
Abstract
Chickpea (Cicer arietinum) is the second most widely grown legume crop after soybean, accounting for a substantial proportion of human dietary nitrogen intake and playing a crucial role in food security in developing countries. We report the ∼738-Mb draft whole genome shotgun sequence of CDC Frontier, a kabuli chickpea variety, which contains an estimated 28,269 genes. Resequencing and analysis of 90 cultivated and wild genotypes from ten countries identifies targets of both breeding-associated genetic sweeps and breeding-associated balancing selection. Candidate genes for disease resistance and agronomic traits are highlighted, including traits that distinguish the two main market classes of cultivated chickpea—desi and kabuli. These data comprise a resource for chickpea improvement through molecular breeding and provide insights into both genome diversity and domestication.
Similar content being viewed by others

Main
The staple crop chickpea (Cicer arietinum) (2n = 2x = 16) is the world's second most widely grown legume. Its cultivation is of particular importance to food security in the developing world where, owing to its capacity for symbiotic nitrogen fixation, chickpea seeds are a primary source of human dietary protein1. Chickpea is a member of the Papilionoid subfamily of legumes, a clade that contains essentially all of the important legume crops. Within this subfamily, chickpea is most closely related to crops such as alfalfa (Medicago sativa), clover (Trifolium spp.), pea (Pisum sativum), lentil (Lens culinaris), and the model legumes barrel medic (Medicago truncatula) and Lotus japonicus. Soybean (Glycine max) and its allied species are more distant relations of chickpea. Originating in southeast Turkey and Syria, chickpea was one of the founder crops of modern agriculture2,3. There are two main types of chickpeas: small-seeded desi and larger-seeded kabuli. Consumption of desi is restricted primarily to the Middle East and Southeast Asia, whereas kabuli is a popular and valuable global commodity.
In common with many other widely grown crops, chickpea has a narrow genetic base that has resulted from domestication. Recent breeding efforts over the past 60 years have been restricted to the limited introduction of diverse germplasm4. In much of the world, chickpea is cultivated in semi-arid environments and on soils of poor agricultural quality, which, combined with its susceptibility to drought and debilitating fungal diseases, have restricted yields to <1 ton/ha, which is considerably below the theoretical potential. Genetic improvement, either by traditional or molecular methods, has been hampered by the limited genomic resources coupled with narrow genetic diversity in the elite gene pool4.
Here we report the draft whole genome shotgun (WGS) sequence of the genotype CDC Frontier, a Canadian kabuli chickpea variety (Supplementary Fig. 1). This inbred line is widely cultivated and is resistant to several important fungal diseases, including Ascochyta blight, and insects like pod borer5. To understand the genetic history among chickpea accessions, we resequenced 29 elite varieties from both desi and kabuli genotypes grown around the world, and we conducted genotyping by sequencing of 61 Cicer accessions from ten countries (Supplementary Table 1).
Genome assembly and annotation
We obtained 153.01 Gb of sequence data, representing 207.32× genome coverage, by Illumina sequencing of 11 genomic libraries with insert sizes ranging from 180 bp to 20 kb (Supplementary Table 2). After filtering, 87.65 Gb of high-quality sequence data were assembled into 544.73 Mb of genomic sequence scaffolds with 50% of all bases in scaffolds larger than 645.3 kb (N50) and a maximum of 6.17 Mb (Table 1 and Supplementary Table 3). Based on k-mer statistics, the chickpea genome is estimated to be 738.09 Mb in size (Supplementary Table 4 and Supplementary Fig. 2), which indicates that 73.8% of the genome is captured in scaffolds. The remaining 36.3% (nonassembled genome) is enriched for repetitive sequences, as suggested both by the increased read depth in repeat-containing regions compared to nonrepeat regions (161-fold versus 74-fold) and a fourfold lower k-mer diversity in the nonassembled fraction compared to the nonrepetitive assembled fraction.
An improved assembly, that spans 532.29 Mb (N50 = 39.99 Mb) and contains 7,163 scaffolds, was produced with the aid of 46,270 repeat-masked paired bacterial artificial chromosome (BAC) end sequences http://cicar.comparative-legumes.org/gb2/gbrowse/Ca1.0/. We anchored 65.23% of this assembly to eight genetic linkage groups using 1,292 previously published genetic markers6,7. We used the combined data to identify eight pseudomolecules, Ca1 to Ca8 (Fig. 1 and Supplementary Table 5). The placement of 93.4% of these scaffolds was verified using 5,953 polymorphic restriction site–associated DNA (RAD) single-nucleotide polymorphism (SNP) markers that were analyzed in two segregating recombinant inbred line populations. Among the mapped scaffolds/contigs, 75% contained a minimum of 3 SNPs (an average of 15 SNPs/scaffold), which enabled validation of scaffold structure based on the coherence of genotype calls. Using this approach, we identified a low proportion of chimeric scaffolds (1.7% of total scaffolds) that contained 4.6 Mbp of misassembled sequence. These chimeric scaffolds were split and the erroneous portion of scaffold sequences were removed from the pseudomolecule models. We also included in the pseudomolecules 18 scaffolds that contained 4.6 Mb of sequence (0.8% of the assembly) that lack genetic support, but for which the M. truncatula genome predicts precise locations based on conserved synteny. These synteny-based placements are hypothetical regions within the pseudomolecules that will be updated as additional genetic data become available for chickpea, or if the M. truncatula genome assembly is modified. The RAD genotyping data were sufficient to orient 75% of scaffolds. We used comparisons to M. truncatula to presumptively orient the remaining 25% of scaffolds.

