptenpc−/−, Pgc1apc+/+; DKO = Ptenpc−/−, Pgc1apc−/−. Stdv: standard deviation of the mean. All error bars represent median with interquartile range. p = p-value a.u: arbitrary units. Statistic tests: one-tailed Mann-Whitney U test (A,B), two-tailed Mann-Whitney U test (D–F); ANOVA (O, right panel); Pearson’s coefficient (O, left panel). ∗p < 0.05, ∗∗∗p < 0.001.
Supplementary Figure 3
(A) mRNA expression of PGC1A, ACO2 and HADHA by qRTPCR in PC3 cells transduced with scramble shRNA (shSC) or PGC1A-targeting shRNA (shPGC1A) (n = 3). (B) PGC1A expression in normal (N, n = 29), primary tumour (PT, n = 131), metastasis (Met, n = 19) specimens and metastatic cell lines. Data is shown as Log2 mRNA expression. (C) Densitometry of PGC1α protein expression in MeWo (endogenous) and PC3 TRIPZ-HA-Pgc1α (ectopic) cell lines, relative to β-Actin (n = 3, independent experiments). (D) Effect of Pgc1α induction (+Dox) on ACO2 mRNA expression in PC3 (n = 4, independent experiments), DU145 (n = 7, independent experiments) and LnCaP cells (n = 3, independent experiments). (E,F) Effect of Pgc1α expression on anchorage-independent growth (E, n = 3, independent experiments) and BrdU incorporation (F, n = 3, independent experiments) in DU145 cells (G), Effect of Pgc1α expression on cell cycle progression in PC3 cells (n = 4, independent experiments). (H) Effect of doxycycline treatment (0.5 μg/ml) on cell growth of non-transduced PC3 cells (n = 3, independent experiments). (I,J) Pgc1α protein expression and cell proliferation by Ki67 immunoreactivity in xenograft samples from Fig. 3f (−Dox n = 14 tumours, +Dox n = 6 tumours). (K) mRNA expression of ACO2 and HADHA in xenograft samples from Fig. 3f. (−Dox n = 9 tumours, +Dox n = 6 tumours). (L,M) Analysis of VEFGA mRNA expression upon Pgc1α induction in PC3 cells (L, n = 4, independent experiments) and xenograft samples (M, −Dox n = 9 tumours and +Dox n = 6 tumours). (N) Quantification of microvessel density (MVD) in xenograft samples (−Dox n = 9 tumours and +Dox n = 7 tumours). Right panels show representative CD31 staining micrographs. Error bars indicate s.e.m (A,C–H,L) and median with interquartile range (J,K,M,N). Statistic tests: two-tailed Student T test (A–H,L) and one-tailed Mann-Whitney U test (J,K,M,N). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Supplementary Figure 4
(A,B) Validation of the microarray by qRTPCR in DU145 (n = 4, independent experiments) and LnCaP (n = 3, independent experiments) TRIPZ-HA-Pgc1a cells. Gene expression values relative to −Dox cells are represented (reference −Dox gene expression values are indicated with a dotted line) (C), mRNA expression of PGC1α target genes in doxycycline-treated (0.5 μg/ml) non-transduced PC3 cells (n = 3, independent experiments). Error bars represent s.e.m. Statistic test: One tail Student T test.∗p < 0.05, ∗∗p < 0.01,∗∗∗p < 0.001.
