
Abstract
Castor bean (Ricinus communis) is an oilseed crop that belongs to the spurge (Euphorbiaceae) family, which comprises ∼6,300 species that include cassava (Manihot esculenta), rubber tree (Hevea brasiliensis) and physic nut (Jatropha curcas). It is primarily of economic interest as a source of castor oil, used for the production of high-quality lubricants because of its high proportion of the unusual fatty acid ricinoleic acid. However, castor bean genomics is also relevant to biosecurity as the seeds contain high levels of ricin, a highly toxic, ribosome-inactivating protein. Here we report the draft genome sequence of castor bean (4.6-fold coverage), the first for a member of the Euphorbiaceae. Whereas most of the key genes involved in oil synthesis and turnover are single copy, the number of members of the ricin gene family is larger than previously thought. Comparative genomics analysis suggests the presence of an ancient hexaploidization event that is conserved across the dicotyledonous lineage.
Similar content being viewed by others
Main
The castor bean plant is a tropical perennial shrub that originated in Africa, but is now cultivated in many tropical and subtropical regions around the world. It can be self- and cross-pollinated and worldwide studies reveal low genetic diversity among castor bean germplasm1,2. Approximately 90% of the oil from castor bean seeds is composed of the unusual hydroxylated fatty acid ricinoleic acid3. Because of the nearly uniform ricinoleic acid content of castor oil, and the unique chemical properties that this fatty acid confers to the oil, castor bean is a highly valued oilseed crop for lubricant, cosmetic, medical and specialty chemical applications. Castor bean has also been proposed as a potential source of biodiesel; the high oil content of its seeds4 and the ease with which it can be cultivated in unfavorable environments contribute to its appeal as a crop in tropical developing countries. It is believed that castor oil was first used as an ointment 4,000 years ago in Egypt, from where it spread to other parts of the world, including Greece and Rome, where it was used as a laxative 2,500 years ago5.
An important obstacle to widespread cultivation of castor bean is the high content of ricin, an extremely toxic protein6, in its seeds. Ricin is considered one of the deadliest natural poisons when administered intravenously or inhaled as fine particles. Ricin was first isolated more than a century ago7. It has been reportedly used as a weapon6 and attempts to use ricin as a specific immunotoxin for therapeutic purposes in different cancers have been reported8,9. Its biochemical activity has been characterized as a type 2 ribosome-inactivating protein (RIP), composed of two subunits linked by a disulfide bond: a 32 kDa ricin toxin A (RTA) chain that harbors the ribosome-inactivating activity, and a 34 kDa ricin toxin B (RTB) chain, with a galactose-binding lectin domain. RTA is an N-glycosidase that depurinates adenine in a specific residue of the 28S ribosomal RNA10,11. The RTB chain allows ricin to enter eukaryotic cells by binding to cell surface galactosides and subsequent endocytosis. Other RIPs are common in plants, although they are not toxic because they are usually monomeric and lack a lectin domain. These proteins constitute the type 1 RIPs12.
Ricin is synthesized as a precursor encoding both subunits in the endoplasmic reticulum of endosperm cells and is translocated and accumulated in protein bodies13. The precursor is proteolytically processed in the endoplasmic reticulum and in the protein bodies, where it is stored as the mature heterodimer. Ricin is very similar to the R. communis agglutinin (RCA)14. However, whereas ricin is a weak hemagglutinin, RCA has low toxicity and strong hemagglutinin activity. In addition, RCA is a tetrameric protein composed of two RTA- and two RTB-like subunits.
The relative ease with which ricin can be purified has raised concerns about its possible use in bioterrorism. For this reason, the United States produces only limited amounts of castor oil and is among the world's largest importers of castor oil and its derivatives. Moreover, much of the West's supply relies on importing castor oil from developing countries periodically threatened by political and economic instability. Therefore, knowledge of the genetics and enzymology of fatty acid metabolism in castor bean seeds is important in efforts to ensure a sustained supply of hydroxy fatty acids without the complications posed by the toxicity of ricin. A better understanding of the biology of ricin accumulation may permit the development of less toxic varieties, and more developed genomic information about the species may improve public safety by tracing the origins of samples used in potential bioterror attacks.
Results
Genome sequencing and annotation
The castor bean genome, which is distributed across ten chromosomes, is estimated by flow cytometry to be ∼320 Mb15. Especially as there is, to our knowledge, no available genetic map and limited genomic information for the species, we set out to generate a draft sequence of the castor bean genome by producing ∼2.1 million high-quality sequence reads from plasmid and fosmid libraries (Online Methods), and then using the Celera assembler to build consensus sequences or contigs that were linked to form 25,800 scaffolds using the two end-sequences from individual clones (mate-paired reads). The assembly covered the genome ∼4.6×, spanning 350 Mb, which is consistent with previous genome size estimations. If only the 3,500 scaffolds larger than 2 kb are considered, the assembly spans 325 Mb with an N50 of 0.56 Mb (Table 1).
We searched the genome sequence assembly for repetitive DNA using a combination of sequence alignment to databases of repetitive sequences and RepeatScout to identify repeats de novo. Overall, >50% of the genome was identified as repetitive DNA (excluding low-complexity sequences), most of which could not be associated with known element families. One-third of the repetitive elements were retrotransposons, and <2% were DNA transposons (Table 2). The most abundant known repeats are long terminal repeat elements (22.7% Gypsy-type and 9.5% Copia-type).
Protein-coding genes were annotated using multiple gene-prediction programs, homology searches against sequence databases and the cDNA spliced-alignment tool PASA (program to assemble spliced alignments). To aid the genome annotation, we also generated 52,165 expressed sequence tags (ESTs) from five cDNA non-normalized libraries. Using PASA, these and other castor bean cDNA sequences from GenBank could be aligned to 5,491 predicted genes and to 688 genomic regions where no gene had been predicted, allowing the creation of additional gene models. Once all gene-prediction programs and homology searches had been run, these data were consolidated into consensus gene predictions using the program Evidence Modeler (EVM; Online Methods). EVM showed better sensitivity and specificity than any of the individual gene finders used (Supplementary Table 1). In this way, we identified 31,237 gene models (Table 1). Using TIGR's paralogous families pipeline, 58.5% of the castor bean gene models were grouped in 3,020 predicted protein families, each comprising at least two members (Supplementary Fig. 1 and Supplementary Table 2).
