
Abstract
The planarian Schmidtea mediterranea is an important model for stem cell research and regeneration, but adequate genome resources for this species have been lacking. Here we report a highly contiguous genome assembly of S. mediterranea, using long-read sequencing and a de novo assembler (MARVEL) enhanced for low-complexity reads. The S. mediterranea genome is highly polymorphic and repetitive, and harbours a novel class of giant retroelements. Furthermore, the genome assembly lacks a number of highly conserved genes, including critical components of the mitotic spindle assembly checkpoint, but planarians maintain checkpoint function. Our genome assembly provides a key model system resource that will be useful for studying regeneration and the evolutionary plasticity of core cell biological mechanisms.
Similar content being viewed by others


Main
Rapid regeneration from tiny pieces of tissue makes planarians a prime model system for regeneration. Abundant adult pluripotent stem cells, termed neoblasts, power regeneration and the continuous turnover of all cell types1,2,3, and transplantation of a single neoblast can rescue a lethally irradiated animal4. Planarians therefore also constitute a prime model system for stem cell pluripotency and its evolutionary underpinnings5. The taxonomic clade Platyhelminthes (‘flatworms’) also includes parasitic lineages that have substantial effects on human health, such as blood flukes (Trematoda) and tape worms (Cestoda)6. Here, the phylogenetic position of planarians as free-living flatworms7 provides a reference point towards an understanding of the evolution of parasitism8.
Despite the modest genome sizes of planarians (mostly in the range of 1–2 gigabase pairs (Gb)), genome resources relating to these animals are limited. Although the model species S. mediterranea was sequenced by Sanger sequencing, even 11.6× coverage of around 600-bp Sanger reads yielded only a highly fragmented assembly (N50 19?kb)9. Recent high-coverage, short-read approaches yielded similarly fragmented assemblies10,11. The high A–T content (about 70%) represents one known assembly challenge. Furthermore, standard DNA isolation procedures perform poorly on planarians, which has so far precluded the application of long-read sequencing approaches or BAC-clone scaffolding.
We here report a highly contiguous PacBio SMRT long-read sequencing12 assembly of the S. mediterranea genome. Giant gypsy/Ty3 retroelements, abundant AT-rich microsatellites and inbreeding-resistant heterozygosity collectively provide an explanation for why previous short-read approaches were unsuccessful. We find a loss of gene synteny in the genome of S. mediterranea and other flatworms. In analysis of highly conserved genes, we find a loss of MAD1 and MAD2, suggesting a MAD1–MAD2-independent spindle assembly check point (SAC)13,14. Our S. mediterranea genome assembly provides a resource for probing the evolutionary plasticity of core cell biological mechanisms, as well as the genomic underpinnings of regeneration and the many other phenomena that planarians expose to experimental scrutiny.
De novo long read assembly of the planarian genome
In preparation for genome sequencing, we inbred the sexual strain of S. mediterranea (Fig. 1a) for more than 17 successive sib-mating generations in the hope of decreasing heterozygosity. We also developed a new DNA isolation protocol that meets the purity and high molecular weight requirements of PacBio long-read sequencing12 (Extended Data Fig. 1a–d, Supplementary Information S1, S2). We used MARVEL, a new long-read genome assembler developed for low complexity read data15 (Supplementary Information S3). An initial de novo MARVEL assembly of reads of more than 4?kb with approximately 60× genome coverage showed an improvement over the PacBio assembly tool (Canu16) and substantial improvements over existing S. mediterranea assemblies based on short read sequencing (Extended Data Table 1). We further made use of the Chicago/HiRise in vitro proximity ligation method17 for scaffolding (Extended Data Fig. 1e, Supplementary Information S4). The polished haplotype-filtered (see below) and error-corrected (Supplementary Information S5) S. mediterranea assembly consists of 481 scaffolds with an N50 length of 3.85?Mb (Extended Data Table 1).

