
Abstract
Bacillus amyloliquefaciens FZB42 is a Gram-positive, plant-associated bacterium, which stimulates plant growth and produces secondary metabolites that suppress soil-borne plant pathogens. Its 3,918-kb genome, containing an estimated 3,693 protein-coding sequences, lacks extended phage insertions, which occur ubiquitously in the closely related Bacillus subtilis 168 genome. The B. amyloliquefaciens FZB42 genome reveals an unexpected potential to produce secondary metabolites, including the polyketides bacillaene and difficidin. More than 8.5% of the genome is devoted to synthesizing antibiotics and siderophores by pathways not involving ribosomes. Besides five gene clusters, known from B. subtilis to mediate nonribosomal synthesis of secondary metabolites, we identified four giant gene clusters absent in B. subtilis 168. The pks2 gene cluster encodes the components to synthesize the macrolactin core skeleton.
Similar content being viewed by others


Main
Bacteria that are associated with plant roots and exert beneficial effects on plant development are referred to as plant growth–promoting rhizobacteria1. They competitively colonize plant roots and can simultaneously act as biofertilizers and as antagonists (biopesticides) of recognized root pathogens, including bacteria, fungi and nematodes. Plant growth–promoting rhizobacteria, most of which are Pseudomonas and Bacillus spp., are applied to a wide range of agricultural species to enhance growth, for example, by promoting seedling emergence, plant biomass and disease control. Plant growth–promoting rhizobacteria antagonize soil pathogens by competing for resources such as iron, or by producing antibiotics or lytic enzymes. The complete genome sequence of the plant growth–promoting and biocontrol agent Pseudomonas fluorescens Pf-5 was published recently2. Although biocontrol strains of fluorescent pseudomonades have contributed greatly to the understanding of the mechanisms that are involved in phytostimulation and disease suppression, biological preparations from spore-forming Bacillus spp. are preferred because their long-term viability facilitates the development of commercial products3. Compared to plant growth–promoting Pseudomonas rhizobacteria, relatively little is known about the lifestyle of plant-associated Bacillus spp., which were originally considered as typical soil bacteria, despite their well-established advantages for beneficial action on plant growth and biocontrol4,5. Owing to obvious differences in the physiology between these representatives of Gram-negative and Gram-positive bacteria, respectively, the two species may exhibit specific mechanisms that benefit plant-microbe interactions.
The plant root–colonizing B. amyloliquefaciens strain FZB42 is a naturally occurring isolate, distinguished from the model organism Bacillus subtilis 168 by its abilities to stimulate plant growth and suppress plant pathogens6. Previous analyses of this strain revealed numerous gene clusters involved in nonribosomal synthesis of cyclic lipopeptides7 and polyketides8 with distinct antimicrobial action.
In this study, we report the complete genome sequence of B. amyloliquefaciens FZB42, and through comparison with the domesticated strain B. subtilis 168, highlight genes that may contribute to its plant-associated lifestyle.
Results
General genomic features and DNA islands
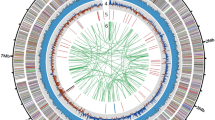
The principal features of the B. amyloliquefaciens FZB42 genome are summarized in Table 1. The circular chromosome (3,918,589 bp) is somewhat smaller than that of the closely related B. subtilis9 and Bacillus licheniformis10,11, primarily owing to the absence of prophage islands, which are abundant in the B. subtilis 168 genome. Deletions, altogether spanning 495 kb, were detected in regions equivalent to B. subtilis prophages 1 to 7, the phage SPβC2 and the skin element (Supplementary Table 1 online). The majority of the B. amyloliquefaciens FZB42 protein-encoding sequences were found conserved in the closely related B. subtilis (3,181 b.p.) and B. licheniformis (2,857 b.p.), and most of them are arranged in a collinear manner in all three strains.
Many of the 214 genes unique in B. amyloliquefaciens FZB42 are clustered in the 17 DNA islands (Supplementary Fig. 1 online, circle 3), which are defined by local deviations in the tetranucleotide usage patterns12 from the signature of the whole B. amyloliquefaciens FZB42 genome. In some of the islands, additional features, such as adjacent tRNAs, remnants of phages, transposase-like sequences and direct repeats, are indicative of horizontal gene transfer (Supplementary Table 1 online). Certain DNA islands appear to be linked with the plant-associated lifestyle of B. amyloliquefaciens FZB42. For example, the location of island 7 between 1,164 and 1,193 kb corresponds to a region occupied by prophage 4 in B. subtilis 168. The region is flanked on the left side by tRNA-Val, followed by two genes of the remnant B. subtilis prophage 4 in which direct repeats of 29 nucleotides are inserted. Several of the genes clustered in this 28,745-bp DNA island exhibit striking similarity to genes involved in extracellular arabinogalactan hydrolysis, galactose uptake by a sugar-specific phosphotransferase system IIABC and galactose catabolism (Leloir pathway) in enterococci, lactobacilli and Erwinia carotovora (Fig. 1). It can be assumed that acquisition of this molecular toolbox, comprising several elements derived from other soil- and plant-associated bacteria unrelated to bacilli, has enhanced the ability of B. amyloliquefaciens FZB42 to exploit plant-derived polysaccharides in the rhizosphere.

