
Abstract
The genus Sorangium synthesizes approximately half of the secondary metabolites isolated from myxobacteria, including the anti-cancer metabolite epothilone. We report the complete genome sequence of the model Sorangium strain S. cellulosum So ce56, which produces several natural products and has morphological and physiological properties typical of the genus. The circular genome, comprising 13,033,779 base pairs, is the largest bacterial genome sequenced to date. No global synteny with the genome of Myxococcus xanthus is apparent, revealing an unanticipated level of divergence between these myxobacteria. A large percentage of the genome is devoted to regulation, particularly post-translational phosphorylation, which probably supports the strain's complex, social lifestyle. This regulatory network includes the highest number of eukaryotic protein kinase–like kinases discovered in any organism. Seventeen secondary metabolite loci are encoded in the genome, as well as many enzymes with potential utility in industry.
Similar content being viewed by others

Main
Natural products and their derivatives provide the basis for medicines targeting a wide range of human diseases. The Gram-negative myxobacteria, members of the δ-subgroup of proteobacteria, are an important source of novel classes of secondary metabolites1. Of these, the genus Sorangium is particularly valuable, as 46% of metabolites isolated from myxobacteria1, including the potent anti-tumor compound epothilone2, derive from this group. The majority of myxobacterial metabolites are polyketides, nonribosomal polypeptides or hybrids of the two structures, many of which are synthesized on gigantic molecular assembly lines composed of polyketide synthase (PKS) and nonribosomal polypeptide synthetase (NRPS) multienzymes3. Sorangium strains exhibit additional characteristic features, including 'social behavior', cell movement by gliding, biofilm formation and morphological differentiation culminating in complex multicellular structures called fruiting bodies4. Three myxobacterial suborders are known5 and the availability of the genome sequence of Myxococcus xanthus (Cystobacterineae)6 enables comparative analysis with the Sorangium cellulosum (Sorangiineae) genome to illuminate the basis for several important behavioral and metabolic differences. These include the ability of Sorangium strains to degrade complex plant materials (Fig. 1). S. cellulosum So ce56, an obligate aerobe, was established previously as a model Sorangium strain7 by virtue of its favorable growth characteristics and ability to differentiate reproducibly under laboratory conditions. It synthesizes the cytotoxic chivosazoles7 and the catecholate-type siderophores myxochelins8. Comparison of the complete genome sequence of strain S. cellulosum So ce56 with the genomes of M. xanthus and other δ-proteobacteria provides new insights into the complex regulatory networks governing myxobacterial lifestyles and secondary metabolism. The sequence also opens opportunities to engineer high-titer producers of clinically relevant natural products.

(a) S. cellulosum colonies. (b,c) Swarming growth patterns. (d) Fruiting bodies. (e) Growth on cellulose as sole carbon source. (f) Adherence to crystalline cellulose.
Results
Architecture and features of the S. cellulosum So ce56 genome
The genome sequence of S. cellulosum So ce56 was obtained by whole genome shotgun sequencing, and the assembly was validated by a complete fosmid map (Supplementary Fig. 1 online). The main features of the genome sequence are shown in Figure 2 and Table 1. At 13,033,799 base pairs, the circular chromosome is the largest bacterial genome described to date by ∼4 Mb. The S. cellulosum genome contains 9,367 predicted protein coding sequences (CDS), numbered from the initiation codon of the dnaA gene. The average GC content of the chromosome is 71.4%, consistent with the strong GC bias observed in other δ-proteobacteria. In the absence of the GC-skew inversion typically seen at the replication origin of bacterial chromosomes (Fig. 2), it was not possible to discern the location of oriC. Simultaneous analysis of the genome for GC content, dinucleotide frequency and codon usage using the PAI-IDA Program 1.1 (ref. 9) revealed a relatively uniform pattern over the first two-thirds of the chromosome (to 8.5 Mb), but nine anomalous sections in the last third of the genome (Supplementary Fig. 2 online). A majority of insertion sequences (16 of 28) occur in this region, suggesting that foreign elements may have been preferentially introduced by horizontal transfer into this section of the chromosome.

From the outer to the inner concentric circle: circle 1, genomic position in kb (the start of the dnaA gene was defined as the zero point of the chromosome); circles 2 and 3, predicted protein-coding sequences (CDS) on the forward (outer wheel) and the reverse (inner wheel) strands, colored according to the assigned COG classes (leading strand 4,725 CDS, 50.4%; lagging strand 4,642 CDS, 49.6%); circle 4, eukaryotic protein kinase-like kinases (green); circle 5, secondary metabolites biosynthetic genes (red); circle 6, GC content showing deviations from the average (71.4%); circle 7, GC skew; no clear bias toward G of the leading strand is visible.
A putative function could be assigned to 4,895 (52.2%) of the encoded proteins on the basis of manual annotation. Of the rest, 1,224 (13.1%) showed similarity to conserved hypothetical proteins in other genomes, whereas the remaining 3,248 (34.7%) had no significant similarity to predicted proteins in the public database. Thus, the number of apparently new genes in S. cellulosum So ce56 is comparable to the total coding capacity of many smaller bacterial genomes, such as those of Escherichia coli K12 (4,488 genes) and the δ-proteobacterium Desulfovibrio vulgaris (3,039 genes). Comparisons of all predicted 9,367 CDS to each other using the basic alignment search tool (BLASTP) indicate that 36% (3,402) constitute 772 families of paralogous genes (where each family has ≥2 members), which probably arose by gene duplication (Supplementary Table 1 online). The first and third largest families encode regulatory proteins: serine/threonine/tyrosine protein kinases and histidine kinases (498 members), and putative σ54-dependent regulators (80 members), respectively. The second largest family (124 members) encompasses hypothetical proteins and other proteins annotated as polymorphic GC-rich repetitive sequences that participate in cell surface interactions in mycobacteria10. The 63 members of the fourth largest paralogous family belong to the functional category of secondary metabolism.
Phylogenomic analysis of S. cellulosum So ce56
We attempted to use phylogenomic analysis to deduce functions for the ∼35% of unassigned genes in the S. cellulosum So ce56 genome. Phylogenomic maps are a three-dimensional landscape in which mountains correspond to proteins with a shared evolutionary history. As CDSs that cluster have a high likelihood of being functionally linked, such maps can be used to make functional predictions for uncharacterized CDSs on the basis of coinheritance. A phylogenomic map of S. cellulosum So ce56 was constructed by aligning the predicted S. cellulosum proteome against each of the predicted proteomes from 446 sequenced bacterial species. To permit meaningful comparisons, we retained predicted proteins in the map only if their phylogenetic profiles matched those from at least five other bacteria. The present iteration contains 5,862 CDSs, representing ∼62% of the predicted proteome. Retained proteins were clustered using a combination of force-directed placement and multidimensional scaling11 into 39 mountains, as visualized by VxInsight12 (Supplementary Fig. 3 online). The breadth and height of each mountain signify the tightness of clustering and the number of proteins, respectively. Each mountain was then assigned an overall function by consideration of the Gene Ontology (GO) categories to which the proteins within each mountain belong. The top three functional assignments for each mountain are shown in Supplementary Table 2 online. In the current version of the map, the 1,416 unassigned CDS, representing 32% of the total in S. cellulosum, are distributed among almost all of the mountains. The putative roles assigned to the majority of these proteins on the basis of the map are shown in Supplementary Table 3 online.