Polyploidization analysis
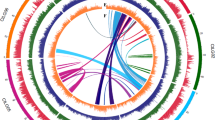
Although the castor bean genome assembly is fairly fragmented, it contains several megabase-sized scaffolds. We took advantage of these to investigate the extent of genome duplications in castor bean and contribute to the elucidation of the evolutionary history of the dicotyledonous lineage. Different models have been proposed to explain the origin of genome duplications in dicots. Whereas one supports the occurrence of an ancestral hexaploidization event common to all dicots16, the other model suggests that all dicot genomes share one duplication event17. As analysis of genomic duplications in the castor bean genome provides an opportunity to contribute to resolving this controversy, we searched for putative paralogous genes using reciprocal best BLAST matches between all castor bean genes. We then selected the 30 pairs of scaffolds that contained the largest numbers of paralogous gene pairs, and displayed the 22 unique scaffolds containing those 30 pairs of scaffolds in a dot plot. This approach identified six triplicated regions (regions for which two additional paralogous regions exist in the genome). We also identified nine duplicated regions (unmarked strings of dots) for which we cannot determine whether or not a third paralogous region exists (Fig. 1). We then carried out a more precise and comprehensive search for evidence of genomic triplications by first building Jaccard clusters of paralogous genes using an all-versus-all BLASTP search. We identified and displayed blocks of syntenic genes using Sybil18 and manually inspected the results to identify triplicated regions. Using this method, we identified 17 triplicated regions (Supplementary Fig. 2) that included those found using the reciprocal best BLAST matches method. The fact that the triplications were found in multiple groups of scaffolds suggests that the castor bean genome underwent a hexaploidization event.

Strings of paralogous genes that correspond to triplicated regions are highlighted in the same color. The 30 pairs of scaffolds that contained the highest numbers of paralogous gene pairs are shown.
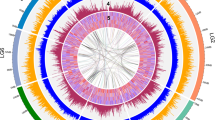
To determine whether the triplication of the castor bean genome corresponds to ancestral polyploidization events previously described in the dicot lineage, we compared triplicated regions in the castor bean genome with the Arabidopsis thaliana19, poplar20, grapevine16, and papaya21 genomes by generating Jaccard clusters in a pairwise manner between castor bean and each of the other genomes. Of the 17 triplications, 8 (including 5 of the 6 triplications identified by reciprocal best BLAST matches) contained blocks of five or more syntenic gene pairs between each of the three castor bean regions and all of the other dicot genomes. Castor bean paralogous gene blocks generally showed a one-to-one, one-to-two and one-to-four relationship with their grapevine, poplar and A. thaliana orthologs, respectively (Fig. 2 and Supplementary Fig. 3). Some exceptions were observed in the comparison with A. thaliana that were expected due to the further re-arrangements that exist in its genome19. Comparison between the castor bean and papaya genomes is less clear due to the fragmentation of both genome assemblies. Our results support the presence of a hexaploidization event common to all dicots, as well as one additional genome duplication in poplar, and two further duplications in the A. thaliana genome.

(a) An example of a conserved paralogous triplication in the castor bean genome. (b–e) Putative orthologous gene pairs are shown as colored lines connecting the castor bean scaffolds (noted as Rc:scaffold number) to chromosomes or scaffolds in the other dicot genome. In most cases, one copy of the paralogous castor bean genes corresponds to two genes in poplar (b), one gene in grapevine (c) and four genes in A. thaliana (d). The castor bean–papaya relationship (e) is inconclusive. Numbers around the circles correspond to linkage group numbers (b), chromosome numbers (c and d) or scaffold numbers (e). Grapevine scaffolds that were mapped to chromosomes but their exact location is unknown are noted with an 'r' (random). The size of the castor bean genomic regions is proportional in all circles. Additional castor bean paralogous regions and their corresponding orthologs from other dicots are shown in Supplementary Figure 3.
The ricin gene family
As the presence of ricin makes castor bean an important subject for biosecurity research, we analyzed the lectin gene family that includes the genes for ricin and RCA. The ricin gene encodes three domains: an N-terminal RIP domain and two C-terminal lectin domains. It has been reported that this gene family comprises 6–8 members, detected by Southern-blot hybridization using a ricin cDNA probe22,23. However, our draft of the castor bean genome reveals 28 putative genes in the family, including potential pseudogenes or gene fragments. To increase the reliability of our analysis of this gene family by improving the sequence and assembly quality, we manually finished sequence gaps or ambiguities inside the ricin-like gene models. In this way, the sequence and assembly of eight scaffolds was improved and the 28 gene family members (Fig. 3) were contained in a total of 17 scaffolds, each containing 1–5 ricin-agglutinin gene family members (Supplementary Table 3). These results suggest that the members of this lectin gene family tend to be clustered in the castor bean genome. The largest cluster spans 70 kb and includes a group of five family members interrupted by one gene that does not belong to the gene family. The other clusters contain two or three gene family members in regions ranging between 0.7 and 17 kb. Ten scaffolds contained only a single gene-family member, and four of them were longer than 250 kb, suggesting that these four genes were not part of clusters. However, it is uncertain if the other six scaffolds that contain only one member of the family are part of clusters because they are shorter than 12 kb. Probably some of these tandem duplications were not discriminated in previous studies using Southern-blot analysis, resulting in an underestimation of the gene family size. Furthermore, although we did not manually curate structural annotation, we found two cases in which adjacent ricin-like gene fragments could belong to pseudogenes that accumulated frame shifts and stop codons (Fig. 3).

Ricin protein domains are represented at the top by blue boxes, and gray boxes represent protein sequences from this gene family aligned to the ricin precursor protein sequence used as reference. The ruler indicates the amino acid coordinates. The ricin and RCA genes are indicated and the amino acid sequence length for each gene model is shown in parenthesis. Pairs of adjacent gene models that could belong to a single pseudogene are shown in gray.