Pseudomolecules (A), gene density (red, B); repeat density (blue, C); retrotransposon density (violet, D); transposon density (magenta, E); average pair-wise nucleotide diversity (theta pi) across 17 desi chickpea varieties (green, F), 12 kabuli chickpea varieties (orange, G) and 29 leading chickpea varieties (black, H).
Tandem Repeat Finder was used to identify 127,377 regions of tandem repeats in the assembly. We found that 84.9% of repeats occur in tracts of <1 kb (average 300 bp), whereas analysis of gap-spanning clones revealed that 0.8% of repeat regions are predicted to be from tracts of 10–103 kb. We could not assemble 29,018 repetitive regions (32.77 Mb in total) owing to low sequence complexity, and in these cases the occurrence of repeats was masked by the insertion of NNs within the pseudomolecules (Supplementary Table 6).
Using a combination of ab initio modeling, and homology-based searches with gene sets from six plant species, including legumes, and the CaTA transcript sequences8, we predicted a nonredundant set of 28,269 gene models, with average transcript and coding sequence sizes of 3,055 bp and 1,166 bp, respectively (Supplementary Table 7). Most of these genes have homology with gene models in TrEMBL9 (89.58%) and Interpro10 (70.03%) databases. Functions were tentatively assigned to 25,365 (89.73%) of genes with 2,904 genes (10.27%) unannotated (Supplementary Table 8). As expected, gene density increases toward the ends of the pseudomolecules (Fig. 1). For nonprotein coding genes, we predict 684 tRNA, 478 rRNA, 420 microRNA (miRNA) and 647 small nuclear RNA (snRNA) genes in the genome assembly (Supplementary Table 9).
To assess the proportion of the gene space captured in this draft genome assembly, we mapped a 454/Roche transcriptome data set (>500 bp read length), produced from the same CDC Frontier line and comprising 60,802 reads, to the genome assembly. On the basis of this analysis, we estimate gene coverage to be 90.8% (Supplementary Table 10). Analysis of the draft genome assembly for core eukaryotic genes11 reveals homologs for >98% of conserved genes in the assembly (Supplementary Table 11). To evaluate the conservation of chickpea gene models in other plant species, we used BLASTP to query the chickpea proteome against the proteomes of A. thaliana, M. truncatula, G. max, Cajanus cajan and L. japonicus. Using this analysis, proteins predicted for chickpea were most similar to those from M. truncatula (89.7% of predicted chickpea proteins were similar to M. truncatula proteins) and least similar to those from A. thaliana (79.2% had similarity with A. thaliana proteins) (Supplementary Table 12). We also observed five instances in which organelle genome segments of >10 kb had been integrated into chickpea pseudomolecules, consistent with findings in both plant and animal genomes12.
Genome organization and evolution
Approximately half (49.41%) of the chickpea genome is composed of transposable elements and unclassified repeats (Fig. 1), which is comparable to other sequenced legumes: M. truncatula (30.5%)13, pigeonpea (C. cajan, 51.6%)14 and soybean (59%)15. Long-terminal repeat (LTR) retrotransposons are the most abundant transposable element class, and cover >45% of the total nuclear genome (Fig. 1 and Table 2).
Centromere regions are composed of microsatellites that are arranged as tandem repeats. The most abundant tandem repeats in the genome are 163-bp, 100-bp and 74-bp unit repeats, and account for 18%, 30% and 13% of identified tandem repeats, respectively. The 163-bp and 100-bp repeats are similar to the previously identified chickpea microsatellites, CaSat1 and CaSat2, respectively, whereas the 74-bp repeat is homologous to CaRep2, a dispersed highly repetitive element16. The 74-bp tandem repeats were organized 'head to tail' with multiple copies within a previously identified LTR16 that we conclude was misannotated. The fluorescence in situ hybridization (FISH) experiments with oligonucleotide probes for the 100-bp tandem repeat revealed centromeric and pericentromeric distribution on pro-metaphase chromosomes in agreement with previous reports16,17. The 74-bp tandem repeats are distributed along all chromosomes but excluded from regions containing the 100-bp tandem repeat (Supplementary Fig. 3).
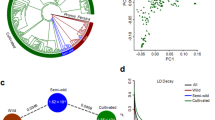
Analysis of the genome sequence for segmental duplications provided evidence for 110 syntenic blocks that ranged in size from 5 to 62 gene pairs (Supplementary Table 13). The rates of synonymous substitution per synonymous site (Ks) in these blocks indicate a divergence time of 58 million years (Myr) ago, consistent with the genome duplication event that occurred at the base of the Papilionoideae18. In this family, the galegoid (M. truncatula, L. japonicus and chickpea) and millettioid (soybean, pigeonpea) clades separated ∼54 Myr ago19. Genome analysis of the galegoid species using genetic distance–transversion rates at fourfold degenerate sites (4DTV) revealed that chickpea diverged from L. japonicus ∼20–30 Myr ago and from M. truncatula ∼10–20 Myr ago (Fig. 2).

(a) Age distribution of 4DTV for genes from three legume species (chickpea, M. truncatula and L. japonicus) genomes. (b) Synteny blocks shared between chickpea and other sequenced legume genomes, including M. truncatula, L. japonicus, soybean and pigeonpea. (c) Shared and unique gene families in legume species chickpea, M. truncatula, L. japonicus, soybean, pigeonpea; in millettioid and galegoids, and in legumes, A. thaliana and grape. (d) Phylogenetic tree of seven species.