M. xanthus is the only other myxobacterium analyzed by phylogenomic mapping11. The latest iteration, also based on 446 sequenced bacterial genomes, contains 4,880 proteins (67% of the predicted proteome) and 34 mountains. The overall topology of the M. xanthus map is very similar to that of S. cellulosum: the majority of mountains are associated with transcription and translation, metabolism, transport, signal transduction, energy production or conversion and chemosensing. Accordingly, both maps have mountains enriched for basic cellular machinery. However, the maps also highlight the secondary metabolic potential of these strains, with the PKS and NRPS proteins divided into distinct mountains (23 and 27, respectively, in the case of S. cellulosum). Mountain 27 contains a substantial number of transcriptional regulators, including extracytoplasmic sigma factors. This observation suggests that the biosynthetic proteins also found in this mountain may be controlled by these regulators (Supplementary Fig. 3 and Supplementary Table 4 online).
Coordination of cell motility and cell-to-cell signaling are crucial behaviors performed by myxobacteria to form multicellular fruiting bodies. Notably, the M. xanthus map contains two chemosensory and motility-associated mountains, whereas only one is present in the S. cellulosum map. This difference probably reflects the alternative lifestyles of the strains: M. xanthus actively hunts prey, whereas the more sedentary S. cellulosum degrades cellulose. Indeed, M. xanthus contains 146 proteases6, compared to only 70 in S. cellulosum.
Comparison of S. cellulosum So ce56 and M. xanthus DK1622 genomes
On the basis of classical morphological studies and phylogenetic analysis of 16S rRNA, S. cellulosum and M. xanthus species have been assigned to the myxobacterial suborders Sorangiineae and Cystobacterineae5, respectively. This relatedness was expected to be reflected in overall genome architecture. However, direct comparison of the S. cellulosum So ce56 and M. xanthus genomes reveals very significant divergence in both organization and content. In addition, the S. cellulosum genome is larger than that of M. xanthus by ∼4 Mb. Reciprocal BLASTP comparison of the two genomes shows that only 2,857 of the predicted CDSs (30.5%) in the S. cellulosum So ce56 genome are homologous to predicted CDSs of M. xanthus6. Analysis of the relative ortholog positions in S. cellulosum and M. xanthus reveals the complete absence of synteny on a genome-wide scale (correlation coefficient for relative gene positions, r = −0.009; Fig. 3a). The genomic distribution of gene conservation scores between S. cellulosum and M. xanthus is shown in Supplementary Figure 4 online.

(a) Relative ORF positions of orthologs in M. xanthus versus S. cellulosum (linear regression shown, r = −0.009). (b) COG category distributions of predicted ORFs in S. cellulosum (light gray), M. xanthus (dark gray), B. bacteriovorus (black), and among orthologs that are locally syntenic between S. cellulosum and M. xanthus (white). Information storage and processing: translation, ribosomal structure and biogenesis (J), RNA processing and modification (A), transcription (K), replication, recombination and repair (L), chromatin structure and dynamics (B). Cellular processes and signaling: cell cycle control, cell division, chromosome partitioning (D), defense mechanisms (V), signal transduction mechanisms (T), cell wall/membrane/envelope biogenesis (M), cell motility (N), intracellular trafficking, secretion and vesicular transport (U), post-translational modification, protein turnover, chaperones (O), cytoskeleton (Z). Metabolism: energy production and conversion (C), carbohydrate transport and metabolism (G), amino acid transport and metabolism (E), nucleotide transport and metabolism (F), coenzyme transport and metabolism (H), lipid transport and metabolism (I), inorganic ion transport and metabolism (P), secondary metabolites biosynthesis, transport and catabolism (Q). Poorly characterized: general function prediction only (R), function unknown (S).
In contrast to the global pattern of gene organization, however, the overall distribution of gene functions is similar in both species (Fig. 3b). Thus, there may be a set of genes that is characteristic of myxobacteria. This view is supported by a three-way comparison of clusters of orthologous groups (COG) category distributions using S. cellulosum, M. xanthus and the δ-proteobacterium Bdellovibrio bacteriovorus HD100 (GenBank accession no. NC_005363), one of the closest relatives of myxobacteria. Although the COG distributions differ significantly when all pair-wise comparisons are made (χ2, goodness-of-fit test), the difference between S. cellulosum and M. xanthus (χ2 = 174) is significantly smaller than that between both myxobacterial species and B. bacteriovorus (χ2 = 3,094 for S. cellulosum versus B. bacteriovorus; χ2 = 1,108 for M. xanthus versus B. bacteriovorus). Furthermore, despite the complete absence of global synteny, there is a high degree of local synteny between S. cellulosum and M. xanthus, with 1,474 genes occurring in locally syntenic clusters. The average sequence conservation score for these clustered genes (calculated as described in ref. 13) is twice the average for all genes (0.4 versus 0.2, respectively), indicating that local synteny correlates with sequence conservation. Local gene organization is often a crucial feature of gene regulation, particularly for prokaryotic operons14. An initial analysis of syntenic genes in the COG motility category N suggests that most locally syntenic genes are located within operons based on operon predictions made for M. xanthus (http://www.cbcb.umd.edu/cgi-bin/operons/taxon_list.cgi). In addition to ribosomal genes (COG category J), which are known to be syntenic across species, other classes represented among the syntenic clusters are involved in energy production and conversion (C), inorganic ion transport and metabolism (P), and intracellular trafficking, secretion and vesicular transport (T).
The most significant difference in genome content occurs in the functional category of carbohydrate transport and metabolism (G; 5.4% of CDSs in S. cellulosum (504 genes) versus 3.7% in M. xanthus (270 genes)). The higher proportion of genes devoted to carbohydrate metabolism in S. cellulosum probably underlies its ability to use cellulose and other sugars as carbon sources (Supplementary Note online), a capability not reported for M. xanthus15. For example, S. cellulosum has the full complement of glycolytic enzymes, whereas M. xanthus lacks glucokinase16. Furthermore, although M. xanthus cannot make branched-chain amino acids, So ce56 can grow on minimal media, indicating its capacity to synthesize all amino acids and vitamins as reflected in the genome content4. The differences in amino acid metabolism also affect nitrogen metabolism in the two species; whereas M. xanthus probably obtains its nitrogen from amino acids, So ce56 appears to contain a different set of genes dedicated to providing nitrogen, including several involved in the use of inorganic nitrite and nitrate.