The length of the different members of the family identified by automatic annotation was variable, ranging from 66 to 584 amino acids. Although some of the shorter genes could be nonfunctional or pseudogenes, start and stop codons could be predicted, making it difficult to determine whether they are functional or not. Moreover, four of them were truncated as a consequence of their location at the end of a contig or scaffold. Sequence comparison to ricin and RCA coding sequences in GenBank uncovered one full-length gene model (60629.m00002) identical to the ricin-coding sequence and another full-length gene model (60637.m00004) showing 99% identity to the sequence encoding RCA. These gene models likely correspond to the reported ricin and RCA sequences, respectively. An additional predicted gene (60628.m00003) shows complete identity to the ricin-coding sequence, although presumably, the sequence coding for about 150 of the 576 amino acids is missing from this gene model because it is located at the end of a scaffold. Three other gene models are truncated in a similar way (60626.m00001; 60639.m00003; 60627.m00002) and show 100% identity to the ricin-coding sequence, although the available sequences are much shorter (149 to 188 amino acids). Thus, it is uncertain whether these genes represent complete identical copies of the gene encoding ricin. The rest of the gene family members showed different degrees of similarity to the ricin- or RCA-coding sequences. Overall, 7 of the 28 genes of the lectin family encode proteins that contain the RIP and the two lectin domains, 9 encode proteins with only the RIP domain and 9 encode proteins with one or two lectin domains only (Fig. 3). cDNA alignments showed evidence of expression of the genes encoding ricin and RCA as well as one of the homologs (60638.m00018) for which a putatively complete gene was modeled (data not shown). Furthermore, evidence of RIP activity has been recently reported for the proteins encoded by the seven full-length ricin-like genes24.
Oil metabolism genes
In light of the importance of castor bean as an oilseed crop, we examined the annotation of 71 gene models that showed similarity to known genes involved in the biosynthesis of fatty acids and triacylglycerols, which in castor bean correspond mainly to ricinoleic acid and triricinolein25. Of these 71 gene models, the annotation of 67 was manually improved (Supplementary Table 4). Castor bean has not only evolved an oleic acid hydroxylase to synthesize ricinoleic acid, but has also developed the capacity to efficiently accumulate high levels of ricinoleic acid in its seed oil. Therefore, we focused on a few key genes in the ricinoleic acid biosynthetic and metabolic pathways. The oleic acid hydroxylase gene (FAH), which produces ricinoleic acid from oleoyl-phosphatidycholine, likely evolved from the widely occurring FAD2 gene for the Δ12-oleic acid desaturase26. BLAST searches of these genes against the entire castor genome confirmed that there is only one copy of each of these genes (28035.m000362 and 29613.m000358, respectively). Among the key enzymes involved in the incorporation of ricinoleic acid into oils are diacylglycerol acyltransferases (DGATs), which catalyze the final step in triacylglycerol assembly. Two classes of endoplasmic reticulum–associated DGATs (DGAT1 and DGAT2) occur in castor bean, as well as a homolog of a soluble DGAT27,28,29. The gene models coding for these enzymes are also single copy (29912.m005373, 29682.m000581 and 29889.m003411, respectively). In addition to DGAT-coding genes, it is likely that other genes have evolved to maintain high and specific flux of ricinoleic acid from its synthesis on phosphatidylcholine to its storage in triacylglycerols in castor bean seeds.
Remarkably, even though ricinoleic acid accounts for nearly 90% of the fatty acids in castor bean seeds, it represents <5% of the fatty acids in phosphatidylcholine30. Although the mechanism for ricinoleic acid flux among lipid classes is not clear, a number of specialized acyltransferase and phosphatidylcholine metabolic enzymes likely participate in these reactions, including phospholipid:diacylglycerol acyltransferase 1 (PDAT1; 29912.m005286)31 and the recently identified phosphatidylcholine:diacylglycerol cholinephosphotransferase32 (PDCT; 29841.m002865). Information on copy number, genomic context and regulatory regions of these and other metabolic genes will be important for the biotechnological transfer of ricinoleic acid production to established oilseed crops that lack ricin and its associated health risks. In addition, it is likely that the correct combination of specialized metabolic genes identified from the castor bean genome sequence will enable the engineering of triricinolein accumulation to amounts substantially higher than the modest levels achieved to date in model oilseeds33,34.
Disease resistance genes
To contribute to research aimed at understanding and improving biotic stress resistance in members of the Euphorbiaceae, especially for cassava35, we compiled a list of predicted castor bean proteins with a functional annotation related to disease resistance. One hundred and twenty-one predicted disease-resistance proteins were identified (Supplementary Table 5) using our automated annotation pipeline. The majority of these predicted proteins belong to the nucleotide binding–leucine-rich repeat class, followed by the less common extracellular leucine-rich repeat–containing proteins36, and dirigent-like proteins that have been associated with disease resistance37. The castor bean gene models coding for these resistance genes were found distributed in 69 scaffolds and were often found in clusters of genes from the same class. However, in some cases (for example, scaffold 30190), different resistance gene classes are found in the same cluster (Supplementary Table 5). These data will be useful for comparative studies on resistance genes in cassava, as well as other crop members of the Euphorbiaceae.
Discussion
The sequence of the castor bean genome constitutes an important resource to study genome evolution, not only in the Euphorbiaceae family but also in plants in general. Besides its value for comparative genomics, and the insights it has yielded regarding synthesis of the highly toxic protein ricin38 and the accumulation of castor oil, the castor bean genome promises to be invaluable in developing improved diagnostic and forensic methods for ricin detection and cultivar identification for tracing sample origins. Molecular diagnostic methods39 and worldwide analyses of castor bean populations1,2 have been reported and the availability of the castor bean genome sequence will accelerate efforts to advance such studies and technologies.
In addition to its relevance for biosecurity, availability of the castor bean genome could have implications for the production of biofuels and thus contribute to reducing greenhouse gas production. The industry of castor oil as a biodiesel component is being developed in Brazil4, where the use of biofuels is highly advanced. Furthermore, castor oil can also be used as lubricity additive to replace sulfur-based lubricant components in petroleum diesel, helping to reduce sulfur emissions40.