Supplementary Figure 5
(A) Analysis of differential abundance in metabolites involved in fatty acid catabolism by untargeted LC-HRMS in DU145 TRIPZ-HA-Pgc1α cells (n = 4, independent experiments). (B,C) Evaluation of the dehydrogenation of 3H-palmitate (readout of β-oxidation) in DU145 cells upon Pgc1α expression (B, n = 3, independent experiments) and, in doxycycline-treated (0.5 μg/ml) non-transduced PC3 cells (C, n = 3, independent experiments). Values relative to −Dox cells are presented. (D) Effect of Pgc1α expression on citrate abundance measured by LC-HRMS metabolomics in DU145 cells (n = 4, independent experiments). (E,F) ATP-producing OCR (upon complex V inhibition by oligomycin injection) in PC3 (E, n = 3, independent experiments) and DU145 (F, n = 3, independent experiments) cells upon Pgc1α expression. (G) Basal mitochondrial ATP production in DU145 cells upon Pgc1α expression (n = 10, independent experiments). (H,I) LC-HRMS quantification of ADP (H) and AMP (I) abundance in PC3 Pgc1α (n = 4, independent experiments), DU145 Pgc1α (n = 4, independent experiments), xenografts (−Dox n = 8 tumours; +Dox n = 4 tumours) and GEMMs (Pten KO n = 3 mice; DKO n = 5 mice). (J) Quantification of area under the curve (AUC, relative to −Dox) of Palmitate labelling from 13C-U6-Glucose in PC3 TRIPZ-HA-Pgc1α cells (data related to Fig. 5j, n = 3, independent experiments). (K) Determination of extracellular lactate in PC3 TRIPZ-HA-Pgc1α cells (n = 3, independent experiments) L-M, Lactate production (L) and OCR (M) in doxycycline-treated (0.5 μg/ml) non-transduced PC3 cells (n = 3, independent experiments). Error bars represent s.e.m., except xenograft and GEMM data in H,I, that represent median with interquartile range. Statistic tests: two tailed Student T test (A–H (PC3 and DU145), I (PC3 and DU145), J–M) and one tailed Mann Whitney U test (H (Xenografts and GEMMs), I (Xenografts and GEMMs)) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Supplementary Figure 6
(A) Analysis of mitochondrial morphology (mitochondrial volume) in PC3 cells upon Pgc1α expression (n = 5, independent experiments). (B) Expression of PGC1αWT and PGC1αL2L3M in PC3 cells after treatment with 0.5 μg/ml doxycycline (Dox) (a representative experiment with technical replicates is presented, similar results were obtained in three independent experiments). (C) Basal mitochondrial ATP production in PGC1αWT and PGC1αL2L3M PC3 cells (n = 11, independent experiments). (D) Expression of Pgc1α and ERRα in doxycycline-treated PC3 TRIPZ-HA-Pgc1α cells transduced with sh-scramble (shSC) or shERRα (a representative experiment with technical replicates is presented, similar results were obtained in three independent experiments). (E) mRNA expression of PGC1α target genes in doxycycline-treated PC3 TRIPZ-HA-Pgc1α cells transduced with shSC or shERRα (n = 4 for ACAT1 and n = 5 for IDH3A, ATP1B1, ISCU, GOT1 and ACADM; independent experiments). (F–I) mRNA expression of PGC1α target genes (F, n = 3 for ACAT1 and n = 4 for ATP1B1 and IDH3A; independent experiments), cell number (G, n = 4, independent experiments), basal oxygen consumption (H, n = 3, independent experiments) and basal mitochondrial ATP production (I, n = 7 for −Dox + XCT790; n = 8 the rest; independent experiments) in vehicle (Veh) or XCT790-treated Pgc1α-inducible PC3 cells. (J) Evaluation of cellular (DCF) and mitochondrial-specific (Mitosox) ROS production in Pgc1α-expressing PC3 (left panel; n = 4, independent experiments) and DU145 (right panel; n = 6, independent experiments) cells. (K) Evaluation of lipid peroxidation in xenograft tissues from Fig. 3f (−Dox n = 4 tumours; +Dox n = 5 tumours). (L) Effect of the indicated antioxidant treatments on cell number (relative to day 0) of Pgc1α-expressing PC3 cells (n = 3, independent experiments). DCF: 2′,7′-dichlorodihydrofluorescein. n.s: not significant. Error bars represent s.e.m. (A,C,E–J,L) or median with interquartile range (K). Statistic tests: two tailed Student T test (A,C,E–J,L) or one tailed Student T test (comparison between +Dox conditions in C,E–I) and two tailed Mann-Whitney U test (K). ∗/$p < 0.05, ∗∗/$$p < 0.01,∗∗∗/$$$p < 0.001. Asterisks indicate statistic between −Dox and +Dox conditions (unless represented otherwise) and dollar symbol between either, vehicle (Veh) and XCT790-treated Pgc1α-expressing cells, shSC and shERRa-transduced Pgc1α-expressing cells or Pgc1αWT and Pgc1αL2L3M.