Although many chemosensory and developmental genes are similar (Supplementary Table 5 online), crucial developmental genes in M. xanthus (e.g., asgA, csgA, fruA, sdeK and actA) were not identified in S. cellulosum. It will thus be difficult to extrapolate insights into fruiting body formation and the regulation of morphogenesis in M. xanthus17,18 directly to S. cellulosum, as extracellular signaling appears to occur by a different mechanism in the two myxobacteria.
The fraction of paralogous genes in S. cellulosum (36%) is also considerably lower than in M. xanthus (48%), although the pattern of gene duplication in both species is similar, with preferential amplification of selected functions including regulatory proteins6 (Supplementary Table 1). The extent of noncoding regions is higher in S. cellulosum (14% versus 10%, amounting to ∼1 Mb of extra DNA in S. cellulosum), as is the average gene size (401 aa versus 376 aa), with both lengths being more characteristic of eukaryotic than prokaryotic genes19. Genome-wide analysis of direct and palindromic repeats with GenAlyzer20 reveals more repetitive sequences in S. cellulosum So ce56 (233 directly repeated elements with a minimum length of 500 bp in S. cellulosum versus 59 in M. xanthus; 1,544 palindromic repeat elements with a minimum length of 100 bp in S. cellulosum versus 231 in M. xanthus). These direct and palindromic repeats together account for ∼2% (253,701 bp) of the S. cellulosum genome.
Complex regulation in S. cellulosum So ce56
S. cellulosum contains an extensive regulatory network, including enhancer binding proteins, two-component regulatory systems, extracytoplasmic function family sigma factors, and serine/threonine/tyrosine protein kinases (eukaryotic protein kinase–like kinases, ELKs). Two-component regulatory systems respond to a broad range of extracellular and intracellular signals, and typically comprise a sensor histidine protein kinase and a response regulator protein. S. cellulosum encodes 119 response regulator proteins, 98 histidine protein kinases and 49 hybrid kinases, containing a total of 182 receiver domains and 153 output domains. Therefore, even when compensating for genome size, both S. cellulosum and M. xanthus possess far more two-component regulatory systems than all other sequenced microorganisms (Supplementary Fig. 5 online). Like M. xanthus6, S. cellulosum contains surprisingly few one-component regulators relative to other soil bacteria, suggesting that one-component regulators have been superseded by multicomponent regulators. The largest group of one-component systems is the LysR-type transcriptional regulators (26% of the 202 total).
ELKs from M. xanthus and other bacteria participate in diverse cellular functions including pathogenicity, regulation of primary and secondary metabolism, and cell differentiation21,22. In total, the S. cellulosum genome encodes 317 ELKs (6.2% of the genome; 3.2% of predicted CDSs) distributed throughout the chromosome (Fig. 2), all of which contain the 11 subdomains known to be critical for activity. Thus, S. cellulosum So ce56 devotes a larger percentage of its genome to ELKs than any other sequenced organism, including all eukaryotes (Fig. 4). In fact, the amount of DNA coding for ELKs is larger than the complete genome of Buchnera aphidicola subsp. Baizongia pistaciae (0.616 Mb), and similar to that of Mycoplasma pneumoniae M129 (0.816 Mb). Like S. cellulosum, three other prokaryotes with high numbers of ELKs, M. xanthus21 (99), the cyanobacterium Anabena sp. strain PCC 7120 (52; ref. 23), and the Gram-positive soil bacterium Streptomyces coelicolor24 (34), also produce a large variety of natural products and have complex developmental cycles. These data support the emerging view that ELKs may play an equally important role to histidine protein kinases in modulating prokaryotic behaviors22. To verify experimentally the importance of phosphorylation in gene regulation by S. cellulosum So ce56, we performed phosphoproteome analysis using two-dimensional (2D) electrophoresis coupled with matrix-assisted laser desorption ionization/tandem time-of-flight mass spectrometry (MALDI-TOF-TOF) (Fig. 5). Analysis of 53 randomly selected proteins revealed that ∼40% are phosphorylated under the conditions tested (Fig. 5 and Supplementary Table 6 online).

Only the organisms with the highest hits after adjustment of genome size are shown.

The numbers indicate a selection of the protein spots used for identification and shown to be phosphorylated. All 53 proteins identified by our analysis, including those found to be phosphorylated, are listed in Supplementary Table 6 online.
Interestingly, many of the regulatory proteins exhibit unique modular architectures comprising several regulatory, input and output domains (Supplementary Tables 7 and 8 online), suggesting that their physiological roles may be unique to S. cellulosum.
Secondary metabolism and biotechnological potential
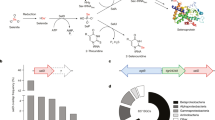
Although secondary metabolites have been used in human therapy for centuries, their functions for the producing organisms often remain unclear. However, the finding that most compounds produced by the Sorangiineae show activity against fungi and other bacteria suggests that they confer a competitive advantage in the soil environment. S. cellulosum So ce56 produces the natural products chivosazol and myxochelin7,8 (Fig. 6b). Although it shares the myxochelin pathway with the myxobacterium Stigmatella aurantiaca, the organization of the cluster in So ce56 is different8 (Supplementary Fig. 6 online).

(a) Genes putatively involved in secondary metabolism biosynthesis. Blue, genes encoding proteins with PKS domains; red, genes encoding proteins with NRPS domains; gray, other genes likely to be involved in biosynthesis. Genes encoding two type III PKS (sce2133, sce2182), two PKS-NRPS hybrid proteins (sce0345, sce6823), four proteins with PKS or NRPS domains (sce0436, sce2387, sce4888, sce8219) and three terpene cyclases (sce1440, sce6369, sce8552) have also been identified in the genome. (b) The chemical structures of known compounds isolated from S. cellulosum So ce56. The structure of flaviolin was determined after heterologous expression of the rppA gene in Pseudomonas putida. (c) Analysis of culture extracts from mutants in the etnangien cluster by HPLC reveals that etnangien is no longer produced. (d) Analysis of mutants in the etnangien cluster by bioassay using the indicator strain Micrococcus luteus. Zones of inhibition due to etnangien were observed with extracts of the wild-type strain (1), and several chivosazol-negative mutant strains (2–5), but inhibition was not observed for the etnangien mutant strain (6).