Unfortunately, the presence of ricin poses a problem for castor bean as a widely cultivated oilseed crop. Therefore, considerable effort has been directed to engineering ricinoleic acid production in seeds of the model plant A. thaliana as a prelude to transferring the required genes to an established ricin-free oilseed crop such as soybean. The initial strategy has involved the seed-specific expression of the castor bean FAH gene for the FAD2-related Δ12 oleic hydroxylase26, the key enzyme in ricinoleic acid synthesis41,42. However, transgenic expression of FAH resulted in the accumulation of ricinoleic acid and other hydroxy fatty acids to only 15–20% of the total fatty acids in A. thaliana seeds41,42. Even co-expression of FAH with one additional ricinoleic acid metabolic gene, including the castor bean gene for DGAT2, yielded only small increases in ricinoleic acid accumulation in seeds of transgenic A. thaliana that were far less than the levels typically found in castor bean seeds33,43. These results also reflect the modest production of other unusual fatty acids that has been achieved by expression of FAD2 variants such as the Δ12 epoxygenase and fatty acid conjugases in seeds of transgenic plants44,45. These results suggest that expression of a single biosynthetic gene, such as FAH alone or together with a gene involved in the metabolism of a given unusual fatty acid, is insufficient to reproduce the oil composition observed in castor bean seeds. Thus, additional information on regulatory and metabolic genes is needed to fully transfer high levels of unusual fatty acid production and accumulation to engineered oilseed crops43,46,47. We believe that the castor bean genome sequence and its annotation constitute the foundation for identifying the regulatory and metabolic networks controlling castor-oil biosynthesis. When combined with metabolomics studies, these castor bean genome resources will enable metabolic engineering for improving castor oil production in crop plants lacking ricin.
Our analysis of the castor bean genome contributes to the debate on the polyploidization events that occurred in dicotyledonous genomes, supporting the presence of an ancestral hexaploidization event. Extending this type of analysis to cassava will benefit the cassava research community as it will synergize with the recently released genome sequence of cassava (http://www.phytozome.net/cassava), which is an important food and, more recently, industrial crop in poor, tropical countries. It has been proposed that cassava is an allopolyploid48, and preliminary comparative genomics analyses between cassava and castor bean showed evidence of genomic duplications in cassava relative to castor bean (S. Rounsley, University of Arizona, Tucson, personal communication). These analyses suggest that the allopolyploidization event may have occurred in the cassava genome relatively recently, after the split between the two lineages. Further genome-wide comparative studies will provide insights on the genome evolution of cassava and the Euphorbiaceae family. Such information will help advance cassava breeding, which is a key means for developing countries to generate improved cassava lines with increased levels of stress resistance and nutritional content.
Methods
Whole genome shotgun sequencing.
Castor bean inbred cultivar Hale49 (NSL 4773) seeds were obtained from the National Center for Genetic Resources Preservation (NCGRP) at Ft. Collins, Colorado (Germplasm Resources Information Network). Nuclear DNA from etiolated castor bean seedlings grown in a growth chamber was purified as described50 and was randomly sheared by nebulization, end-repaired with consecutive BAL31 nuclease and T4 DNA polymerase treatments and size-selected using gel electrophoresis on 1% low-melting-point agarose. After ligation to BstXI adapters, DNA was purified by three rounds of gel electrophoresis to remove excess adapters, and the fragments were ligated into the vector pHOS2 (a modified pBR322 vector) linearized with BstXI. The pHOS2 plasmid contains two BstXI cloning sites immediately flanked by sequencing-primer binding sites. Six libraries with small average insert size (3.5–9 kb) were constructed by electroporation of the ligation reaction into E. coli. strain GC10. In addition, two fosmid libraries were constructed using 30 μg of DNA that was sheared by bead beating and end-repaired (as described above). Fragments between 39 and 40 kb were isolated with a pulse field electrophoresis system and ligated to the blunt-end CopyControl pCC1FOS vector (Epicentre). Lambda phage packaging and infection were performed following the manufacturer's instructions. All clones were plated onto large format (16 × 16 cm) diffusion plates prepared by layering 150 ml of antibiotic-free Luria Bertani (LB)-agar onto a previously set 50-ml layer of LB-agar containing ampicillin or chloramphenicol as required by the vector. Colonies were picked for template preparation using Qbot or QPix colony-picking robots (Genetix), inoculated into 384-well blocks containing liquid medium and incubated overnight with shaking. High-purity plasmid DNA was prepared using the DNA purification robotic workstation custom-built by Thermo CRS and based on the alkaline lysis miniprep51 and isopropanol precipitation. The DNA precipitate was washed with 70% ethanol, dried and resuspended in 10 mM Tris HCl buffer containing a trace of blue dextran. The typical yield of plasmid DNA from this method is ∼600–800 ng per clone, providing sufficient DNA for at least four sequencing reactions per template. Sequencing was carried out using the di-deoxy sequencing method52. Two 384-well cycle-sequencing reaction plates were prepared from each plate of plasmid template DNA for opposite-end, paired-sequence reads. Sequencing reactions were completed using the Big Dye Terminator chemistry (Applied Biosystems) and standard M13 forward and reverse primers. Reaction mixtures, thermal cycling profiles and electrophoresis conditions were optimized to reduce the volume of the Big Dye Terminator mix and to extend read lengths on the AB3730xl sequencers (Applied Biosystems). Sequencing reactions were set up using a Biomek FX (Beckman Coulter) pipetting workstation. Robotics was used to aliquot and combine templates with reaction mixes consisting of deoxy- and fluorescently labeled di-deoxy-nucleotides, DNA polymerase, sequencing primers and reaction buffer in a 5 μl volume. Bar-coding and tracking systems promoted error-free template and reaction mix handling. After 30–40 consecutive cycles of amplification, reaction products were precipitated with isopropanol, dried at 25 °C, resuspended in water and transferred to an AB3730xl DNA analyzer.