Gene arrangement of the 28-kb island and the G+C profile are shown on the bottom. Flanking genes, conserved in B. subtilis, are shown in green. A tRNA (Val) (blue) is found at the left border of the island. Parts of the island are distinguished by their unique GC content, deviation in oligonucleotide usage pattern and the presence of direct repeat sequences (DR, red-filled triangles). An extended gene cluster consisting of genes for extracellular arabinogalactane hydrolysis (ganA), a specific sugar-uptake system by membrane-bound PtsIIABC, phospho-β-galactosidase (LacG) and enzymes of the Leloir pathway (Gal K, E, T) is controlled by one promoter element (P), with a CcpA-binding region (cre) and a binding site for divergently arranged LacRFZB42 transcription regulator. A MarR-related transcription regulator and a membrane protein of unknown function belonging to the major facilitator superfamily are localized at the opposite flanking region. The upper part shows uptake and metabolism of the galactosides derived from extracellular arabinogalactane hydrolysis. All reactions are catalyzed by the products of the gene cluster described above and the pgm gene located downstream of LacR. The resulting product, glucose-6-phosphate, is further catabolized by central routes of carbon metabolism (glycolysis and pentose phosphate cycle).
Of the thirty genes for sensor kinases present in B. amyloliquefaciens FZB42, five encode potential intramembrane-sensing histidine kinases (IM-HK) involved in cell envelope stress13. Two of these are absent in B. subtilis 168, but display weak similarity to BceRS-BceAB and YxdJK-YxdLM (Supplementary Table 2 online). Cell-envelope stress is also sensed by a specific group of membrane-bound σ factors with extracytoplasmic function14. B. amyloliquefaciens FZB42 possesses genes for 16 σ factors, six of which are predicted to have extracytoplasmic function and five of which are shared by B. subtilis 168 and B. amyloliquefaciens FZB42. The latter lack Bs168 σY and σZ, but instead possess a novel putative extracytoplasmic function σ factor, RBAM00641, together with its cognate anti-σ factor, RBAM00642 (Supplementary Table 3 online).
B. amyloliquefaciens FZB42 can use a wide range of substrates and analysis of the genome revealed 75 putative ATP-binding cassette (ABC) transporters, 29 amino acid permeases and at least 17 phosphotransferase system sugar transporters (Supplementary Table 4 online).
Small functional RNAs (sRNAs) and riboswitches are essential for a number of cellular processes such as regulation of gene expression, tRNA processing and protein secretion15. In B. subtilis, 2% of all genes are under riboswitch control alone16. Based on phylogenetic relationships (Supplementary Fig. 2 online), the Bacillus clausii, Bacillus cereus and Bacillus halodurans genome sequences were chosen for a comparative genomics-based scan of putative sRNAs, riboswitches and other RNA elements. Applying stringent parameters, 84 hits were found in the intergenic regions of the B. amyloliquefaciens FZB42 genome. By comparison with the contents of the Rfam database, three of these were predicted to encode RNase P RNA, tmRNA and the 6S RNA. Eighteen others were annotated as riboswitches (ten binding S-adenosylmethionine, three specific for thiamine pyrophosphate, two for purines and one each for glycine, flavin mononucleotide and lysine). A detailed report can be found at http://www.cyanolab.de/prediction/bacillus_fzb42/summary.html and entries have been included in the associated GenBank file.
Genes involved in bacterium-plant interactions
The ability of B. amyloliquefaciens FZB42 to efficiently colonize surfaces of plant roots is a prerequisite for phytostimulation. Rhizosphere competence is linked to the capability to form sessile, multicellular communities (biofilms). In liquid culture without shaking, B. amyloliquefaciens FZB42 forms robust pellicles at the liquid-air interface, whereas domesticated B. subtilis strains usually form thin, fragile pellicles17. The genome of B. amyloliquefaciens FZB42 contains the complete set of genes implicated in biofilm- and fruiting body–formation in B. subtilis, including the 15-gene exopolysaccharide operon epsA-O, apparently required for producing an exopolysaccharide that holds chains of cells together in bundles18. An additional gene cluster in the B. amyloliquefaciens FZB42 genome, which probably participates in exopolysaccharide and/or lipopolysaccharide biosynthesis, has no counterpart in B. subtilis. The unique genes RBAM00750, RBAM00751 and RBAM00754 encode proteins with a collagen-related GXT structural motif, and are probably involved in surface adhesion or biofilm formation (Supplementary Table 5 online).
B. amyloliquefaciens FZB42 displays a robust swarming phenotype. Both the protein encoded by swrA and the lipopeptide surfactin, previously demonstrated for B. amyloliquefaciens FZB42 (ref. 5), are thought to be essential for swarming motility. These proteins permit colonization of surfaces and nutrient acquisition through their surface wetting and detergent properties18. Nonswarming mutants, whose surfactin biosynthesis is blocked, are also severely impaired in biofilm formation. Furthermore, an swrA gene homolog, sharing 88% identity to swrA wild-type alleles present in environmental B. subtilis isolates, was detected in the B. amyloliquefaciens FZB42 genome, together with other genes known to be necessary for swarming motility (Supplementary Table 5).