Examination of synteny with other legume and selected nonlegume dicot genomes revealed extensive conservation between chickpea and six other plant species (Supplementary Table 13), with 87–90% of the chickpea assembly showing evidence of conservation with one or more of these six genomes. The largest number of extended (>10 kb) conserved syntenic blocks was observed for M. truncatula, whereas synteny with L. japonicus was considerably more fragmented (Fig. 2). Among legumes, soybean had the highest number of synteny blocks, reflecting its recent polyploid ancestry15, whereas the fractured colinearity with pigeonpea likely reflects the incomplete status of the pigeonpea genome assembly14.
Reciprocal pair-wise comparisons20 of the 28,269 chickpea gene models with 230,161 gene models from four sequenced legumes (M. truncatula13, L. japonicus21, pigeonpea14, soybean15) and two nonlegume species (A. thaliana22 and grape23) identified 15,441 orthologous groups (Supplementary Table 14). On the one hand, 5,940 of these orthologous groups contain only a single chickpea gene, suggestive of simple orthology (Supplementary Table 15 and Supplementary Fig. 4). On the other hand, 4,468 chickpea genes occur in species-specific groups, with identifiable chickpea paralogs but lacking genes from other species; such groups might arise from structural rearrangements that obscure simple orthology (e.g., nonallelic recombination or gene conversion) followed by duplication, as occurs among nucleotide-binding site leucine-rich repeat (NBS-LRR) disease resistance genes.
Adjusting for 2,871 genes that were not classified by OrthoMCL20, a minimum of 69% of predicted chickpea genes have a history of duplication after the divergence of the legumes from A. thaliana and grape. This same time interval includes the whole genome duplication event at the base of the Papilionoideae so the chickpea genome has been shaped by a combination of gene loss and duplication. Interestingly, several thousand genes from each of the seven species analyzed could not be placed into orthologous groups. This might be due to heterogeneity in gene prediction in the seven species (Supplementary Table 15), but it might also reflect lineage-specific evolution.
Comparisons at higher taxonomic levels (Fig. 2) revealed 16,098 orthologous groups conserved between any two galegoid species (M. truncatula, L. japonicus and chickpea), 15,503 orthologous groups conserved between millettioid species (pigeonpea and soybean), and 16,380 orthologous groups derived from the galegoid-millettioid split near the base of the Papilionoideae. Similarly, 10,667 orthologous groups are common to legumes, A. thaliana and grape. This catalog of homologous relationships provides an important foundation for comparative biology and functional inference in chickpea, as well as other species, because genes with simple orthologous relationships often exhibit conserved functions, whereas genes duplicated recently relative to speciation often underlie functional diversification.
Disease resistance genes
Among the largest gene families in plants are the NBS-LRR genes, which confer resistance to a broad range of plant pests and pathogens. The chickpea genome assembly contains 187 disease resistance gene homologs (RGHs), of which 153 are anchored in pseudomolecules. These numbers are considerably less than those observed in other legume species using comparable methods (e.g., M. truncatula, 764; soybean, 506; and pigeonpea, 406).
To explore the possibility that the low number of chickpea RGHs results from their disproportionate representation in unassembled regions of the genome, we compared nucleotide binding site (NBS) domains from CaGA v1.0 (the version 1.0 of C. arietinum Genome Assembly) with 132 NSB domains derived from PCR amplification of the C. arietinum genome. Phylogenetic analysis of these sequences, including M. truncatula RGHs to root sequence divergence, reveal no evidence of missing RGH loci in the genome assembly, further supporting the completeness of the assembly. Whereas 74 CaGA v1.0 RGHs reside in clades lacking PCR-derived RGHs, in no case is there a PCR-derived NBS domain that does not reside in close proximity to a representative from CaGA v1.0 (Supplementary Figs. 5 and 6). Furthermore, C. arietinum and M. truncatula RGHs are evenly distributed across the pseudomolecules, indicating that if there has been RGH gene loss, it is likely a gradual process rather than an acute loss of one or more major sequence clades.
Polymorphisms for chickpea breeding and genetics
Simple sequence repeat (SSR) and SNP markers are valuable tools for molecular breeding. The chickpea genome assembly contained 81,845 SSRs, of which 48,298 SSRs were suitable for PCR primer design for use as genetic markers (Supplementary Tables 16 and 17). SNP discovery using 1,073 million Illumina transcript reads and 15 million 454-sequencer transcript reads generated in an earlier study8 from four desi genotypes of chickpea (ICC 506, ICC 1882, ICC 4958, ICCC 37) and one accession (PI 489777) of the progenitor species C. reticulatum identified a total of 76,084 SNPs in 15,526 genes (Supplementary Table 18 and Supplementary Fig. 7). Of these gene-associated SNPs, 27,117 were present in the cultivated accessions, with a maximum of 22,505 between CDC Frontier and any one of the four cultivated desi genotypes. By contrast 54,178 SNPs were identified in the comparison of CDC Frontier with C. reticulatum PI 489777, most of which (49,957) were monomorphic among the four cultivated accessions. This result is consistent with the basal nature of C. reticulatum and a reduction of diversity in the cultivated gene pool. Although a portion of the cultivated SNPs may represent novel variation in cultivated germplasm, this situation can only be properly assessed through a more extensive survey of wild genotypes.