A total of 2,276,000 paired-end sequence reads were attempted yielding 2,079,000 high-quality sequences, of which 12% correspond to fosmid clones (40 kb insert size), 60% to 9 kb insert size clones, 10% to 5 kb insert size clones and 18% to 3.5 kb insert clones. The average read-length was 839 bp. All reads were assembled into contigs using the Celera assembler53 version 3.20 that utilizes an 'overlay-layout-consensus' approach to produce consensus sequences or contigs. Celera also uses mate-pair read information to build scaffolds where contigs are ordered and oriented relative to each other. The Celera assembler was run using the default parameters for large genomes. In addition to the normal contigs, the assembler creates so-called 'degenerate contigs' which have some kind of problem, such as excessive deviation from the expected level of coverage. We manually inspected the degenerate contigs and recovered ∼12.4 Mb of sequences that contained plant gene-like sequences as determined by BLAST analysis. The consensus sequences were entered in an in-house genome annotation relational database called RCA1.
As the genomic DNA used for sequencing was purified from non-axenic seedlings, plant-associated bacteria were likely to be present in our sequence. Therefore, contigs smaller than 2 kb that did not show a high level of identity to plant organelle sequences (BLASTN E value cutoff < 10−50), and showed sequence similarity to bacterial proteins from available bacterial genome sequences with BLASTX E values < 10−20 were removed.
Closure of sequence gaps.
To increase the quality of the ricin gene family annotation, we performed finishing work on eight scaffolds that contained members of this gene family to close sequence gaps or ambiguities within the corresponding gene models. Closure was conducted by editing the ends of sequence traces, primer walking on plasmid templates, sequencing genomic PCR products that spanned the gaps or by transposon insertion and sequencing of selected fosmids clones54.
Gene prediction and genome annotation.
All R. communis scaffolds were processed through the TIGR eukaryotic annotation pipeline. Before running the gene prediction software, we used RepeatMasker to mask the genomic sequence using a library of known plant repeats from an in-house plant repeat database and novel castor bean repeats identified by running RepeatScout, an algorithm that identifies sequences that are overrepresented in the assembly55. To prevent incorrect annotation of repeats as genes, we took a conservative approach and any sequence repeated at least ten times in the genome was considered repetitive. Manual inspection of the list of repeats generated by RepeatScout was carried out to remove members of known gene families that were wrongly reported as repeats. Further screening by manual review was carried out to remove putative gene families that were mistakenly identified as repeats, resulting in a final set of 1,517 consensus repeat sequences. With the so-constructed repeat library, 50.33% of the castor bean genome was masked as repetitive sequences. Low complexity sequences and tandem repeats were identified but not masked because they are often part of protein coding sequences. The RepeatScout library masked 49.88% of the genome whereas the known plant repeat library masked 8.24% of the genome. Repeats were classified using 2,994 Viridiplantae repeats from RepBase56 and the consensus repetitive sequences identified by RepeatScout (Table 2).
Four gene finders were run on the masked genome: FgenesH gene prediction algorithm trained with a dicotyledonous matrix57; Augustus trained with Arabidopsis58; GlimmerHMM trained with Arabidopsis59; and SNAP trained with Arabidopsis60.
We used PASA61 to align 53,516 castor bean cDNA sequences to the castor bean genome. We used all available castor bean cDNA sequences from GenBank at the time, and 52,165 ESTs from five cDNA non-normalized libraries constructed from mRNAs from leaves, flowers, roots and two different seed developmental stages. cDNA clones were sequenced from the 5′ end, except for the root cDNA clones, which were sequenced from both ends to increase the chances of obtaining full-length cDNA sequences. PASA also assembles the aligned cDNA sequences into so-called 'PASA assemblies'. Using the unmasked castor genome sequence, PASA aligned and assembled ∼73% of the castor bean cDNA sequences. For a cDNA sequence to be aligned to the genome, it should have at least 95% identity along 90% of its length, and consensus splice sites should be present at all inferred exon/intron boundaries. After alignment, PASA generated 8,132 nonredundant cDNA assemblies, of which 5,491 overlapped predicted gene models and 688 identified nonannotated regions. These PASA assemblies were used for identification of new gene models as well as to validate or update existing ones. Other PASA assemblies were not incorporated into gene models owing to intron/exon structure conflicts or because the fragmentary nature of the genome assembly precluded the alignments to meet the stringency criteria.
Sequence homology to nucleotide and protein datasets was computed using the Analysis and Annotation Tool (AAT) package62 on the unmasked castor bean genome. AAT utilizes a two-step approach consisting of a fast database homology search followed by a rigorous, splice-aware local alignment. The datasets used for AAT analyses included: (i) Oryza sativa peptides (October 2006 release); (ii) Arabidopsis proteins (TAIR 6, September 2006 release); (iii) an in-house nonredundant amino acid database; (iv) a database of transcript assemblies that contains clustered and assembled ESTs and other cDNA sequences from plant species63 for which over 1,000 sequences are available in GB (http://plantta.jcvi.org/).
Proteins having the highest scoring amino acid alignment to our gene models were incorporated into the gene models using GeneWise64 to increase protein prediction reliability.
All gene structures predicted by the methods described above as well as the alignments to protein and nucleotide databases were combined into consensus gene models using EVM65, a software package developed at The Institute for Genomic Research (TIGR, now the J.C. Venter Institute or JCVI) that integrates data from multiple gene prediction programs as well as protein and cDNA similarity searches, to achieve the most accurate annotation possible with automated tools. It uses a nonstochastic, weighted-evidence combining technique that accounts for both the type and abundance of evidence to compute weighted consensus gene structures. All potential gene structure components were scored based on manually set weights so that exon and intron structures supported by PASA alignments and high-quality protein alignments had the highest relevance in determining a gene model's final structure, and the structure predicted by ab initio gene-finding software were given lower weights according to their accuracy for castor bean. Evidence from transcript assemblies alignments, protein alignments and gene prediction software were given a weight of 1, whereas GeneWise protein alignments received a weight of 5, and the weight of PASA assemblies was set at 20. Dynamic programming then was applied by EVM to find the highest scoring consensus gene structure, supported by all available evidence.