The polyketide metabolite etnangien, an inhibitor of bacterial and viral nucleic acid polymerases, has been isolated from S. cellulosum strains So ce750 and So ce1045 (ref. 25). Analysis of culture broths of So ce56 demonstrate that the strain also produces etnangien (Fig. 6). The candidate biosynthetic gene locus was readily identified in the genome, as it is the only PKS-encoding region of the appropriate size (in addition to the chivosazol cluster). We confirmed the identity of the cluster by insertional mutagenesis in a PKS gene, resulting in the abolition of etnangien production as judged independently by both high-performance liquid chromatography (HPLC)–mass spectrometry analysis and bioassay (Fig. 6c). Although the structures of many actinomycete-derived polyketides can be predicted from the domain organization of their associated PKSs, inspection of the etnangien genes reveals many deviations from this architectural colinearity. This observation further illustrates that attempts to predict structures of myxobacterial compounds based on sequence information must be performed with caution26.
Sequencing revealed several additional regions of the chromosome that encode proteins containing PKS and/or NRPS domains, whose products have not been isolated to date (Figs. 2 and 6a; the domain organization and predictions for substrate specificity are given in Supplementary Fig. 6); among them is a large gene cluster (sce8254–sce8262) spanning 44.5 kb. As none of the clusters has obvious homologs in other bacteria, the associated metabolites are likely to be novel and therefore attractive targets for genome mining. Sets of genes potentially involved in carotenoid and terpenoid biosynthesis (three terpene cyclases sce1440, sce6369 and sce8552) were also identified, giving a total of 17 unique loci involved in secondary metabolism. Thus, as with other soil bacteria6, genome sequencing has revealed that the metabolic potential of S. cellulosum So ce56 is much richer than was suggested from traditional fermentation studies. Furthermore, analysis of the M. xanthus proteome27 has shown that most of the clusters of unknown function are expressed in this strain, indicating that these cryptic gene sets are also active in S. cellulosum.
The putative carotenoid cluster contains seven genes, several of which have homologs in cartenoid gene sets found in M. xanthus and S. coelicolor, although the gene order and orientations differ among the clusters (Supplementary Fig. 6). The carotenoids produced by S. cellulosum So ce56 have not been identified to date, but comparison to known carotenoid biosynthetic pathways28 suggests that the strain should assemble phytoene, and further convert it into phytofluene, ζ-carotene and neurosporene. There also appear to be genes present for the further transformation of neurosporene to unknown products. Gene sce1440, encoding a terpene cyclase, has substantial similarity to genes in other bacteria that are involved in biosynthesis of geosmin29,30, a sesquiterpene-derived natural product that gives soil its characteristic smell. The genome also encodes an unusual L-dopa decarboxylase31 and two type III PKSs; a gene encoding one of the PKSs, sce2133, shows similarity to bacterial RppA-encoding genes (61% identity)32. Although the compound has not yet been isolated from S. cellulosum So ce56, expression of sce2133 in Pseudomonas putida resulted in biosynthesis of flaviolin33, supporting the utility of heterologous hosts for identifying the products of cryptic myxobacterial clusters and dissecting gene function. S. cellulosum So ce56 also contains 22 cytochrome P450 enzymes, which often modify specific functional groups of secondary metabolites. The sequencing also helps to reveal the basis for S. cellulosum's broad intrinsic antibiotic resistance, as the genome contains 18 efflux pumps, 11 of which are associated with the functional category of secondary metabolism.
The S. cellulosum genome encodes numerous other enzymes with biotechnological potential, including lipolytic activities, proteases, cellulases, nitrilases, amidases and hydantoinases. Lipases represent an important class of biocatalysts with applications both in industry and organic synthesis34. The genome contains nine CDSs for putative lipases, three of which (sce7029, sce4066 and sce2331) show the highest sequence homology to enzymes from higher plants. The product of sce2981 exhibits greatest similarity to proteins from Myxococcus and a second myxobacterial genus, Stigmatella, suggesting that this family of lipases may play particular roles in myxobacterial physiology. The genome also houses 51 genes potentially encoding members of the short-chain dehydrogenase/reductase superfamily of enzymes, which are used industrially for the production of chiral alcohols35. A similar number of short-chain dehydrogenase/reductase genes is present in the biotechnologically valuable strain Pseudomonas putida KT2240, suggesting that S. cellulosum could become an important source of short-chain dehydrogenase/reductase enzymes with novel properties. S. cellulosum also appears to encode a rich supply of S8 family peptidases, subtilisin-like enzymes that are widely exploited as additives in laundry detergents. Both S. cellulosum and M. xanthus harbor genes encoding at least nine putative proteases of this type, the same number found in established sources of hydrolases such as Pseudomonas aeruginosa.
S. cellulosum represents one of the rare groups of aerobic Gram negative bacteria that degrade cellulose for use as a carbon source (Fig. 1), a capability which underlies its importance in soil ecology. Hemicellulose, a component of the cellulose matrix, also serves as a substrate for S. cellulosum, and its degradation products, mannose and arabinose, are superior to glucose as carbon sources. Pathways for cellulose, hemicellulose and pectin degradation were identified in the genome (Supplementary Table 9 online). In contrast to Bacillus licheniformis, in which genes for cellulose degradation are clustered36, the corresponding genes in S. cellulosum are distributed throughout the genome. The enzymes involved in cellulose degradation comprise endoglucanases and exoglucanases, some of which incorporate carbohydrate-binding domains, consistent with previous studies of extracellular lytic enzymes produced by Sorangium strains37. A detailed analysis of the enzymes involved in polysaccharide metabolism in So ce56 is included as a Supplementary Note online.
Discussion
As the myxobacteria S. cellulosum and M. xanthus have many similar morphological and behavorial characteristics, it was assumed that they would share a common genetic architecture. However, our analysis reveals that the overall composition and organization of the two genomes are strikingly different. In M. xanthus, genomic expansion appears to have arisen largely through gene duplication6. Whereas gene duplication in S. cellulosum also contributed to genome growth, particularly within kinase families (more than 0.8 Mb), the overall number of paralogous genes is lower than in M. xanthus. We find no evidence for acquisition of plasmids or phages, whereas the smaller number of secondary metabolic gene clusters in the strain compared to M. xanthus and their distribution across the genome (Fig. 2) suggest that horizontal gene transfer did not contribute substantially to genome expansion6. Nonetheless, although we can ascribe at least one additional megabase in the S. cellulosum genome to more noncoding regions, a longer average gene length and more repetitive sequences, further investigation will be required to account for the remaining ∼3 Mb difference. Like M. xanthus, however, S. cellulosum So ce56 dedicates a substantial portion of its genome to regulatory proteins, many of which appear to have arisen from gene duplication and divergence. This observation suggests that the complex, multicellular lifestyle of the myxobacteria requires a similarly complex regulatory apparatus. Our genomic, phylogenomic and proteomic analyses further show that post-translational phosphorylation plays a particularly important role in regulation by S. cellulosum. The genus Sorangium produces ∼50% of all known myxobacterial metabolites. Sequencing of this model Sorangium therefore opens the way to understanding the molecular details of the regulatory processes governing cell differentiation and secondary metabolism in other S. cellulosum strains, as demonstrated recently for chivosazol biosynthesis38. Such information should in future facilitate increased exploitation of the metabolic potential of the more than 1,800 Sorangium strains available from the German Collection of Microorganisms and Cell Cultures, including the epothilone producer S. cellulosum So ce90.