Genetic diversity among cultivated varieties and germplasm
With the objective of understanding genetic diversity in chickpea, we used whole genome resequencing (WGRS) of 29 (17 desi and 12 kabuli) chickpea breeding lines and released varieties collected from the leading chickpea-growing countries. WGRS yielded 204.52 Gb of high-quality sequence data with an average coverage of 9.5×, from which we calculated the average pairwise nucleotide diversity within population (θπ), Watterson's estimator of segregating sites (θw) and Tajima's D, commonly used metrics of genetic diversity. Although the sample size was small, diversity in the desi group was slightly higher than the kabuli group across all pseudomolecules except Ca4 (Supplementary Table 19). These results are consistent with the fact that kabuli is defined primarily on the basis of traits derived after domestication including large and light colored seeds, so kabuli varieties have likely undergone a more recent, secondary bottleneck.
Plotting diversity metrics in sliding windows across the genome (Figs. 1 and 3) reveals high θπ and θw, which are usually associated with repeat-rich, gene-poor genome intervals. We noted several intervals of Tajima's D > 2 or D < −2, which are consistent with either recent balancing selection for diverse allele content (D > 2) or selective sweeps and/or purifying selection (D < −2). Regions of elevated Tajima's D encompass 4.8% of the genome, yet contain 12% of the anchored NBS-LRR disease resistance genes. NBS-LRR genes are known to be targets of diversifying selection24; however, the identity of possible pathogen targets in chickpea remains uncertain. Among the 0.7% of the genome with Tajima's D < −2 is a region of Ca4 that contains a tandem array of three co-orthologs of the MATE family transporter TT12. The M. truncatula ortholog of TT12 functions in condensed tannin formation, which results in pigmented seeds25. This signature of purifying selection was evident in dark-colored desi but not light-colored kabuli genotypes, consistent with ongoing purifying selection for seed color. Interestingly, Ca4 is the most differentiated between kabuli and desi types and Ca4 also contains most of the mapped traits that distinguish kabuli from desi genotypes26. Establishing precise genetic correlations and testing the biological significance of the underlying genes represent key opportunities for chickpea breeding and biotechnology.

Diversity metrics, presented as average pair-wise nucleotide diversity (current [θπ] and historical [θw]) and Tajima's D, are shown across all eight pseudomolecules in 29 elite (17 desi and 12 kabuli) chickpea varieties. #, six regions with increased Tajima's D and high FST are present on pseudomolecules Ca2, Ca3 and Ca4. +, pseudomolecule Ca4 has a region with reduced Tajima's D (–2.32) that conatins 11 genes including 3MATE transporter TT12 orthologs. *, NBS-LRR disease resistance genes are associated with regions of elevated Tajima's D on five pesudomolecules namely Ca1, Ca2, Ca4, Ca7 and Ca8.
To gain a genome-wide view of genetic structure, we sequenced an additional 61 genotypes using restriction site–associated DNA (RAD) protocols, which produced an additional 34.77 Gb of data. Combined with the 29 genotypes used for WGRS, the total set of 90 Cicer accessions includes 60 improved chickpea lines, 25 landraces, 4 accessions of C. reticulatum and 1 accession of C. echinospermum (Supplementary Fig. 8). Within this set we identified 4.4 million variants (SNPs and INDELs) (Supplementary Table 20). Principal component analysis (PCA) reflects limited genetic diversity in two distinct groups of cultivated genotypes that are mixtures of desi and kabuli types (Fig. 4b, principal component 1), and a more diverse spread of wild genotypes. Analyses based either on pair-wise dissimilarity using neighbor joining (NJ) or allele frequencies using structure27 (Fig. 4) also revealed several distinct groups. Again, these major groups do not reflect the traditionally held separation of desi and kabuli genotypes, and this observation holds true for both advanced breeding lines and landraces. Group I contains all 5 wild-species genotypes and 8 landraces (6 desi and 2 kabuli), group II includes 31 cultivars and/or breeding lines and 17 landraces (12 desi and 5 kabuli) and group III contains all 29 elite varieties (19 desi and 10 kabuli) (Fig. 4c). Pedigree analysis reveals three genotypes (ICCV 96029, ICCV 94954 and ICCV 96970) that share ICCC 42 as a common parent but that were placed into two different groups (ICCV 96029 in group II and ICCV 94954 and ICCV 96970 in group III). This situation could have arisen from diverse nonrecurrent parents that have been used by breeders in developing these genotypes. Important targets for chickpea breeding are disease resistance to Fusarium oxysporum fsp ciceri and drought tolerance. Among a total of 17 Fusarium-resistant lines, 16 were assigned to group II, whereas three drought-tolerant genotypes namely ICC 4918, ICC 8261 and ICC 4958 were also assigned to group II. These observations suggest recurrent use by breeders of common lines to breed these valuable traits. More generally, analysis with the Structure program27 reveals numerous admixed genotypes, which highlights the mixed use of desi and kabuli genotypes in the breeding of both types of varieties. It seems that breeding has obscured the true genetic history of the desi and kabuli varieties.

(a) Structure analysis of 29 elite varieties (19 desi and 10 kabuli) based on whole-genome resequencing data. (b) Principle component analysis (PCA) of 90 chickpea genotypes including 29 elite varieties and 61 germplasm lines using 1.96 million SNPs as datapoints. (c) Neighbor-joining (NJ) tree analysis of 90 genotypes based on 1.96 million SNPs.