Gene models produced by EVM were then updated by new PASA assembly alignments. PASA extended untranslated regions and added small missed exons. This resulted in a total of 31,237 gene models of which 19,768 have either EST or protein support (5,316 gene models have castor bean EST support determined by PASA, and 16,848 have protein evidence support determined by AAT searches). 3,150 models were labeled as 'partial' because they missed either start and/or stop codons. 354 gene models contained an internal gap, which is represented by 'Ns' in the nucleotide sequence and 'Xs' in protein sequence, indicating the location and predicted size of the gap.
A dataset of 60 castor bean genes manually modeled based on highly conserved cDNA and protein alignments across multiple plant species were used as reference to evaluate the gene prediction algorithms' performance in comparison with EVM consensus predictions (Supplementary Table 1). Although this is a small set of genes, we used the exons to estimate the specificity and sensitivity of exon prediction by the different gene-finder programs as described65. Future iterations of the annotation can be improved by using a larger set of genes for training and evaluation of the gene prediction software, as more castor bean cDNA sequences become available.
Gene models were automatically named and their function was assigned by computationally extracting this information from BLASTP searches against the TAIR6 Arabidopsis peptides, Uniprot-Swissprot and experimentally verified Panda (http://www.ebi.ac.uk/panda), Panther (http://www.pantherdb.org/) and Interpro (http://www.ebi.ac.uk/interpro) databases. Gene models whose hits in those databases were defined as “unknown function” were labeled “conserved hypothetical protein” in our genome annotation. Gene models with no match in these databases above the selected threshold were labeled “hypothetical protein.”
Automated Gene Ontology GO term assignments were done by extrapolating GO terms from matching Arabidopsis proteins using BLASTP with an E value threshold of 10−40. Castor bean gene models with no match to Arabidopsis were screened against Pfam domains and assigned the Pfam associated GO term, if matches were above the selected cutoff. Altogether, this resulted in the assignment of 43,657 GO terms to 14,991 R. communis proteins.
Putative signal peptide sequences were identified using SignalP66 and TargetP (http://www.cbs.dtu.dk/services/TargetP), and transmembrane regions were predicted by tmHMM67. Castor bean protein domains were also compared against the Pfam database of conserved families68. Proteins were organized into putative paralogous families based on conserved domain composition, taking into account both previously identified domains from public databases and potential novel domains identified using independent methods69,70.
Noncoding RNAs were identified by searching against various RNA libraries. tRNAscan-SE71 was run on the assembled genomic sequence to identify tRNAs. All 20 tRNAs were found in the genome with a total of 717 copies. rRNA sequences were annotated based on homology to previously published rRNA sequences in plants. snRNA were searched by blasting against the NONCODE database72.
We assigned Enzyme Commission (EC) classification developed by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology, to provide metabolic pathway annotation. Castor bean proteins were searched against PRIAM profiles73 using PSI-BLAST, and EC numbers were assigned for hits with an E value < 10−10.
Annotation data are displayed in the project website (http://castorbean.jcvi.org/), which includes a generic genome browser (http://gmod.org/wiki/GBrowse), where gene models can be viewed in their sequence and genomic context. We used a gene model nomenclature that is composed by the scaffold ID number, followed by a period and the gene model number that consists of the letter 'm' followed by the gene model number. This number can be used to locate genes in the castor bean genome browser. Gene models in the genome browser are linked to Manatee pages, which include additional annotation information (http://manatee.sourceforge.net).
The castor bean predicted proteome could be matched to over 3,000 protein domains from Pfam68, several of which are not present in Arabidopsis or poplar, including secondary metabolism genes (Supplementary Fig. 1). However, these results may have a substantial error due to inaccuracies of the automatic annotation both in poplar and castor bean.
We also searched for tandem gene duplications and found a total of 2,610 (8% of the total) genes forming part of tandem arrays.
Identification of genome duplications.
A total of 167,984 predicted polypeptides from R. communis, Vitis vinifera, Populus trichocarpa, Arabidopsis thaliana and Carica papaya were subjected to an all-versus-all BLASTP analysis using WU-BLASTP 2.0MP, with the default BLOSUM62 substitution matrix, no low-complexity sequence filter, and an E-value cutoff of 10−5. The castor bean subset of the BLAST results was analyzed to extract 5,536 pairs of castor genes that are reciprocal best hits and reside on distinct sequence contigs.
Each of the 721 (of 25,828) castor scaffolds with at least five annotated protein-coding genes was examined for runs of five or more genes that are collinear and are reciprocal best hits of collinear genes in another castor bean scaffold. Images were generated from these results and inspected manually for the presence of regions that appear to be triplicated in the castor bean genome, on the basis of overlapping runs of collinear matching gene pairs. The regions thus determined were also cross-checked against dot plots showing the relative positions of the paralogous gene pairs.
To further analyze these putative triplications four sets of Jaccard74 orthologous (protein) clusters18 were computed between castor and each of the four other genomes: Jaccard clusters were first defined within each genome by taking all BLASTP matches with E value ≤ 10−10, ≥80% identity and ≥70% sequence coverage and then forming clusters by transitively merging all pairs of proteins with Jaccard coefficient ≥0.6. In the second step, pairs of Jaccard clusters in distinct genomes were merged if each contained a protein with a best hit in the other cluster, taking into consideration only BLASTP matches with E value ≤ 10−10 and ≥70% sequence coverage (but imposing no other restriction on percent identity). For each triplication, Circos75 was used to display the three castor regions and any collinear cluster matches between genes in those regions and those in the respective target genomes.
Accession codes.
GenBank nuccore: AASG02000000; and GenBank gene: XP_002509419.1–XP_002540639.1. (The annotation data can also be freely accessed through the project's website (http://castorbean.jcvi.org/), which includes a genome browser and a BLAST server.)
Accession codes
References
Allan, G. et al. Worldwide genotyping of castor bean germplasm (Ricinus communis L.) using AFLPs and SSRs. Genet. Resour. Crop Evol. 55, 365–378 (2008).