Methods
Whole genome shotgun sequencing.
Several DNA shotgun libraries were constructed in pTZ18R, pUC18, pTOPO and pUC19 with insert sizes between 1 kb and 8 kb, and end-sequenced by the German Research Center for Biotechnology (GBF), Integrated Genomics and Qiagen. Base-calling was carried out using PHRED39. High-quality reads were defined by a minimum length of 200 base pairs, with an average quality value of ≥20. Overall, 207,009 high-quality reads were obtained, covering at least 98.87 Mb (7.6 genome equivalents).
Sequence assembly and assembly validation.
Basecalling, quality control and elimination of vector DNA from the shotgun-sequences were performed using the software package BioMake (Bielefeld University, unpublished methodology) as previously described40. Sequence assembly was performed using the PHRAP assembly tool (http://www.phrap.org/). Genome finishing was accomplished using the CONSED/AUTOFINISH software package41, supplemented by the in-house tool BACCardI42. A fosmid library was constructed to perform gap closure and assembly validation by the Max Plank Institute (MPI) for Developmental Biology using the EpiFOS Fosmid Library Production Kit (Epicentre). End-sequencing of 2880 fosmids was carried out by the MPI for Developmental Biology. Remaining gaps within the whole genome shotgun assembly were closed by IIT and GBF, by sequencing of shotgun and fosmid clones. To obtain a high quality genome sequence, we polished all regions of the consensus sequence to at least phred40 quality by primer walking; in total, 1,444 sequencing reads were added to the shotgun assembly. Repetitive elements, such as rRNA operons, were sequenced completely by primer walking on fosmid clones. To validate the assembly, we mapped fosmid end sequences onto the genome sequence by employing the BACCardI software42.
Genome analysis and annotation.
Initially, automatic gene prediction and annotation were performed using the genome annotation system GenDB 2.0 (ref. 43). All genes were then manually curated. Intergenic regions were then rechecked using the BLAST programs44 for any CDS that may have been missed by the automated annotation. Analysis of paralogous genes was performed by using BlastP, applying a threshold for the e value of 1e-30.
Genomic comparison.
The annotated genome sequence of Myxococcus xanthus (accession no. NC_008095) was imported into the genome annotation system GenDB43. Bidirectional BLASTP analyses were carried out with GenDB, using a threshold e value of 1e-05 for identifying reciprocal best blast hits between S. cellulosum and M. xanthus.
Investigation of repetitive sequences.
Direct and palindromic repeat sequences were detected using the GenAlyzer genome explorer version 0.81 beta20. The following parameters were chosen for direct and palindromic repeats, respectively: minimum size 500 bp, 100 bp; edit distance 10, 5; seed length 14, 14.
Detection of regions with atypical GC content.
Analysis for anomalous gene clusters or putative pathogenicity islands in the genome was performed using the PAI-IDA program 1.1 (http://compbio.sibsnet.org/projects/pai-ida/) based on an iterative discriminant analysis9.
Construction of the S. cellulosum phylogenomic map.
A phylogenomic map was constructed for S. cellulosum So ce56 as described by Srinivasan et al.11. Briefly, BLASTP44 was used to align each predicted protein in the S. cellulosum genome against a local database of predicted proteins from 352 sequenced bacteria, obtained from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi; accessed June 20, 2006). Results were retained for each protein match that registered a bit score >50 and an e-value <1e-05 in five or more of the sequenced genomes. A raw data matrix of bit scores was constructed with each row representing a protein and each column corresponding to a different sequenced genome. Correlations for each pair of proteins were calculated using Spearman's rank correlation. For each protein, the top 50 correlates that had the highest positive correlation scores were retained. Each protein was then assigned an (x, y) coordinate in the plane using a combination of force-directed placement and multi-dimensional scaling11,12. These proteins were then visualized as a three-dimensional topographical map using the computer program VxInsight12. Each mountain on the map represents those proteins that share similar phylogenetic history, and the height of each mountain is proportional to the density of the proteins within that area. A total of 5,647 out of 9,417 proteins (or 60%) were retained and visualized in this manner.
In addition to the visualization of each retained protein, we also corrected for paralogy. Proteins with similar sequences have a tendency to exhibit similar BLASTP scores, and hence, similar phylogenetic profiles. For each pair of proteins on the map, we extracted their corresponding sets of GenInfo Identification (GI) numbers and calculated the Jaccard coefficient of similarity (defined as A ∩ B/A ∪ B) between these two sets. We retained those protein pairs that had a Jaccard coefficient >0.50 and superimposed this network onto the map. The resulting co-location of paralogous proteins is thus visualized on the map.
GO functional annotation of each mountain.
Mountain discretization was carried out through the application of an expectation maximization algorithm based on Gaussians with spherical covariance matrices seeded with the gap statistic11. A total of 32 mountains were discretized in this manner. To assign Gene Ontology functional annotations45 to each discretized mountain, a GO database file for all proteins in the S. cellulosum genome was constructed. We used the GO::TermFinder software package46 to determine the GO functional annotation enrichment for each mountain. The top three GO functional annotations for each mountain were retained (Supplementary Table 2).
Colinearity analysis.
To derive a quantitative measure of genome wide colinearity between M. xanthus and S. cellulosum, the x and y coordinates of the orthologous pairs were used, applying an approach similar to that used in Eppinger et al.13. For each pair of neighboring ORFs (xi, xi+1) on the query genome, the position of the orthologs (yj, yj+1) on the target genomes was found. The difference Min(|yi+1−yi|, no. ORFs−|yi+1−yi|) between the positions on the target genome was calculated, taking into account the circularity of the genome. The difference values for all the ORF pairs were summed and divided by the number of ORFs. However, this average value is still dependent on the genome size, because for a larger genome, the positions of the ORFs can vary to a greater extent. To obtain a colinearity factor independent of genome size, we divided the resulting value by the value that would be expected for a random permutation of a genome of the same size. The resulting colinearity factor for the comparison of M. xanthus with S. cellulosum is 0.8742, which reflects a low-level of colinearity (1 = no colinearity, 0 = perfect colinearity)13.
Syntenic cluster analysis.
We defined a syntenic mini-cluster as at least two neighboring ORFs in M. xanthus whose orthologs in S. cellulosum are also neighboring, and a syntenic cluster as a group of at least four neighboring ORFs, the orthologs of which are also neighboring in the other species. In both cases, 'neighboring' was defined as two genes that have a maximum of two other ORFs between them. Of the 9,367 genes in S. cellulosum and the 7,338 genes in M. xanthus, respectively, 1,162 genes are located in 451 mini-clusters and 312 are located in 56 clusters.