We identified 262 genes encoding proteins with putative secretion signals (Supplementary Table 6 online) recognized by signal peptidases type I (150 gene products), type II (98 gene products) and the Tat system (14 gene products). The B. amyloliquefaciens FZB42 alkaline serine protease AprE displays high similarity (98% identity) to cuticle-degrading extracellular proteases that eventually kill plant-pathogenic nematodes19. Many of the 55 proteins we have experimentally verified as constituents of the extracellular proteome of B. amyloliquefaciens FZB42 by two-dimensional (2D gel) electrophoresis and subsequent MALDI-TOF mass spectrometry (http://www.mpiib-berlin.mpg.de/2D-PAGE) possess counterparts in the secretome of B. subtilis 168 (refs. 20,21). The wide range of extracellular macromolecular depolymerases, including the arabinogalactanase discussed above, enables B. amyloliquefaciens FZB42 to grow on plant surfaces. Phytase was detected as a prominent member of the B. amyloliquefaciens FZB42 secretome and its concencentration increases in the presence of plant root exudates. By contrast, a promoter mutation prevents phytase expression in B. subtilis22. The phytate-degrading activity of B. amyloliquefaciens FZB42 could generate phytate phosphorus to nourish plants under conditions of phosphate starvation and thus promote their growth. This is consistent with the finding that the transcription of the related B. amyloliquefaciens FZB45 phytase is regulated by the PhoP/R two component system induced by phosphate starvation22.
Root exudates also stimulated upregulation of enzymes probably involved in response to oxidative stress generated in plant roots by, for example, thiol peroxidase or enzymes catabolizing compounds secreted by plant roots. Enzymes of this type include bacillopeptidase F, γ-glutamyl transpeptidase (which generates peptides, oligopeptides and amino acids) and phosphotransacetylase (which acts on organic acids) (Supplementary Table 5).
Our proteomic analysis suggests that the hook-associated flagellar proteins HAP1 and HAP2 and the HAG flagellin are differently affected by exudates secreted by plant roots. Although levels of the flgK gene product HAP1 were reduced in the presence of root exudates, expression of the fliD and hag gene products (HAP2, HAG) was upregulated (Supplementary Table 5). Flagellin proteins are thought to elicit a host basal defense against potential pathogens. It is likely that variations of the flagellins and other exposed bacterial proteins during colonization at surfaces of plant roots might enhance the ability of B. amyloliquefaciens FZB42 to tolerate unfavorable plant responses, and thereby contribute to its competence in the rhizosphere23.
It has been shown that a blend of volatile compounds—especially 3-hydroxy-2-butanone (acetoin) and 2,3-butanediol, emitted by the rhizobacteria B. subtilis and B. amyloliquefaciens—can enhance plant growth24. These volatiles have also been implicated in eliciting induced systemic resistance by Bacillus strains GB03 and INR937a24. Acetolactate synthase catalyzes the condensation of two pyruvate molecules into acetolactate, which is decarboxylated by AlsD to acetoin. B. amyloliquefaciens FZB42 harbors the genes that catalyze the 2,3-butanediol pathway (Supplementary Table 5).
Bacillus spp. are thought to enhance plant growth through synthesis of the plant growth hormones gibberellic acid and an auxin, indole-3-acetic acid (IAA), although the genetic basis for the synthesis of any plant growth–regulating compound in bacilli has yet to be reported25. We have recently shown that representatives of the B. subtilis/B. amyloliquefaciens group produce substances with IAA-like bioactivity26 and that B. amyloliquefaciens FZB42 produces reasonable amounts of IAA when fed tryptophan27. A careful examination of the whole genome sequence of B. amyloliquefaciens FZB42 revealed three candidate genes with apparent homology to genes previously reported to be involved in IAA metabolism: ysnE (encoding a protein similar to IAA acetyltransferase from Azospirillum brasilense28), dhaS (similar to indole-3-acetaldehyde dehydrogenase from Ustilago maydalis29) and yhcX (a putative nitrilase similar to nitrilase2 from Arabidopsis thaliana30) (Supplementary Table 5). YsnE and YhcX, but not DhaS, participate in IAA synthesis27.
Nine giant gene clusters are involved in biocontrol
B. amyloliquefaciens FZB42 posseses a previously unrecognized potential to synthesize bioactive secondary metabolites. The nine gene clusters that direct the synthesis of bioactive peptides and polyketides by modularly organized mega-enzymes define both nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKS) (Fig. 2). Four (bmyD, pks2, pks3 and nrs) are not found in B. subtilis 168. Except for the gene cluster encoding bacilysin synthesis, the functional activities of the remaining gene clusters depend on Sfp, an enzyme that transfers 4′-phosphopantetheine from coenzyme A to the carrier proteins of nascent peptide or polyketide chains (Table 2). Recently, we have assigned biological functions to three NRPS-encoding (bmyD, fen, srf) and two PKS-encoding (pks1, pks3) gene clusters in the genome of B. amyloliquefaciens FZB42 (refs. 7,8).