Impact of breeding on genetic diversity
During breeding, phenotypically and agronomically important traits are selected to develop superior varieties with improved crop productivity. As a result, genetic diversity is lost through fixation and genetic sweeps, and through breeders' increased dependence on smaller sets of superior genotypes, creating successive bottlenecks. Excluding the 5 wild species accessions, RAD-based SNP data were available for 25 landraces and 31 breeding lines or elite cultivars. After filtering for missing data, we used 4,696 high-quality segregating sites to assess genetic differentiation (that is, fixation index, FST) between landraces and cultivars, compared using FDIST28. Although the data were sparse, we identified 6 genomic regions of 50–200 kb characterized by >5 segregating sites from multiple RAD tags, each of which were in the top 5% of FST values. In all cases, these genome intervals correspond to regions identified as having Tajima's D values >2. Together these genome regions comprise 122 genes that are candidates for selection during modern breeding efforts, including a large set of 54 genes on Ca3 that contains a homolog of the flowering time gene CONSTANS. Furthermore, a functional flowering time quantitative trait locus is roughly mapped to the same location on Ca3 as this 54-gene set and breeding-based manipulation of flowering time has been a crucial factor in adapting elite chickpea germplasm to different agro-climatic zones.
Discussion
This draft whole genome sequence of chickpea (CDC Frontier, a kabuli chickpea variety) adds to the genomic resources available for legume research. The Papilionoideae subfamily now has the draft or complete genome sequences of two model species (M. truncatula13 and L. japonicus21) and three crop legume species (chickpea, soybean15 and pigeonpea14). The availability of these genome sequences should facilitate de novo assembly of the genomes of other important but less-studied galegoid legume crops such as pea (Pisum sativum), lentil (Lens culinaris) and faba bean (Vicia faba).
In addition to identifying SSRs and SNPs based on genome scanning and RNA-seq analysis, our analysis of 90 genomes reveals numerous additional chickpea genome polymorphisms including both SNPs and INDELs. These resources will assist genomics-based breeding approaches such as genotyping by sequencing, genome-wide association studies and genomic selection. Furthermore, population structure, diversity and phylogenetic analyses not only document the mixed use of desi and kabuli genotypes in breeding, but also serve to identify regions (and candidate genes) across the genome that might have been greatly affected by selection during domestication and/or breeding. Combined with knowledge of germplasm diversity and candidate gene regions, the analyses presented here should accelerate future breeding of elite cultivars. This will eventually move us closer to the goal of improving the livelihood and productivity of chickpea farmers worldwide, with particular emphasis on the resource-poor, marginal environments of sub-Saharan Africa and Southeast Asia.
Methods
Whole-genome shotgun sequencing and assembly.
Whole-genome shotgun sequencing was performed with the HiSeq 2000 Sequencing System. A total of 11 paired-end sequencing libraries were constructed with insert sizes of ∼170, 500, 800, 2,000, 5,000, 10,000 and 20,000 bp. In total, 153.01 Gb data were generated of paired-ends, ranging from 49 to 100 bp (Supplementary Table 2). After stringent filtering and correction steps, only 87.65 Gb data were used for de novo genome assembly (Supplementary Table 2).
The genome size was estimated using the total length of sequence reads divided by sequencing depth, as described14. SOAPdenovo was used for assembly, scaffold construction and gap closure, as described14. Sequences of genetic markers6,7 were placed on genomic scaffold assemblies using BLASTN from the BLAST+ package29, or using e-PCR30 where only flanking primers were available. For the BLAST searches, the top match was accepted, where the E-value was <1e-25 and percent identity was >85. For e-PCR, the best-scoring match was accepted, allowing up to three mismatches and one gap per primer. Provisional pseudomolecules were assembled based on marker order, and refined locally based on synteny with the Medicago truncatula genome13. Synteny was computed using promer from the MUMmer 3 package (v. 3.23)31, with the promer–mum option for “maximum unique matches” between pseudomolecule pairs.
Assessing genome assembly and gene space.
The genome assembly was checked for microbial contamination by searching against databases of bacterial genomes and fungal genomes using Megablast (E-value < 1e-5). To check for contamination with organellar DNA, the C. arietinum chloroplast (NC_011163.1) and L. japonicus mitochondrion (NC_016743.2) genomes were screened against the chickpea genome assembly using BLAT32 (default parameters). To check the completeness of the assembly, we mapped a transcriptome assembly comprising 48,668 transcriptome assembly contigs assembled from 139,241 Sanger ESTs, 7.12 million 454/FLX and 134.95 million Illumina transcript reads, to the genome assembly using BLAT32 at various sequence homology and coverage parameters (Supplementary Table 6). Core eukaryotic genes identified by CEGMA v.2.3 (ref. 11) were also mapped to the genome assembly by BLAT32 to predict exome coverage. CEGMA data were downloaded from http://korflab.ucdavis.edu/datasets/cegma/#SCT6.
Repeat annotation.
There are two main types of repeats in the genome: tandem and interspersed. Tandem Repeats Finder33 was applied to the genome assembly, filtering for >3 copies and >60-bp consensus length. Tandem repeat abundance was estimated using counts of all unique 25-mer sequences in the genome. 25-mer sequences occurring >3,000 times were used for BLAST searches to identify abundant repeats. FISH with 100-bp and 74-bp tandem repeat probes was undertaken to study repeat distribution in the genome (Supplementary Fig. 3).