Statistics.
Chi-square tests were performed on expected versus observed numbers of predicted ORFs in each COG category (other than V–Z, due to zero values) for all pairwise comparisons between M. xanthus, S. cellulosum and B. bacteriovorus. To test for significant differences between these three data sets, the difference in percentage of ORFs in each COG category was first calculated for each pairwise comparison and the resulting value sets were tested for significance differences in their mean values with a paired t-test.
Inactivation of the etnangien PKS.
Primers OP73 (5′-aagcttGATCCCGAAGCGATTGTAGA-3′) and OP74 (5′-ggatcCTCAGCAGGATGTCGAACAA-3′) were designed based on PKS fragments from the genome data (inserted restriction sites are indicated in lower case). A PCR product containing a PKS-encoding fragment was obtained from the chromosome of So ce56, and cloned into a pSUP102 derivative as described previously7 to generate plasmid pOPB45. Plasmid pOPB45 was introduced into So ce56 by conjugational transfer7. HPLC analysis of the mutants was performed as described previously7, using authentic etnangien as a reference standard. Bioassays for etnangien production were performed using Micrococcus luteus as an indicator strain, as described by Perlova et al. for chivosazol47.
Protein extraction and two-dimensional PAGE.
50 ml of culture of S. cellulosum So ce56 grown in M medium7 to late exponential phase was harvested, and the cells resuspended and washed with 10 ml 4 °C washing buffer (10 mM Tris hydroxymethylaminoethane (Tris) (pH 7); Sigma Aldrich), 250 mM sucrose (Roth)). After centrifugation (10 min at 4 °C, 3,220g) using an Eppendorf 5810R centrifuge, the cell pellet was stored at −20 °C. Cell lysis was performed by adding 1.5 ml 4 °C lysis buffer (5 M urea (Roth), 2 M thiourea (Roth), 10 mM Tris (pH 7), 175 mM dithiothreitol (DTT; MP Biomedical), 0.5 M etheylenediaminetetraacetic acid (Roth), 0.05% Triton X-100 (Sigma Aldrich), 0.5% vol/vol pharmalyte 311 (Amersham), protease inhibitor (Roche), phosphatase inhibitor cocktail I+II (Roche), 0.1 units RNAse/DNAse (Roth)), and incubation at 4 °C for 1 h. Cell debris was removed by centrifugation and the protein concentration was estimated using Bradford reagent (Biorad). The proteins were then separated by 2D-PAGE.
Sample application: eight IPG strips (Amersham, pH range 3−11NL (nonlinear)) were incubated for 14 h in rehydration buffer (227 μl; 8 M urea, 1% 3-[3-(cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS; Sigma Aldrich), 0.4% DTT, 0.5% vol/vol pharmalyte 3−11, 0.005% Bromophenol blue (Sigma Aldrich)) containing 250 μg protein in 113 μl lysis buffer under a layer of mineral oil. The strips were transferred to IPGphor (Amersham), and isoelectric focusing carried out for 80 kVh 24 h. The strips were then washed with each of equilibration (EQ) buffers 1 and 2 for 15 min (buffer base: 6 M urea, 2% SDS, 50 mM Tris (pH 8.8), 30% glycerol (Merck); EQ1: 10 ml buffer+100 mg dithiothreitol; EQ2: 10 ml buffer+250 mg iododacetamide (Sigma Aldrich)). Subsequently, the strips were transferred to SDS polyacrylamide gels (prepared according to the manufacturer's recommendations) and fixed with 1% agarose (Sigma Aldrich) in 2 × running buffer (10 × : 1% SDS, 250 mM Tris (pH 8.8), 1.92 M glycine (Sigma Aldrich)). The gels were inserted into an Ettan DALTtwelve (Amersham) which was filled with running buffer. Electrophoresis was performed at 2 W/gel for 1 h, followed by 15 W/gel for 6 h.
The gels were then fixed for 2 h in fixation solution (40% ethanol, 10% acetic acid (Acros), 50% Milli-Q water). Two gels were stained with silver stain (Amersham PLUSone Silver staining kit) for detection of the maximal spot number, and four with colloidal Coomassie (Sigma Aldrich); spots were then excised from the Coomassie-stained gels and analyzed by mass spectrometry. For this, the gels were washed for 2 h in Coomassie fixation solution (50% ethanol, 3% phosphoric acid (Acros), 47% Milli-Q water) and for 2 h in preincubation solution (34% methanol, 3% phosphoric acid, 17% ammonium sulfate (Sigma Aldrich), 46% Milli-Q water). 10 ml of 3.5% Coomassie solution in methanol was added per liter preincubation solution. The gels were incubated for 3−4 d and then the background was destained by washing multiple times with Milli-Q water for several hours. All silver- and Coomassie-stained gels were scanned using a gel scanner (Amersham). Images were analyzed by the software Image Master 6.5 (Amersham). The x- and y-coordinates of the individual spots were transferred to a spot-picker (Amersham) and all spots were cut out and placed into individual wells of microtiter plates.
Protein identification and neutral loss detection using mass spectrometry.
Protein spots were excised from colloidal Coomassie-stained gels, in-gel digested with trypsin, and the protein identities determined using a combination of MALDI-TOF peptide mass fingerprint (PMF) and MALDI-TOF/TOF peptide fragmentation fingerprint (PFF) analysis. Excised gel pieces were washed with ammonium hydrogen carbonate (50 μl; 50 mM; Sigma Aldrich) for 15 min, then with ammonium hydrogen carbonate/acetonitrile (1:1) for 15 min and once with acetonitrile (100 μl; Acros). The liquid was then removed, and the spots were air dried before rehydration in ammonium hydrogen carbonate (25 μl; 50 mM) containing trypsin (50 ng; Promega). Digestion was performed at 37 °C overnight and stopped by addition of 0.1% trifluoroacetic acid (5 μl; Merck). The microtiter plates were stored at −20 °C.
Peptide extracts (0.5 μl) were spotted twice onto a 384 OPTI-TOF plate and mixed with equal amounts of matrix. Two different matrices were used for cocrystallization. For protein identification by PMF and MS-MS experiments (PFF), α-cyano cinnamic acid (CCA, FLUKA) was used as a matrix. The three most abundant peptides in the PMF were analyzed by PFF. 2, 5-dihydroxybenzoic acid (DHB, FLUKA) supplemented with 1% phosphoric acid was used for identification of phosphopeptides by detection of the 'neutral loss' of the phosphate group48. CCA was dissolved to 2.5 mg ml−1 in solution (50% Milli-Q water/acetonitrile containing 0.1% trifluoroacetic acid (TFA)). To generate the DHB matrix, 2.5 mg ml−1 DHB was dissolved in solution (50% Milli-Q water/acetonitrile containing 0.1% TFA and 1% phosphoric acid). The MALDI spots, containing the samples and the matrix, were dried at 21°C and analyzed using a 4800 MALDI TOF/TOF analyzer (Applied Biosystems) between 800−4,000 Da in positive ion mode with the reflector on. Only peaks with a signal/noise ratio of ≥15 were used for protein identification and only if they showed a typical isotopic distribution. Identification of proteins in the CCA matrix was performed using MASCOT49 and the 'in-house' database including the S. cellulosum predicted protein sequences (with mass tolerance of 70 p.p.m. in MS analysis and 0.1 Da in MS-MS analysis). Phosphopeptides were identified in spectra obtained using the DHB matrix, based on a mass shift of 79.9 Da (with mass tolerance 200 p.p.m.). To avoid false-positives arising from automated phosphopeptide identification, all spectra obtained using DHB were manually validated. Any remaining questionable peaks were confirmed by measurement in negative, reflective mode50.