Indole-3-acetic acid (IAA) and 2,3-butanediol are shown in green. The acetolactate synthase AlsS (1) and the decarboxylase AlsD (2) catalyze the two-step conversion from pyruvate to acetoin. The genes ysnE and yhcX (3) encoding a putative tryptophan acetyl transferase and a putative nitrilase, respectively, are involved in tryptophan-dependent IAA synthesis27. Antibacterial polyketides are synthesized by membrane-anchored, polyketide-megasynthases (PKS-MS)29. Structures of bacillaene, difficidin and macrolactin are shown in brown. The lipopeptides surfactin, fengycin and bacillomycinD (red) are nonribosomally synthesized by modularly organized, giant peptide synthetases (NRPS), which are either diffusible or membrane anchored. Synthesis of polyketides and lipopeptides is dependent on functional phospho-panthetheinyl-transferase Sfp and often on the membrane protein YczE (yellow-filled circle). NRPSs are also involved in synthesis of the dipeptide bacilysin (red) and the Fe2+ siderophore bacillibactin (blue). A further gene cluster harboring NRPS-encoding genes (NRS-NRPS) was detected in the genome of FZB42, but its putative synthesis product remains to be identified.
PKS gene clusters
The bae (formerly pks1) gene cluster is involved in synthesis of bacillaene8, an inhibitor of prokaryotic protein synthesis. The structure of the bae gene cluster reflects the assembly line of bacillaene synthesis as apparent for the pksX gene cluster of B. subtilis31, with one notable deviation. The observation that the baeM gene of B. amyloliquefaciens FZB42 does not contain the superfluous last module present in the homologous pksM gene of B. subtilis suggests that this module is skipped during synthesis of the antibiotic in B. subtilis. In silico analysis of the pks2 gene cluster, renamed mln here, revealed that it could be attributed to the production of macrolactin, an inhibitor of bacterial peptide deformylase detected in both B. subtilis32 and B. amyloliquefaciens33. The domain structure of the mln gene cluster of B. amyloliquefaciens FZB42 reflects the pathway whereby the 24-membered ring lactone skeleton of macrolactin is synthesized by extension of an acetyl starter unit by 11 successive Claisen condensations with malonyl-CoA (Fig. 3). The polyketide megasynthase encoded by mlnB through mlnH comprises 11 modules, each containing at least three basic domains—a ketosynthase, a ketoreductase and an acyl carrier protein—but lacking an integrated acyl transferase (AT). In the growing family of type 1 PKS systems lacking AT activity, acyl units are transferred by a discrete and iteratively acting acyl transferase8, encoded by the mlnA gene in B. amyloliquefaciens FZB42. MlnA displays striking similarity to malonyl-CoA–specific trans-ATs and its activity should lead to the incorporation of the same extender unit in all modules lacking AT. The completed polyketide chain is released and cyclized by the thioesterase at the C terminus of the last module. Many of the modules are encoded on two separate genes; this split-module arrangement was previously reported in other PKS lacking AT, for example, those of bacilli6,31 and myxobacteria34 (Fig. 3). The chemical structure of the macrolactin ring is in accordance with the domain organization of the mln assembly line with three notable deviations. One enoyl reductase necessary for complete reduction and two dehydratase (DH) domains necessary for formation of double bonds lack modules 2, 7 and 10, respectively (Fig. 3). Deviations from the colinearity rule in PKS without AT are not unusual and the absence of dehydratase and enoyl reductase domains has been reported, for example in the difficidin gene cluster8. Given the three exceptions mentioned above, the number and the overall structure of the modules found in the mln gene cluster agree perfectly with the macrolactin assembly line. In fact, by combining mass spectrometric and ultraviolet-visible data obtained from the culture filtrates of wild-type and dif−/bae double-mutant strains with a database search, we identified four members of the macrolactin family—macrolactins A and D as well as 7-O-malonyl- and 7-O-succinyl-macrolactin—as mln product–specific metabolites. The technical details of identification of the macrolactins in B. amyloliquefaciens FZB42 are described elsewhere35. Currently, at least 17 macrolactins have been described and one of them, 7-O-malonyl macrolactin A, is efficient against Gram-positive bacterial pathogens36. The knowledge of the genetic basis of macrolactin assembly will greatly facilitate future efforts to enhance application of this interesting class of polyketides.

AT activity is delivered by the trans-acting mlnA gene product. Enzyme functions necessary but not detectable inside the corresponding modules are indicated by red filled circles. The thiolation domains (T, ACP) predicted to be inactive are not colored.
Besides the substantial similarity in their domain structure8, the three pks gene clusters present in B. amyloliquefaciens FZB42 share specific features such as iteratively used separate AT domains, unusual split modules at the interface of two separate synthases and the skipping of superfluous modules and domains. As the three pks gene clusters displayed no apparent deviation in GC content and oligonucleotide usage pattern, it seems unlikely that they were acquired by recent events of horizontal gene transfer. Instead, they probably evolved from an ancient pks gene cluster by several duplicative events. A recent report37 describes the existence of a membrane-bound, organelle-like protein complex responsible for bacillaene synthesis in B. subtilis. It is likely that similar superstructures also exist in the three PKS assembly lines encoded by B. amyloliquefaciens FZB42. Moreover, we propose that the membrane-spanning protein YczE, shown to be essential for synthesis of the three polyketides and the lipopeptide bacillomycin D, may anchor the mega-synthases at the membrane (Table 2 and Fig. 3).