Transposable elements in the genome assembly were identified using a combination of de novo and homology-based approaches. Three de novo software programs with default parameters were used, with a minimum repeat length of 50 bp (LTR_Finder v 1.03 (ref. 34), PILER-DF v1.0 (ref. 35) and RepeatScout v 1.05 (ref. 36) to build a chickpea repeat database. We then used RepeatMasker v 3.2.7 (ref. 37) to identify repeats using both the chickpea repeat database and Repbase38. Additionally, RepeatProteinMask (http://repeatmasker.org/, v 3.2.2) was used to search the protein database in Repbase against the genome to identify repeat-related proteins. Identified repeats were classified into known repeat classes using standard methods14,15,36.
Gene prediction and function analysis.
We used three approaches for gene prediction: homology-based (H), de novo (P) and transcript sequences-based (C). These results were integrated by GLEAN39, filtered multiple times and then checked manually (Supplementary Table 6). This resulted in a gene set (GD-set) comprising 28,256 genes. Additionally, we identified a set of 453 core genes that are supposed to be highly conserved in all eukaryotes, using CEGMA11. Based on this analysis, a set of 13 genes out of 453 core genes did not align with any gene defined in the initial GD-set, but were in the genome sequence, so were added to the GD-set. The final “Official Gene Set” (OGSv1.0) set contains 28,269 genes.
Gene functions were assigned according to the best match of the alignments using BLASTP (E-value: 1e-5) to the SwissProt and TrEMBL databases9 (release-2012_03, http://www.uniprot.org/downloads). InterProScan v4.7 (ref. 10) determined motifs and domains of genes against protein databases including Pfam, PRINTS, PROSITE, ProDom, SMART and PANTHER. Gene Ontology IDs for each gene were obtained from the corresponding InterPro entry. All genes were aligned against KEGG (KEGG_release58) proteins using BLASTP (E-value: 1e-5), and the pathway in which the gene might be involved was derived from the matching genes in KEGG. The tRNA genes were predicted by tRNAscan-s.e.m. v1.23 (ref. 40) with eukaryote parameters. Aligning the rRNA template sequences from plants (e.g., Arabidopsis and rice) using BLASTN with E-value 1e-5 identified the rRNA fragments. The miRNA and snRNA genes were predicted by INFERNAL v0.81 (ref. 41) software against the Rfam database (release 9.1).
Analysis of orthologous genes.
All the predicted protein sequences from chickpea, Medicago13, Lotus21, soybean15 and pigeonpea14, together with two out-group species (Arabidopsis22 and grape23), were analyzed using OrthoMCL20 to circumscribe sets of orthologous genes. In a first step, species-by-species as well as within species BLASTP (E-value: 1e-5) was performed to identify reciprocal best hit pairs between species (putative orthologs), as well as sets of genes more closely related within than between species (sets of co-orthologs, also known as in-paralogs). This reciprocal best hit matrix served as the basis for ortholog definition using OrthoMCL20 (inflation [I] parameter = 1.5). Orthologous groups were then organized into species-specific and higher taxonomic level groups by requiring that at least one sequence from each clade under comparison be present in the intersecting set. Sets of single-copy orthologs with representation in all species were selected and fourfold degenerate sites of these genes were used to construct a phylogenetic tree across nine species using MRBAYES42.
Identification and phylogenetic analysis of NBS-LRR genes.
A diverse set of 1,120 protein sequences from the highly conserved NBS domain was built from a panel of cloned legume NBS sequences (B.D.R., unpublished data), as well as published NBS domains from Mt1.0 and Poplar43,44. These sequences were used as a TBLASTN query against CaGA v1.0 to identify all NBS genomic regions.
Nucleotide sequences were translated into amino acids, imported into the CIPRES Science Gateway45 and aligned using the E-INS-i search strategy in MAFFT46. Alignments were converted into relaxed phylip format. Phylogenetic trees were generated with RAxML47 with the JTT substitution matrix, a Gamma distribution (+G), empirical base frequencies (+F) and automatic bootstrap criteria (Maximum Likelihood). Models were evaluated in MEGA5 (ref. 48). Medicago NBS domains were included as an outgroup.
Synteny analysis.
Synteny blocks between the genomes of chickpea and other legumes were computed by SyMAP49,50. Genomic sequences were first aligned using promer/MUMmer31. Raw anchors resulting from MUMmer were clustered into (putative) gene anchors, filtered using a reciprocal top-2 filter and used as input to the synteny algorithm49. The algorithm constructs maximal-scoring anchor chains based on a given gap penalty, and also searches a range of gap penalties to generate the longest chains subject to several quality criteria, which are based on the Pearson correlation coefficient applied to the anchors in the chain as well as the anchors in its bounding box. The chains are not required to be entirely colinear and may incorporate local inversions relative to the overall chain orientation.
Identification of SSRs and SNPs.
MIcroSAtellite51 was used to mine SSRs in the chickpea genome, and used for primer design14 (Supplementary Tables 15 and 16). For identification of SNPs based on transcript sequence data, high-quality transcript reads were mapped to the genome assembly using TopHat, allowing two mismatches. SAMtool v 0.18 (ref. 52) with default parameters was used to call SNPs (Supplementary Table 17).
Resequencing of genotypes.
WGRS was used for 29 elite varieties and RAD-sequencing was used for 61 genotypes (Supplementary Table 1). For WGRS, separately indexed sequencing libraries were prepared for each genotype using Illumina TruSeq library kits (Illumina) and were pooled together for 100 bp pair-end sequencing using established v3 chemistry methodologies on single lanes of an Illumina HiSeq 2000 flow-cell (Illumina). RAD-sequencing, with the standard protocol53 was used for ApekI-digested DNAs of 48 genotypes and on HindIII-digested DNAs of 24 genotypes on the Illumina HiSeq 2000 system. Subsequently, RAD-sequence data for 61 nonredundant genotypes (from ApeKI and HindIII) were compiled. The resulting sequence data were processed using Casava pipeline (Casava v1.8).