Accession code.
The nucleotide sequence of the S. cellulosum So ce56 chromosome was submitted to EMBL under accession number AM746676.
Note: Supplementary information is available on the Nature Biotechnology website.
Accession codes
References
Gerth, K., Pradella, S., Perlova, O., Beyer, S. & Müller, R. Myxobacteria: proficient producers of novel natural products with various biological activities – past and future biotechnological aspects with the focus on the genus Sorangium. J. Biotechnol. 106, 233–253 (2003).
Gerth, K., Bedorf, N., Höfle, G., Irschik, H. & Reichenbach, H. Epothilons A and B: antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties. J. Antibiot. (Tokyo) 49, 560–563 (1996).
Fischbach, M.A. & Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 106, 3468–3496 (2006).
Gerth, K., Perlova, O. & Müller, R. Sorangium cellulosum. in Multicellularity and Differentiation Among the Myxobacteria and Their Neighbors (eds. Kaplan, H.B. & Whitworth, D.) (ASM press, Washington DC, in the press).
Reichenbach, H. Order VIII. Myxococcales Tchan, Pochon and Prévot 1948, 398AL. in Bergey's Manual of Systematic Bacteriology (eds. Brenner, D.J., Krieg, N.R., Staley, J.T. & Garrity, G.M.) 1059–1072 (Springer, New York, 2005).
Goldman, B.S. et al. Evolution of sensory complexity recorded in a myxobacterial genome. Proc. Natl. Acad. Sci. USA 103, 15200–15205 (2006).
Pradella, S. et al. Characterisation, genome size and genetic manipulation of the myxobacterium Sorangium cellulosum So ce56. Arch. Microbiol. 178, 484–492 (2002).
Gaitatzis, N., Kunze, B. & Müller, R. Novel insights into siderophore formation in myxobacteria. ChemBioChem 6, 365–374 (2005).
Tu, Q. & Ding, D.F. Detecting pathogenicity islands and anomalous gene clusters by iterative discriminant analysis. FEMS Microbiol. Lett. 221, 269–275 (2003).
Brennan, M.J. & Delogu, G. The PE multigene family: a 'molecular mantra' for mycobacteria. Trends Microbiol. 10, 246–249 (2002).
Srinivasan, B.S. et al. Functional genome annotation through phylogenomic mapping. Nat. Biotechnol. 23, 691–698 (2005).
Davidson, G.S., Wylie, B.N. & Boyack, K.W. Cluster stability and the use of noise in interpretation of clustering. Proceedings of the IEEE Symposium on Information Visualization 2001 (INFOVIS'01), 23–30 (2007).
Eppinger, M., Baar, C., Raddatz, G., Huson, D.H. & Schuster, S.C. Comparative analysis of four Campylobacterales. Nat. Rev. Microbiol. 2, 872–885 (2004).
Lawrence, J.G. Gene organization: selection, selfishness, and serendipity. Annu. Rev. Microbiol. 57, 419–440 (2003).
Bretscher, A.P. & Kaiser, D. Nutrition of Myxococcus xanthus, a fruiting myxobacterium. J. Bacteriol. 133, 763–768 (1978).
Watson, B.F. & Dworkin, M. Comparative intermediary metabolism of vegetative cells and microcysts of Myxococcus xanthus. J. Bacteriol. 96, 1465–1473 (1968).
Youderian, P., Burke, N., White, D.J. & Hartzell, P.L. Identification of genes required for adventurous gliding motility in Myxococcus xanthus with the transposable element mariner. Mol. Microbiol. 49, 555–570 (2003).
Youderian, P. & Hartzell, P.L. Transposon insertions of magellan-4 that impair social gliding motility in Myxococcus xanthus. Genetics 172, 1397–1410 (2006).
Brocchieri, L. & Karlin, S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 33, 3390–3400 (2005).
Choudhuri, J.V., Schleiermacher, C., Kurtz, S. & Giegerich, R. GenAlyzer: interactive visualization of sequence similarities between entire genomes. Bioinformatics 20, 1964–1965 (2004).
Inouye, S., Nariya, H. & Muñoz-Dorado, J. Protein Ser/Thr kinases and phosphatases in Myxococcus xanthus. in Multicellularity and Differentiation Among the Myxobacteria and Their Neighbors (eds. Kaplan, H.B. & Whitworth, D.) (ASM Press, Washington D.C., in the press).
Kannan, N., Taylor, S.S., Zhai, Y., Venter, J.C. & Manning, G. Structural and functional diversity of the microbial kinome. PLoS Biol. 5, e17 (2007).
Wang, L., Sun, Y.P., Chen, W.L., Li, J.H. & Zhang, C.C. Genomic analysis of protein kinases, protein phosphatases and two-component regulatory systems of the cyanobacterium Anabaena sp. strain PCC 7120. FEMS Microbiol. Lett. 217, 155–165 (2002).
Petrickova, K. & Petricek, M. Eukaryotic-type protein kinases in Streptomyces coelicolor: variations on a common theme. Microbiology 149, 1609–1621 (2003).
Irschik, H. et al. Etnangien, a macrolide-polyene antibiotic from Sorangium cellulosum that inhibits nucleic acid polymerases. J. Nat. Prod. 70, 1060–1063 (2007).
Wenzel, S.C. & Müller, R. Myxobacterial natural product assembly lines: fascinating examples of curious biochemistry. Nat. Prod. Rep. (in the press).
Schley, C., Altmeyer, M.O., Swart, R., Müller, R. & Huber, C.G. Proteome analysis of Myxococcus xanthus by off-line two-dimensional chromatographic separation using monolithic poly-(styrene-divinylbenzene) columns combined with ion-trap tandem mass spectrometry. J. Proteome Res. 5, 2760–2768 (2006).
Schmidt-Dannert, C., Umeno, D. & Arnold, F.H. Molecular breeding of carotenoid biosynthetic pathways. Nat. Biotechnol. 18, 750–753 (2000).