Supplementary Figure 7
(A,B) Representation of the average signal of genes within the PGC1α-upregulated gene set (A) (Fig. 7b, blue circle) and within the PGC1α-dependent ERRα-upregulated gene set (B) (Fig. 7b, yellow circle, Supplementary Table 6) in the indicated datasets1,4,6,7, in normal (N; Taylor n = 29 and Grasso n = 12), primary tumours (PT; Taylor n = 131 and Grasso n = 49) and metastasis (Mets; Taylor n = 19 and Grasso n = 27). (C) qRTPCR mRNA expression analysis of PGC1α target genes from C, in benign prostatic hyperplasia (BPH) and PCa specimens from Basurto University Hospital cohort (BPH n = 14 patient specimens; Prostate cancer n = 16 patient specimens). D, Expression of the indicated genes (from Supplementary Table 7) in different disease states (N: normal Lapointe n = 9, Taylor n = 29 and Grasso n = 12; PT: primary tumour, Lapointe n = 13, Taylor n = 131 and Grasso n = 49; Met: metastasis, Lapointe n = 4, Taylor n = 19 and Grasso n = 27) in three PCa datasets1,4,5. (E) Representation of “PGC1α-ERRα Q1 signature’ frequency within different tumour types (N: normal; PT: primary tumour; Met: metastasis) in two datasets1,4 (Taylor: N, n = 29; PT, n = 131; Met, n = 19; Grasso: N, n = 12; PT, n = 49; Met, n = 27). Error bars represent s.e.m. (A, B), median with interquartile range (C) and maximum and minimum (D). Statistic tests: Statistic tests: ANOVA (A, B, D); two tailed Student T test (A, B), one tailed Mann Whitney U test (C), Chi Square (E). Asterisks in A,B indicates statistics between normal and metastasis and hash between primary tumours and metastasis. p: p-value.∗/#p < 0.05, ∗∗/##p < 0.01, ∗∗∗p < 0.001.
Supplementary Figure 8 Unprocessed blots.
(A) Western blot corresponding to Fig. 3a. (B) Western blot corresponding to Fig. 3b. (C) Western blot corresponding to Supplementary Fig. 3I. (D) Western blot corresponding to Supplementary Fig. 6B. (E) Western blot corresponding to Supplementary Fig. 6D. Precision Plus ProteinTM Dual Color Standards (Ref #1610374) markers was used in A–D. Pink Pre-stained protein ladder, Nippon Genetics, Cat.No. MWP02, was used in E.
Supplementary information
Supplementary Information
Supplementary Information (PDF 1850 kb)
Supplementary Table 1
Supplementary Information (XLSX 95 kb)
Supplementary Table 2
Supplementary Information (XLSX 234 kb)
Supplementary Table 3
Supplementary Information (XLSX 234 kb)
Supplementary Table 4
Supplementary Information (XLSX 152 kb)
Supplementary Table 5
Supplementary Information (XLSX 129 kb)
Supplementary Table 6
Supplementary Information (XLSX 48 kb)
Supplementary Table 7
Supplementary Information (XLSX 9 kb)
Supplementary Table 8
Supplementary Information (XLSX 64 kb)
Supplementary Table 9
Supplementary Information (XLSX 29 kb)
Rights and permissions
About this article

Cite this article
Torrano, V., Valcarcel-Jimenez, L., Cortazar, A. et al. The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat Cell Biol 18, 645–656 (2016). https://doi.org/10.1038/ncb3357
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb3357
This article is cited by
-
Transcending frontiers in prostate cancer: the role of oncometabolites on epigenetic regulation, CSCs, and tumor microenvironment to identify new therapeutic strategies
Cell Communication and Signaling (2024)
-
ERRα: unraveling its role as a key player in cell migration
Oncogene (2024)
-
PGC-1α promotes colorectal carcinoma metastasis through regulating ABCA1 transcription
Oncogene (2023)
-
Knockdown of PGC1α suppresses dysplastic oral keratinocytes proliferation through reprogramming energy metabolism
International Journal of Oral Science (2023)
-
Notch signaling regulates a metabolic switch through inhibiting PGC-1α and mitochondrial biogenesis in dedifferentiated liposarcoma
Oncogene (2023)