Genetic diversity analysis.
Sequence diversity analysis, including PCA, dendrogram, and diversity parameters θπ and θw, were measured as described54. FST values for 31 elite cultivars and 25 landraces were calculated by using population branch statistics55 and these values were compared using FDIST28. This approach has been found to be effective in identifying recent artificial selection considering the very short divergence time between landraces and elite cultivars55.
Accession code.
Genome sequence assembly and annotation data, NCBI Genome: PRJNA175619.
Accession codes
References
Jukanti, A.K., Gaur, P.M., Gowda, C.L. & Chibbar, R.N. Nutritional quality and health benefits of chickpea (Cicer arietinum L.): a review. Br. J. Nutr. 108, S11–S26 (2012).
van der Maesen, L.J.G. Origin, history and taxonomy of chickpea in The Chickpea (eds. Saxena, M.C. & Singh, K.B.) 11–34 (C.A.B. International, 1987).
Zohary, D. & Hopf, M. Domestication of pulses in the old world: legumes were companions of wheat and barley when agriculture began in the Near East. Science 182, 887–894 (1973).
Varshney, R.K., Thudi, M., May, G.D. & Jackson, S.A. Legume genomics and breeding. Plant Breed. Rev. 33, 257–304 (2010).
Warkentin, T. et al. CDC Frontier kabuli chickpea. Can. J. Plant Sci. 85, 909–910 (2005).
Hiremath, P.J. et al. Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol. J. 10, 716–732 (2012).
Thudi, M. et al. Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS ONE 6, e27275 (2011).
Hiremath, P.J. et al. Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol. J. 9, 922–931 (2011).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Zdobnov, E.M. & Apweiler, R. InterProScan–an integration platform for the signature recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Timmis, J.N. et al. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135 (2004).
Young, N.D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).
Varshney, R.K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89 (2012).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010).
Staginnus, C., Winter, P., Desel, C., Schmidt, T. & Kahl, G. Molecular structure and chromosomal localization of major repetitive DNA families in the chickpea (Cicer arietinum L.) genome. Plant Mol. Biol. 39, 1037–1050 (1999).
Zatloukalova, P. et al. Integration of genetic and physical maps of the chickpea (Cicer arietinum L.) genome using flow-sorted chromosomes. Chromosome Res. 19, 729–739 (2011).
Cannon, S.B. et al. Polyploidy did not predate the evolution of nodulation in all legumes. PLoS ONE 5, e11630 (2010).
Lavin, M., Herendeen, P.S. & Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 54, 575–594 (2005).
Li, L., Stoeckert, C.J. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Sato, S. et al. Genome structure of the legume, Lotus japonicus. DNA Res. 15, 227–239 (2008).
The Arabidopsis genome initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Velasco, R . et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2, e1326 (2007).
McDowell, J.M. et al. Intragenic recombination and diversifying selection contribute to the evolution of downy mildew resistance at the RPP8 locus of Arabidopsis. Plant Cell 10, 1861–1874 (1998).
Zhao, J. & Dixon, R.A. MATE transporters facilitate vacuolar uptake of epicatechin 3′-O-glucoside for proanthocyanidin biosynthesis in Medicago truncatula and Arabidopsis. Plant Cell 21, 2323–2340 (2009).
Millan, T. et al. A consensus genetic map of chickpea (Cicer atietinum L.) based on 10 mapping populations. Euphytica 175, 175–189 (2010).
Pritchard, J.K., Stephens, P. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Beaumont, M.A. & Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B 263, 1619–1626 (1996).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Schuler, G.D. Sequence mapping by electronic PCR. Genome Res. 7, 541–550 (1997).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Kent, W.J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Edgar, R.C. & Myers, E.W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Prot. Bioinform. 25, 4.10.1–4.10.14 (2009).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Elsik, C.G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E.P., Kolbe, D.L. & Eddy, S.R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Huelsenbeck, J.P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Ameline-Torregrosa, C. et al. Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 146, 5–21 (2008).
Kohler, A. et al. Genome-wide identification of NBS resistance genes in Populus trichocarpa. Plant Mol. Biol. 66, 619–636 (2008).
Miller, M.A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees (Gateway Computing Environments Workshop, 14 November 2010; IEEEXplore, 2010).
Katoh, K. & Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900 (2010).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Soderlund, C., Nelson, W., Shoemaker, A. & Paterson, A. SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 16, 1159–1168 (2006).
Soderlund, C., Bomhoff, M. & Nelson, W. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39, e68 (2011).
Thiel, T., Michalek, W., Varshney, R.K. & Graner, A. Exploiting EST databases for the development and characterization of gene derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422 (2003).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Baird, N.A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3, e3376 (2008).
Xu, X. et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 30, 1005–1011 (2012).
Xia, Q. et al. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 326, 433–436 (2009).