Dickschat, J.S., Bode, H.B., Mahmud, T., Müller, R. & Schulz, S. A novel type of geosmin biosynthesis in myxobacteria. J. Org. Chem. 70, 5174–5182 (2005).
Jiang, J., He, X. & Cane, D.E. Geosmin biosynthesis: Streptomyces coelicolor germacradienol/germacrene D synthase converts farnesyl diphosphate to geosmin. J. Am. Chem. Soc. 128, 8128–8129 (2006).
Bode, H.B. & Müller, R. Possibility of bacterial recruitment of plant genes associated with the biosynthesis of secondary metabolites. Plant Physiol. 132, 1153–1161 (2003).
Ueda, K., Kim, K.M., Beppu, T. & Horinouchi, S. Overexpression of a gene cluster encoding a chalcone synthase-like protein confers redbrown pigment production in Streptomyces griseus. J. Antibiot. (Tokyo) 48, 638–646 (1995).
Gross, F. et al. Bacterial type III polyketide synthases: phylogenetic analysis and potential for the production of novel secondary metabolites by heterologous expession in pseudomonads. Arch. Microbiol. 185, 28–38 (2006).
Rosenau, F., Tommassen, J. & Jaeger, K.E. Lipase-specific foldases. ChemBioChem 5, 152–161 (2004).
Kroutil, W., Mang, H., Edegger, K. & Faber, K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr. Opin. Chem. Biol. 8, 120–126 (2004).
Rey, M.W. et al. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 5, R77 (2004).
Coucke, P. & Voets, J.P. The study of enzymatic cellulolysis of Sorangium composition. Ann. Inst. Pasteur (Paris) 115, 549–560 (1968).
Rachid, S., Gerth, K., Kochems, I. & Müller, R. Deciphering regulatory mechanisms for secondary metabolite production in the myxobacterium Sorangium cellulosum So ce56. Mol. Microbiol. 63, 1783–1796 (2007).
Ewing, B., Hillier, L., Wendl, M.C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998).
Kaiser, O. et al. Whole genome shotgun sequencing guided-by bioinformatics pipelines – an optimized approach for an established technique. J. Biotechnol. 106, 121–133 (2003).
Gordon, D., Desmarais, C. & Green, P. Automated finishing with autofinish. Genome Res. 11, 614–625 (2001).
Bartels, D. et al. BACCardI – a tool for the validation of genomic assemblies, assisting genome finishing and intergenome comparison. Bioinformatics 21, 853–859 (2005).
Meyer, F. et al. GenDB – an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31, 2187–2195 (2003).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Harris, M.A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Boyle, E.I. et al. GO:TermFinder – open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715 (2004).
Perlova, O., Gerth, K., Hans, A., Kaiser, O. & Müller, R. Identification and analysis of the chivosazol biosynthetic gene cluster from the myxobacterial model strain Sorangium cellulosum So ce56. J. Biotechnol. 121, 174–191 (2006).
Kjellstrom, S. & Jensen, O.N. Phosphoric acid as a matrix additive for MALDI MS analysis of phosphopeptides and phosphoproteins. Anal. Chem. 76, 5109–5117 (2004).
Pappin, D.J., Test, G.-Q., Test, M.-M., Hojrup, P. & Bleasby, A.J. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3, 327–332 (1993).
Tholey, A., Reed, J. & Lehmann, W.D. Electrospray tandem mass spectrometric studies of phosphopeptides and phosphopeptide analogues. J. Mass Spectrom. 34, 117–123 (1999).
Acknowledgements
We would like to thank H. Reichenbach for his support of the S. cellulosum functional genome project. This work was funded by grants from the German Federal Ministry of Education and Research (BMBF) (321066579, 031U113D, 031U213D, 0313105 and 0313805A, 0313805P) in the framework of the GenoMik and the GenoMikPlus network “Genome Research on Bacteria Relevant for Agriculture, Environment and Biotechnology.”
Author information
Authors and Affiliations
Contributions
S.S. contributed to the writing of the manuscript, data analysis, finishing and annotation of the genome; O.P. coordinated the annotation and the analysis of the data and contributed to the writing of the manuscript, the annotation and data analysis; O.K. coordinated the sequencing and finishing of the genome and analysis of the data; K.G. contributed to the annotation and analysis of the data and performed physiological characterization of the strain; A.A., E.B., C.J.B., J.V.C., S.D., B.F., L.G., C.G., F.G., Lars Jelsbak, Lotte Jelsbak, J.K., C.K., T.K., M.K., D.K., T.M., R.M.-A., M.M., F.M., S.M., S.R., C.R., F.S., F.-J.V., S.C.W., C.W. contributed to the annotation; M.O.A. contributed to the proteomic data; D.B. contributed to the assembly of the genome, annotation and data analysis; T.B. contributed to the processing of sequence reads and quality assessment; S.B. contributed to the coordination and initial planning of the project; Y.A.E. contributed to the annotation and proteomic data; A.G., contributed to the automatic and manual annotation and the coordination of software development for genome analysis; S.K. contributed to the genome data analysis; L.K., A.C.M. contributed to the gene prediction; B.L. contributed to the maintenance and improvement of GenDB web interface; J.M.-D., J.B., F.R., A.T.L., D.E.W., S.W., H.B.B. contributed to the annotation and data analysis; S.P. contributed to the BAC sequencing; G.R. contributed to the genomic comparison analysis; M.S. contributed to sequencing; S.C.S. contributed to the fosmid sequencing; G.S. contributed to phylogenomic data analysis; G.J.V. contributed to the genomic comparison analysis; K.J.W. contributed largely to the writing of the manuscript; R.D.W. contributed to phylogenomic data analysis; H.B. coordinated the sequencing; A.P. was coresponsible for the project as a whole and contributed to the writing of the manuscript; R.M. coordinated the project, contributed to the annotation and the data analysis, and was responsible for the writing of the manuscript.
Corresponding author
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6; Supplementary Tables 1–9; Supplementary Note (PDF 2941 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike license (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar license.
About this article
Cite this article
Schneiker, S., Perlova, O., Kaiser, O. et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol 25, 1281–1289 (2007). https://doi.org/10.1038/nbt1354
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt1354
This article is cited by
-
Microenvironment of Landfill-Mined Soil-Like Fractions (LMSF): Evaluating the Polymer Composting Potential Using Metagenomics and Geoenvironmental Characterization
International Journal of Environmental Research (2024)
-
Calcium promotes persistent soil organic matter by altering microbial transformation of plant litter
Nature Communications (2023)
-
The Mycobacterium tuberculosis protein O-phosphorylation landscape
Nature Microbiology (2023)
-
Comparative analysis of genome code complexity and manufacturability with engineering benchmarks
Scientific Reports (2022)
-
Complete genome sequence and identification of polyunsaturated fatty acid biosynthesis genes of the myxobacterium Minicystis rosea DSM 24000T
BMC Genomics (2021)