Peptide antibiotics and siderophores
In addition to the previously identified gene clusters involved in nonribosomal synthesis of the lipopeptides surfactin, bacillomycin D and fengycin, B. amyloliquefaciens FZB42 also harbors the bac and dhb gene clusters responsible for synthesis of the antibacterial dipeptide bacilysin38 and the iron-siderophore bacillibactin in B. subtilis39. Both compounds were expressed in B. amyloliquefaciens FZB42 (Table 2). The 15-kb nrs gene cluster, which probably directs nonribosomal synthesis of a hybrid comprising a cysteine-containing peptide and a polyketide (Supplementary Fig. 3 online), is located at the genomic island 12 within a region with substantially lower GC content and variation in oligonucleotide usage. It seems to be the only NRPS/PKS gene cluster acquired by horizontal gene transfer in B. amyloliquefaciens FZB42. The nrs gene cluster is preceded by a SigA-dependent promoter, generating a transcript that was detected by RT-PCR. Although the product of the nrs gene cluster cannot be assigned yet, it may act as a siderophore and enhance the ability of B. amyloliquefaciens FZB42 to scavenge iron from the rhizosphere. Under conditions of iron starvation, growth of a double mutant with deletions in both of the bacillibactin and nrs gene clusters is more hampered than that of a mutant deficient in bacillibactin alone (X.H.C., unpublished results).
In total, we estimate that B. amyloliquefaciens FZB42 dedicates 344 kb to the synthesis of secondary metabolites used to cope with competing microorganisms in the plant rhizosphere (Table 2). As this bacterium colonizes plant roots, it inhibits growth of phytopathogenic bacterial or fungal competitors either by depriving them of the essential iron (e.g., through the action of bacillibactin and, possibly, the nrs-encoded peptide) or by directly inhibiting their growth (e.g., through antimicrobial lipopeptides and polyketides).
Genetic amenability
B. amyloliquefaciens FZB42 is naturally competent and amenable to genetic transformation using a modified one-step protocol, originally developed for B. subtilis 168 (ref. 40). This property was used to generate a set of mutants impaired in many distinct functions, such as production of extracellular enzymes6, secondary metabolites7,8, biofilm formation, alternative σ factors and plant-growth promotion27, by a gene-replacement strategy involving homologous recombination. B. amyloliquefaciens FZB42 exhibited its maximal competence somewhat earlier than B. subtilis, during late exponential growth27. Not surprisingly, the B. amyloliquefaciens FZB42 genome harbors the complete set of genes necessary for development of genetic competence (Supplementary Table 7 online). The majority of competence genes are highly homologous to their counterparts in B. subtilis 168, but the genes that control the competence quorum-sensing system of B. amyloliquefaciens FZB42 (comQ, comX, comP) exhibit low similarity to the respective genes of B. subtilis 168 (36%, 31% and 55%, respectively). This genetic variability correlates with specificity in the quorum-sensing response, so that each pheromone is sensed only by its strain-specific cognate receptor.
Discussion
Analysis of its 16S rRNA sequence indicates that B. amyloliquefaciens FZB42 is closely related, but not identical, to the B. amyloliquefaciens type strain DSMZ7 (ref. 6). A phylogenetic tree constructed from the tetranucleotide usage patterns of genomes of previously sequenced Bacillus strains confirmed that B. amyloliquefaciens FZB42 represents a separate branch, clearly distant from B. subtilis 168 (Supplementary Fig. 2). The gyrA and cheA gene sequences were used to resolve its taxonomic position more precisely; both genes have efficiently resolved closely related taxa of the B. subtilis group40. The resulting phylogenetic trees (Supplementary Fig. 4 online) revealed that B. amyloliquefaciens FZB42 is rather close to strains recently introduced as endophytic or plant-associated ecomorphs of B. amyloliquefaciens41. Essentially all Bacillus strains that are currently commercialized for their plant growth–promoting and biocontrol activities belong to this ecotype. Moreover, nearly all of these strains produced a wide spectrum of polyketides, including bacillaene, difficidin and macrolactin (Supplementary Fig. 4). This suggests that this plant growth–promoting bacterium can be considered a paradigm for plant-associated bacteria related to the B. amyloliquefaciens type strain DSMZ7. The observation that as much as 8.5% of the genome appears to be devoted to antibiotic production is remarkable, especially in the light of recent estimations that not more than 4–5% of an average Bacillus genome is devoted to antibiotic production42. By comparison, Streptomyces avermitilis, which is well known for producing a broad range of antibiotics, devotes 6.4% of its genome secondary metabolite production43. Surprisingly, the genome of B. amyloliquefaciens FZB42 lacks known biosynthetic gene clusters of ribosomally synthesized antibiotics (lantibiotics), which are common in B. subtilis strains, even though it encompasses the regulatory and immunity genes directed against the lantibiotics mersacidin and subtilin (Supplementary Fig. 1; outmost circle). Nevertheless, B. amyloliquefaciens FZB42 seems to produce other conventionally synthesized lantibiotics or toxins, as it was recently shown that a sigW mutant strain of B. subtilis is extremely sensitive to culture fluids derived both from B. amyloliquefaciens FZB42 and its sfp derivative44.