Foster, J.T. et al. Single nucleotide polymorphisms for assessing genetic diversity in castor bean (Ricinus communis). BMC Plant Biol. 10, 13 (2010).
da Silva Ramos, L.C., Shogiro Tango, J., Savi, A. & Leal, N.R. Variability for oil and fatty acid composition in castorbean varieties. J. Am. Oil Chem. Soc. 61, 1841–1843 (1984).
da Silva Nde, L., Maciel, M.R., Batistella, C.B. & Maciel Filho, R. Optimization of biodiesel production from castor oil. Appl. Biochem. Biotechnol. 130, 405–414 (2006).
Scarpa, A. & Guerci, A. Various uses of the castor oil plant (Ricinus communis L.). A review. J. Ethnopharmacol. 5, 117–137 (1982).
Knight, B. Ricin–a potent homicidal poison. BMJ 1, 350–351 (1979).
Lord, J.M., Roberts, L.M. & Robertus, J.D. Ricin: structure, mode of action, and some current applications. FASEB J. 8, 201–208 (1994).
Schnell, R. et al. A Phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients with refractory CD30+ Hodgkin's and non-Hodgkin's lymphoma. Clin. Cancer Res. 8, 1779–1786 (2002).
Fidias, P., Grossbard, M. & Lynch, T.J. Jr. A phase II study of the immunotoxin N901-blocked ricin in small-cell lung cancer. Clin. Lung Cancer 3, 219–222 (2002).
Endo, Y., Mitsui, K., Motizuki, M. & Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 262, 5908–5912 (1987).
Macbeth, M.R. & Wool, I.G. Characterization of in vitro and in vivo mutations in non-conserved nucleotides in the ribosomal RNA recognition domain for the ribotoxins ricin and sarcin and the translation elongation factors. J. Mol. Biol. 285, 567–580 (1999).
Lord, J.M., Hartley, M.R. & Roberts, L.M. Ribosome inactivating proteins of plants. Semin. Cell Biol. 2, 15–22 (1991).
Lord, J.M. Synthesis and intracellular transport of lectin and storage protein precursors in endosperm from castor bean. Eur. J. Biochem. 146, 403–409 (1985).
Roberts, L.M., Lamb, F.I., Pappin, D.J. & Lord, J.M. The primary sequence of Ricinus communis agglutinin. Comparison with ricin. J. Biol. Chem. 260, 15682–15686 (1985).
Arumuganathan, K. & Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218 (1991).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Velasco, R. et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2, e1326 (2007).
Crabtree, J., Angiuoli, S.V., Wortman, J.R. & White, O.R. Sybil: methods and software for multiple genome comparison and visualization. Methods Mol. Biol. 408, 93–108 (2007).
The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Tuskan, G.A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996 (2008).
Halling, K.C. et al. Genomic cloning and characterization of a ricin gene from Ricinus communis. Nucleic Acids Res. 13, 8019–8033 (1985).
Tregear, J.W. & Roberts, L.M. The lectin gene family of Ricinus communis: cloning of a functional ricin gene and three lectin pseudogenes. Plant Mol. Biol. 18, 515–525 (1992).
Leshin, J. et al. Characterization of ricin toxin family members from Ricinus communis. Toxicon 55, 658–661 (2010).
McKeon, T.A., Chen, G.Q. & Lin, J.T. Biochemical aspects of castor oil biosynthesis. Biochem. Soc. Trans. 28, 972–974 (2000).
van de Loo, F.J., Broun, P., Turner, S. & Somerville, C. An oleate 12-hydroxylase from Ricinus communis L. is a fatty acyl desaturase homolog. Proc. Natl. Acad. Sci. USA 92, 6743–6747 (1995).
He, X., Turner, C., Chen, G.Q., Lin, J.T. & McKeon, T.A. Cloning and characterization of a cDNA encoding diacylglycerol acyltransferase from castor bean. Lipids 39, 311–318 (2004).
Kroon, J.T., Wei, W., Simon, W.J. & Slabas, A.R. Identification and functional expression of a type 2 acyl-CoA:diacylglycerol acyltransferase (DGAT2) in developing castor bean seeds which has high homology to the major triglyceride biosynthetic enzyme of fungi and animals. Phytochemistry 67, 2541–2549 (2006).
Saha, S., Enugutti, B., Rajakumari, S. & Rajasekharan, R. Cytosolic triacylglycerol biosynthetic pathway in oilseeds. Molecular cloning and expression of peanut cytosolic diacylglycerol acyltransferase. Plant Physiol. 141, 1533–1543 (2006).
Thomaeus, S., Carlsson, A.S. & Stymne, S. Distribution of fatty acids in polar and neutral lipids during seed development in Arabidopsis thaliana genetically engineered to produce acetylenic, epoxy and hydroxy fatty acids. Plant Sci. 161, 997–1003 (2001).
Dahlqvist, A. et al. Phospholipid:diacylglycerol acyltransferase: an enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants. Proc. Natl. Acad. Sci. USA 97, 6487–6492 (2000).
Lu, C., Xin, Z., Ren, Z., Miquel, M. & Browse, J. An enzyme regulating triacylglycerol composition is encoded by the ROD1 gene of Arabidopsis. Proc. Natl. Acad. Sci. USA 106, 18837–18842 (2009).
Burgal, J. et al. Metabolic engineering of hydroxy fatty acid production in plants: RcDGAT2 drives dramatic increases in ricinoleate levels in seed oil. Plant Biotechnol. J. 6, 819–831 (2008).
Cahoon, E.B. et al. Engineering oilseeds for sustainable production of industrial and nutritional feedstocks: solving bottlenecks in fatty acid flux. Curr. Opin. Plant Biol. 10, 236–244 (2007).
Hillocks, R.J. & Jennings, D.L. Cassava brown streak disease: a review of present knowledge and research needs. Int. J. Pest Manage. 49, 225–234 (2003).
van Ooijen, G., van den Burg, H.A., Cornelissen, B.J. & Takken, F.L. Structure and function of resistance proteins in solanaceous plants. Annu. Rev. Phytopathol. 45, 43–72 (2007).
Fristensky, B., Horovitz, D. & Hadwiger, L.A. cDNA sequences for pea disease resistance response genes. Plant Mol. Biol. 11, 713–715 (1988).