Acknowledgements
We would like to thank CGIAR Generation Challenge Programme (Theme Leader Discretionary grant to R.K.V., G7009.06 to D.R.C. and R.K.V.); US National Science Foundation (DBI 0605251 to D.R.C., IOS-0965531 to D.R.C. and R.K.V., and DBI 0822258 to S.A.J.); Rural Development Administration of the Republic of Korea (Woo Jang Choon Project No. PJ906910 to D.R.C.); Indo-German Science Technology Centre (R.K.V., G.K. and P.W.); BGI, Shenzhen Key Laboratory of Transomics Biotechnologies (CXB201108250096A); Grains Research Development Corporation (UWA00149 to K.S.B.), Australian Research Council (DP0985953, LP110100200 to D.E.); Saskatchewan Pulse Growers (B.T. and A.G.S.); Saskatchewan Agriculture Development Fund (to A.G.S.); University of Cordoba and National Institute for Agricultural and Food Research and Technology (INIA) (RTA2010-00059 to T.M.); National Research Initiative of US Department of Agriculture's National Institute of Food and Agriculture (#0214643 to M.-C.L.); Ministry of Education, Youth and Sports of the Czech Republic and the European Regional Development Fund (ED0007/01/01 to J.D. and J.A.C.); Indian Council of Agricultural Research (ICAR) and ICRISAT for financial support to undertake parts of research presented in this study. Thanks are due to K. Hobson of Pulse Breeding Australia (PBA) for providing seeds of some genotypes used in this study. We would also like to thank W.D. Dar (director general, ICRISAT), D.A. Hoisington (deputy director general–research, ICRISAT) and J.-M. Ribaut (director, CGIAR GCP) for their helpful advice and assistance, wherever required, during the course of the study.
Author information
Authors and Affiliations
Contributions
R.K.V., C. Song, R.K.S., S.A., S.Y., A.G.S., J.B., B.T., T.M., L.D.R., R.V.P., P.W., N.C.-G., J.A.C., M.T., M.-C.L., K.K.G., J.D., X.X., G.Z., G.K., K.B.S., J.W. and D.R.C. contributed to generation of genome sequence, transcriptome sequence, BAC-end sequencing, genetic mapping and physical mapping data; B.T., T.M., P.M.G., H.D.U., C.L.L.G., N.P.S., J.R., N.N., K.C.B., J.G. and S.K.D. contributed genetic material, C. Song, S.A., S.Y., A.G.S., S.C., X.Z., J.B., Y.W., C.X. and W.H. worked on genome assembly; C. Song, S.Y., R.K.S., A.I., S.Z. and W.N., contributed to genome annotation and gene function analysis; R.K.V., C. Song, S.A., S.Y., B.D.R., W.N., C. Sodurland, S.Z., J.K.H., M.-C.L., J.L., J.D., X.X., D.E., G.Z., G.K., K.B.S., S.A.J., J.W. and D.R.C. worked on genome analysis and comparative genomics; R.K.V., R.K.S., S.Y., B.T., T.M., Y.W., A.D.F., P.M.G., C.X., A.K.B., W.H., P.W., S.Z., H.D.U., C.L.L.G., N.P.S., J.L., J.R., N.N., K.C.B., G.K., J.G., K.B.S., S.K.D., J.W. and D.R.C. worked on germplasm diversity analysis; and R.K.V., D.R.C. and J.W., together with S.A.J., K.B.S., S.K.D., G.Z., T.M., B.T., D.E., S.C., A.G.S., R.K.S., S.A. and C. Song wrote and finalized the manuscript; R.K.V. and D.R.C. conceived and directed the project.
Corresponding authors
Ethics declarations
Competing interests
P.W. is employed by GenXPro.
Supplementary information
Supplementary Text and Figures
Supplementary Tables 1–20 and Supplementary Figures 1–8 (PDF 631 kb)
Supplementary Table 1
Details on 90 Cicer accessions used for re-sequencing (29 cultivars) and RAD genotyping (61 accessions including cultivars/breeding lines, land races and wild relatives) (XLS 50 kb)
Supplementary Table 6
Details on the repeat regions and masking (XLS 2321 kb)
Supplementary Table 13
Synteny blocks of the chickpea genome conserved with genomes of four legume (Medicago truncatula, Lotus japonicus, soybean, and pigeonpea) and two non-legume dicot (Arabidopsis thailiana and grape) genomes (XLS 365 kb)
Supplementary Table 17
Primer sequences for SSR markers (XLS 9825 kb)
Supplementary Figure 5
Maximum likelihood phylogram comparing NBS domains of the non-TIR resistance gene homologs from chickpea (Ca) with Medicago (Mt). (PDF 617 kb)
Supplementary Figure 6
Maximum likelihood phylogram comparing NBS domains of the TIR resistance gene homologs from chickpea (Ca) with Medicago truncatula (Mt). (PDF 478 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Varshney, R., Song, C., Saxena, R. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31, 240–246 (2013). https://doi.org/10.1038/nbt.2491
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.2491
This article is cited by
-
Identification and expression profile of the SMAX/SMXL family genes in chickpea and lentil provide important players of biotechnological interest involved in plant branching
Planta (2024)
-
eQTL-seq: a Rapid Genome-Wide Integrative Genetical Genomics Strategy to Dissect Complex Regulatory Architecture of Gene Expression Underlying Quantitative Trait Variation in Crop Plants
Plant Molecular Biology Reporter (2024)
-
Delineation of genes for a major QTL governing heat stress tolerance in chickpea
Plant Molecular Biology (2024)
-
Genome-wide association study in Chickpea (Cicer arietinum L.) for yield and nutritional components
Euphytica (2024)
-
Transcriptional Regulation of Biotic and Abiotic Stress Responses: Challenges and Potential Mechanism for Stress Tolerance and Chickpea Improvement
Tropical Plant Biology (2024)