The complete genome sequence of B. amyloliquefaciens FZB42, along with its amenability to genetic manipulation, should facilitate exploitation of FZB42's hitherto unappreciated potential to produce secondary metabolites for developing agrobiotechnological agents with predictable features. The genome should be equally valuable in revealing the complex interactions between Gram-positive rhizo-bacteria and plants and in studying biofilm formation and other biological processes that have been lost or attenuated during the intensive use of B. subtilis 168 as an experimental system over the past six decades45.
Methods
Strains, genome sequencing, assembly, annotation.
B. amyloliquefaciens FZB42 was deposited as 10A6 in Bacillus Genetic Stock Center, BGSC. A set of mutant strains is also available from BGSC. Characteristics of regulatory mutants and the mutants used in this study for verifying bacilysin, bacillibactin and macrolactin production are compiled in Supplementary Table 8 online. Genomic DNA prepared from B. amyloliquefaciens FZB42 was used to produce whole shotgun libraries. For the libraries, fragments of 1.5 to 3.0 kbp were separated by gel electrophoresis after mechanical shearing (Nebulizer; Invitrogen), end-repaired and cloned using vectors pTZ19R (Amersham) or pCR2.1-TOPO (TOPO TA Cloning Kit for Sequencing; Invitrogen). More than 20,000 plasmid DNAs were isolated using two BioRobots8000 (Qiagen). A total of 40,000 sequence reactions were automatically analyzed on ABI PRISM models 377-96 and 3730XL (Applied Biosystems) or MegaBace1000 and MegaBace4000 sequencers (GE Healthcare). The 40,000 generated sequences were assembled into contigs using the Phrap assembly tool (http://www.phrap.org/). Primer-walking on plasmids, as well as PCR-based techniques were used to close remaining gaps and to solve misassembled regions caused by the high degree of repetitive sequences. All manual editing steps were performed using the GAP4 software package v4.5 and v4.6 (ref. 46). Prediction of protein-encoding sequences and open reading frames (ORFs) was initially accomplished with YACOP47 using the ORF-finding programs Glimmer, Critica and Z-curve. All ORFs have been manually curated. Initial annotation was done by a close comparison to B. subtilis 168 using Subtilist48 as reference. All predictions were verified and modified manually by comparing the protein sequences with the public databases SwissProt, GenBank, ProDom, COG and Prosite using the annotation software GeneSOAP49.
Prediction of sRNA, riboswitches and genomic islands.
A comparative method similar to that used to predict sRNAs in cyanobacteria50 was applied. After the extraction of intergenic regions and homology detection by BLASTN, homologous sequences were clustered. The resulting (multiple) sequence alignments were scored using RNAz51. Homologies to known sRNAs stored in the Rfam database were identified by BLAST. The gene islands were identified by the specific genetic repertoire and by alterations in relative abundance of tetranucleotides12. Variations in oligonucleotide usage patterns were calculated as described previously12.
Proteomics.
Cells were grown under shaking (210 r.p.m.) at 24 °C for 14 h in medium 1C consisting of 0.7% tryptone, 0.3% peptone, 0.1% glucose, 0.5% NaCl and 10% soil extract. When appropriate, root exudates prepared from maize seedlings were added to the culture (250 mg/l). Preparation of the extracellular protein fraction, 2D-gel electrophoresis and protein identification by in-gel digestion with trypsin, extraction of peptides in the Ettan Spot Handling Workstation (Amersham) and determination of peptide masses in the Proteomics Analyzer 4700 (Applied Biosystems) were done as recently described52. The 2D-gel images were analyzed using the PD-Quest (Bio-Rad) software.
Mass spectrometry.
Polyketides were identified by mass-spectrometric analysis, as described previously8. High-performance liquid chromatography (HPLC)-electrospray ionization (ESI) mass spectrometry (MS) was performed from aliquots of acetonitrile-water extracts of the culture filtrates of the Bacillus strains. Before HPLC-ESI-MS was done, the extracts were desalted by solid-phase extraction. Every sample was measured in two different modes, negative and positive mode, and mass spectra were acquired in the range of m/z 300–800.
Accession number.
GenBank CP000560.
Note: Supplementary information is available on the Nature Biotechnology website.
Accession codes
References
Kloepper, J.W., Leong, J., Teintze, M. & Schroth, M.N. Enhancing plant growth by siderophores produced by plant growth-promoting rhizobacteria. Nature 286, 885–886 (1980).
Paulsen, I.T. et al. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat. Biotechnol. 23, 873–878 (2005).
Haas, D. & Defago, G. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3, 307–319 (2005).
Kloepper, J.W., Ryu, C.-M. & Zhang, S. Induced systemic resistance and promotion of plant growth by Bacillus spp. Phytopathology 94, 1259–1266 (2004).
Compant, S. et al. Use of plant-growth promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl. Environ. Microbiol. 71, 4951–4959 (2005).
Idriss, E.E.S. et al. Extracellular phytase activity of Bacillus amyloliquefaciens FZB45 contributes to its plant-growth-promoting effect. Microbiology 148, 2097–2109 (2002).
Koumoutsi, A. et al. Structural and functional characterization of gene clusters directing nonribosomal synthesis of bioactive cyclic lipopeptides in Bacillus amyloliquefaciens strain FZB42. J. Bacteriol. 186, 1084–1096 (2004).