Musshoff, F. & Madea, B. Ricin poisoning and forensic toxicology. Drug Test Anal 1, 184–191 (2009).
Audi, J., Belson, M., Patel, M., Schier, J. & Osterloh, J. Ricin poisoning: a comprehensive review. J. Am. Med. Assoc. 294, 2342–2351 (2005).
Goodrum, J.W. & Geller, D.P. Influence of fatty acid methyl esters from hydroxylated vegetable oils on diesel fuel lubricity. Bioresour. Technol. 96, 851–855 (2005).
Broun, P. & Somerville, C. Accumulation of ricinoleic, lesquerolic, and densipolic acids in seeds of transgenic Arabidopsis plants that express a fatty acyl hydroxylase cDNA from castor bean. Plant Physiol. 113, 933–942 (1997).
Smith, M.A., Moon, H., Chowrira, G. & Kunst, L. Heterologous expression of a fatty acid hydroxylase gene in developing seeds of Arabidopsis thaliana. Planta 217, 507–516 (2003).
Lu, C., Fulda, M., Wallis, J.G. & Browse, J. A high-throughput screen for genes from castor that boost hydroxy fatty acid accumulation in seed oils of transgenic Arabidopsis. Plant J. 45, 847–856 (2006).
Li, R., Yu, K., Hatanaka, T. & Hildebrand, D.F. Vernonia DGATs increase accumulation of epoxy fatty acids in oil. Plant Biotechnol. J. 8, 184–195 (2010).
Cahoon, E.B. et al. Conjugated fatty acids accumulate to high levels in phospholipids of metabolically engineered soybean and Arabidopsis seeds. Phytochemistry 67, 1166–1176 (2006).
Cernac, A. & Benning, C. WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J. 40, 575–585 (2004).
Thelen, J. & Ohlrogge, J. Metabolic engineering of fatty acid biosynthesis in plants. Metab. Eng. 4, 12–21 (2002).
Umanah, E.E. & Hartmann, R.W. Chromosome numbers and karyotypes of some Manihot species. Am. Soc. Hortic. Sci. 98, 272–274 (1973).
Brigham, R. Registration of castor variety Hale (Reg. No. 3). Crop Sci. 10, 457 (1970).
Rabinowicz, P.D. et al. Differential methylation of genes and retrotransposons facilitates shotgun sequencing of the maize genome. Nat. Genet. 23, 305–308 (1999).
Sambrook, J. & Russell, D.W. Molecular Cloning. A Laboratory Manual 3rd edn., (Cold Spring Harbor Laboratory Press, 2001).
Sanger, F., Nicklen, S. & Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74, 5463–5467 (1977).
Myers, E.W. et al. A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000).
Birren, B., Green, E.D., Klapholz, S., Myers, R.M. & Roskams, J. Genome Analysis. A Laboratory Manual. Analyzing DNA Vol. 1 (Cold Spring Harbor Laboratory Press, 1997).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21 Suppl 1, i351–i358 (2005).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Salamov, A.A. & Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 Suppl 2, ii215–ii225 (2003).
Majoros, W.H., Pertea, M. & Salzberg, S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Haas, B.J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Huang, X., Adams, M.D., Zhou, H. & Kerlavage, A.R. A tool for analyzing and annotating genomic sequences. Genomics 46, 37–45 (1997).
Childs, K.L. et al. The TIGR Plant Transcript Assemblies database. Nucleic Acids Res. 35, D846–D851 (2007).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Bendtsen, J.D., Nielsen, H., von Heijne, G. & Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795 (2004).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Finn, R.D. et al. Pfam: clans, web tools and services. Nucleic Acids Res. 34, D247–D251 (2006).
Haas, B.J. et al. Complete reannotation of the Arabidopsis genome: methods, tools, protocols and the final release. BMC Biol. 3, 7 (2005).
Wortman, J.R. et al. Annotation of the Arabidopsis genome. Plant Physiol. 132, 461–468 (2003).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
He, S. et al. NONCODE v2.0: decoding the non-coding. Nucleic Acids Res. 36, Database issue, D170–D172 (2008).
Claudel-Renard, C., Chevalet, C., Faraut, T. & Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 31, 6633–6639 (2003).
Jaccard, P. The distribution of the flora in the alpine zone. New Phytol. 11, 37–50 (1912).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services, under NIAID Contract N01-AI-30071 (R. communis subproject) awarded to C.M.F.-L., J.R., J.R.W. and P.D.R.; Federal Bureau of Investigation grant J-FBI-04-186 to C.M.F.-L., J.R. and P.D.R.; and National Science Foundation grant DBI 0701919 to E.B.C. We thank the Joint Technology Center at J. Craig Venter Institute for carrying out all sequencing work, and K. Wurdack for his assistance with phylogenetics.
Author information
Authors and Affiliations
Contributions
A.P.C., J.C., H.L., B.J.H. and J.R.W. performed genomic analyses. Q.Z., J.O. and M.S. conducted genome annotation. D.P. worked on the genome assembly. A.M.-B., K.M.J. and J.R. made DNA preparations, library constructions, and closure work. G.C., E.B.C. and M.G. performed manual annotations. C.M.F.-L. and J.R. conceived the project. P.D.R. conceived and directed the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Tables 1–5 and Supplementary Figs. 1–3 (PDF 11751 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike license (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar license.
About this article
Cite this article
Chan, A., Crabtree, J., Zhao, Q. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol 28, 951–956 (2010). https://doi.org/10.1038/nbt.1674
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.1674
This article is cited by
-
A phytochemical and pharmacological review of Ricinus communis L.
Discover Applied Sciences (2024)
-
Dynamic QTL mapping revealed primarily the genetic structure of photosynthetic traits in castor (Ricinus communis L.)
Scientific Reports (2023)
-
Genome-wide identification and expression analysis of Hsp70 family genes in Cassava (Manihot esculenta Crantz)
3 Biotech (2023)
-
Genomic insights into lineage-specific evolution of the oleosin family in Euphorbiaceae
BMC Genomics (2022)
-
Genome-wide identification and characterization of lncRNAs in sunflower endosperm
BMC Plant Biology (2022)