Chen, X.-H. et al. Structural and functional characterization of three polyketide synthase gene clusters in Bacillus amyloliquefaciens FZB42. J. Bacteriol. 188, 4024–4036 (2006).
Kunst, F. et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390, 249–256 (1997).
Veith, B. et al. The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J. Mol. Microbiol. Biotechnol. 7, 204–211 (2004).
Rey, M.W. et al. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biology 5, 1–12/R77 (2004).
Reva, O.N. & Tümmler, B. Differentation of regions with atypical oligonucleotide composition in bacterial genomes. BMC Bioinformatics 6, 251 [online] doi:10.1186/1471–2105–6-251 (2005).
Mascher, T. Intramembrane-sensing histidine kinases: a new family of cell-envelope stress sensors in Firmicutes bacteria. FEMS Microbiol. Lett. 264, 133–144 (2006).
Helmann, J.D. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 46, 47–110 (2002).
Storz, G. An expanding universe of noncoding RNAs. Science 296, 1260–1263 (2002).
Mandal, M. et al. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell 113, 577–586 (2003).
Chu, F., Kearns, D.B., Branda, S.S., Kolter, R. & Losick, R. Targets of the master regulator of biofilm formation in B. subtilis. Mol. Microbiol. 59, 1216–1228 (2006).
Kearns, D.B., Chu, F., Rudner, R. & Losick, R. A master regulator for biofilm formation by Bacillus subtilis. Mol. Microbiol. 55, 739–749 (2005).
Niu, Q. et al. Functional identification of the gene bace16 from nematophagous bacterium Bacillus nematocida. Appl. Microbiol. Biotechnol. 75, 141–148 (2007).
Tjalsma, H. et al. Proteomics of protein secretion in Bacillus subtilis: separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 68, 207–233 (2004).
Antelmann, H. et al. A proteomic view on genome based signal peptide predictions. Genome Res. 11, 1484–1502 (2001).
Makarewicz, O., Dubrac, S., Msadek, T. & Borriss, R. Dual role of the PhoP∼P response regulator: Bacillus amyloliquefaciens FZB45 phytase gene transcription is directed by positive and negative interactions with the phyC promoter. J. Bacteriol. 188, 6953–6965 (2006).
Abramovitch, R.B., Anderson, J.C. & Martin, G.B. Bacterial elicitation and evasion of plant innate immunity. Nat. Rev. Mol. Cell Biol. 7, 601–611 (2006).
Ryu, C.-M. et al. Bacterial volatiles promote growth in Arabidopsis. Proc. Natl. Acad. Sci. USA 100, 4927–4932 (2003).
Wipat, A. & Harwood, C.R. The Bacillus subtilis genome sequence: the molecular blueprint of a soil bacterium. FEMS Microbiol. Ecol. 28, 1–9 (1999).
Idris, E.E.S., Bochow, H., Ross, H. & Borriss, R. Use of Bacillus subtilis as biocontrol agent. 6. Phytohormone-like action of culture filtrates prepared from plant-growth promoting Bacillus amyloliquefaciens FZB24, FZB42, FZB45 and Bacillus subtilis FZB37. J. Plant Disease Protection 111, 583–597 (2004).
Idris, E.E.S., Iglesias, D., Talon, M. & Borriss, R. Tryptophan dependent production of indole-3-acetic acid (IAA) affects level of plant growth promotion by Bacillus amyloliquefaciens FZB42. Mol. Plant Microbe Interact. 20, 619–626 (2007).
Zimmer, W., Aparicio, C. & Elmerich, C. Relationship between tryptophane biosynthesis and indole-3-acetic acid production in Azospirillum: identification and sequencing of a trpGDC cluster. Mol. Gen. Genet. 229, 41–51 (1991).
Basse, C.W., Lottspeich, F., Steglich, W. & Kahmann, R. Two potential indole-3-acetaldehyde dehydrogenases in the phytopathogenic fungus Ustilago maydis. Eur. J. Biochem. 2242, 648–665 (1996).
Hillebrand, H., Bartling, D. & Weiler, E.W. Structural analysis of the nit2/nit1/nit3 gene cluster encoding nitrilases, enzymes catalyzing the terminal activation step in indole-acetic acid biosynthesis in Arabidopsis thaliana. Plant Mol. Biol. 36, 89–99 (1998).
Butcher, R.A. et al. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 104, 1506–1509 (2007).
Yoo, J.-S. et al. Macrolactin N, a new peptide deformylase inhibitor produced by Bacillus subtilis. Bioorg. Med. Chem. Lett. 16, 4889–4892 (2006).
Lee, S.-J. et al. Isolation and characterization of antimicrobial substance macrolactin A produced from Bacillus amyloliquefaciens CHO104 isolated from soil. J. Microbiol. Biotechnol. 14, 525–531 (2004).
Perlova, O. et al. Identification and analysis of the chivosazol biosynthetic gene cluster from the myxobacterial model strain Sorangium cellulosum So ce56. J. Biotechnol. 121, 174–191 (2006).
Schneider, K. et al. Macrolactin is the polyketide biosynthesis product of the pks2 cluster of Bacillus amyloliquefaciens FZB42. J. Nat. Prod. (in the press).
Romero-Tabarez, M. 7-O-malonyl macrolactin A, a new macrolactin antibiotic from Bacillus subtilis active against methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci and a small-colony variant of Burkholderia cepacia. Antimicrob. Agents Chemother. 50, 1701–1709 (2006).
Straight, P.D. et al. A singular enzymatic megacomplex from Bacillus subtilis. Proc. Natl. Acad. Sci. USA 104, 305–310 (2006).
Steinborn, G., Hajirezaei, M.-R. & Hofemeister, J. bac genes for recombinant bacilysin and anticapsin production in Bacilllus host strains. Arch. Microbiol. 183, 71–79 (2005).
Miethke, M. et al. Ferri-bacillibactin uptake and hydrolysis in Bacillus subtilis. Mol. Microbiol. 61, 1413–1427 (2006).
Kunst, F. & Rapoport, G. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J. Bacteriol. 177, 2403–2407 (1995).
Reva, O.N., Dixelius, C., Meijer, J. & Priest, F.G. Taxonomic characterization and plant colonizing abilities of some bacteria related to Bacillus amyloliquefaciens and Bacillus subtilis. FEMS Microbiol. Ecol. 48, 249–259 (2004).
Stein, T. Bacillus subtilis antibiotics: structure, syntheses and specific functions. Mol. Microbiol. 56, 845–857 (2005).
Omura, S. et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. USA 98, 12215–12220 (2001).
Butcher, B.G. & Helmann, J.D. Identification of Bacillus subtilis sigma-dependent genes that provide intrinsic resistance to antimicrobial compounds produced by Bacilli. Mol. Microbiol. 60, 760–782 (2006).
Burkholder, P.R. & Giles, N.H. Induced biochemical mutations in Bacillus subtilis. Am. J. Bot. 34, 345–348 (1947).
Staden, R. The Staden sequence analysis package. Mol. Biotechnol. 5, 233–241 (1996).
Tech, M. & Merkl, R. YACOP: Enhanced gene prediction obtained by a combination of existing methods. In Silico Biol. 3, 441–451 (2003).
Moszer, I. et al. SubtiList: the reference database for the Bacillus subtilis genome. Nucleic Acids Res. 30, 62–65 (2002).
Pohlmann, A. et al. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24, 1257–1262 (2006).
Axmann, I.M. et al. Identification of cyanobacterial non-coding RNAs by comparative genome analysis. Genome Biol. 6, R73 (2005).
Washietl, S., Hofacker, I.L. & Stadler, P.F. Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. USA 102, 2454–2459 (2005).
Voigt, B. et al. The extracellular proteome of Bacillus licheniformis grown in different media and under different nutrient starvation conditions. Proteomics 6, 268–281 (2006).
Acknowledgements
This project was supported by funds of the competence network Genome Research on Bacteria (GenoMik, GenoMikPlus), financed by the German Ministry for Education and Research (R.B. and G.G.) and by grants from the Lower Saxony Ministry of Science and Culture (G.G.). Steffen Porwollik (Sidney Kimmel Cancer Center) is thanked for valuable hints to improve the manuscript. Stefanie Rauschke and Ulrike Leppert are thanked for their contributions to the secretome analysis. Support given by Uwe Völker (Greifswald University) in the initial stage of our proteomic experiments is gratefully acknowledged. Rainer Cramm, and Anne Pohlmann (Humboldt University) are thanked for introducing software GeneSOAP. Yueqiu He (Yunnnan Agricultural University), Xuewen Gao (Nanjing Agricultural University), Joseph Kloepper (Auburn University) and Brian McSpadden Gardener (Ohio State University) are thanked for kindly providing Bacillus strains with plant growth promoting activities.
Author information
Authors and Affiliations
Contributions
R.B. and G.G. designed research and wrote the manuscript; X.H.C. did the main part of the experimental and annotation work including contig assembling; H.L. and A.S. performed sequencing and data management; H.J. was involved in strain isolation and characterization of its plant growth properties; X.H.C., A.K., R. Scholz, K.S., J.V. and R. Süssmuth investigated secondary metabolites in FZB42; A.E., B. Voigt and P.R.J. analyzed the extracellular proteome; I.H., B.M., B. Voss, W.R.H. and O.R. predicted sRNA, riboswitches and genomic islands.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–4 and Supplementary Tables 1–8 (PDF 688 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike license (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar license.
About this article
Cite this article
Chen, X., Koumoutsi, A., Scholz, R. et al. Comparative analysis of the complete genome sequence of the plant growth–promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol 25, 1007–1014 (2007). https://doi.org/10.1038/nbt1325
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt1325
This article is cited by
-
Host genotype, soil composition, and geo-climatic factors shape the fonio seed microbiome
Microbiome (2024)
-
Rhizosphere inoculation of PGPR strain Bacillus cereus BC56 enhances salt tolerance of cucumber
Plant Growth Regulation (2024)
-
A novel biocontrol agent Bacillus velezensis K01 for management of gray mold caused by Botrytis cinerea
AMB Express (2023)
-
Biocontrol and molecular characterization of Bacillus velezensis D against tobacco bacterial wilt
Phytopathology Research (2023)
-
Genomic and metabolomic insights into the antimicrobial compounds and plant growth-promoting potential of Bacillus velezensis Q-426
BMC Genomics (2023)