a, Top, individual of the sequenced sexual strain. Bottom left, egg cocoons. Bottom right, karyotype (2N?=?8). Scale bars, 2?mm (top) and 2.5?μm(bottom right). b, Chicago quality control of the assembly. c, Treemap comparison between the MARVEL S. mediterranea assembly and the most contiguous existing Sanger S. mediterranea assembly10. Squares encode the relative contributions of individual scaffolds or contigs to assembly size.
To assess the quality of this genome assembly, we back-mapped a transcriptome of the sequenced strain (Supplementary Information S6) and found that more than 99% of transcripts were mapped, thus confirming that the assembly was both near-complete and accurate (Supplementary Information S7, Extended Data Fig. 1f, g). To assess the contiguity of the global assembly, we analysed structural conflicts between the MARVEL assembly and Chicago/HiRise scaffolding. Out of 51 such events across the 782.1?Mb of assembled genome sequence, only two represented unambiguous MARVEL assembly mistakes (Fig. 1b, Supplementary Information S4.3). Furthermore, high-stringency back-mapping of high-confidence cDNA sequences (Supplementary Information S7.3) confirmed assembly contiguity below the approximately 1-kb resolution limit of the Chicago/HiRise method, with small-scale sequence duplications near assembly gaps as only minor inconsistencies (Extended Data Fig. 2).
Our S. mediterranea genome assembly represents a major improvement over existing S. mediterranea assemblies10 (Fig. 1c) and, to our knowledge, is the first long-range contiguous assembly of the genome of a non-parasitic flatworm species. A UCSC genome browser instance with supplementary quality control, annotation and experimental data tracks (Supplementary Information S8) is available at PlanMine18 (http://planmine.mpi-cbg.de). All analyses in this manuscript refer to the assembly release version dd_Smed_g4. The current source code of the MARVEL assembler is available at https://github.com/schloi/MARVEL. The execution scripts used for S. mediterranea can be found in the smed subfolder of the examples folder.
Assembly challenges in the S. mediterranea genome
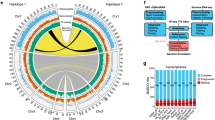
To understand why the S. mediterranea genome was recalcitrant to earlier short-read assembly, we first analysed its repeat content (Supplementary Information S9). The genome has a repetitive fraction of 61.7% (Fig. 2a), substantially exceeding the 38% or 46% repeat content of the mouse or human genomes, respectively19. We detected more than 7,000 insertions of 11 distinct families of long terminal repeat (LTR) retroelements (Fig. 2b, Extended Data Fig. 3a, Supplementary Information S10). These do not cluster with known Metaviridae (Fig. 2b), suggesting that they represent either extremely divergent or so far undescribed retroelement families. Three LTR families were more than 30?kb long—an exceptional size that is more than three times longer than the 5–10?kb typically observed in vertebrates (Fig. 2c, Extended Data Fig. 3b). The only known similar-sized LTRs are the plant-specific Ogre elements20, which is why we refer to the giant S. mediterranea repeat families as Burro (big, unknown repeat rivalling ogre; Supplementary Information S10.3). Burro elements are pervasively transcribed (Extended Data Fig. 3c, d, Supplementary Information S10.4), yet their high degree of intra-family sequence divergence suggests a relatively ancient invasion (Supplementary Table 1, Supplementary Information S10.5, Extended Data Fig. 3e). Burro-1, the most abundant giant retroelement with 130 fully assembled copies, is highly overrepresented at contig ends, and 50% of all current scaffolds terminate in a Burro-1 element (Fig. 2d, Supplementary Information S10.6). Therefore, these abundant, over 30-kb repeat elements still limit the size of the current assembly. In addition, abundant AT-rich microsatellite regions disrupt the alignment of spanning reads and thus also reduce contig contiguity (Extended Data Fig. 4, Supplementary Information S11). Finally, the S. mediterranea assembly graphs showed substantial structural heterogeneity (Supplementary Information S12) in the form of bubbles (transient divergences in sequencing read alignments) and spurs (divergences without re-connection), which were largely absent from a comparable genome assembly (Drosophila melanogaster using PacBio sequencing and MARVEL assembly; Fig. 2e, Supplementary Information S12.1) or assemblies of 17 other species (Supplementary Table 2). Heterozygous mobile element insertions and microsatellite tracts were prominent causes of assembly divergences (Fig. 2f, Extended Data Fig. 4d, Supplementary Information S12.3). The persistence of substantial genomic heterozygosity in spite of more than 17 successive sib-mating generations confirms that meiotic recombination is inefficient in S. mediterranea21.

a, Repeat content of the assembly. b, LTR family phylogeny. Known LTR families are shown in colour, S. mediterranea LTR families in black. Red arcs delimit clusters for consensus calculation. Scale bar: 0.2 substitutions per site. c, Domain annotation of the 11 S. mediterranea LTR families (SLFs). d, Enrichment analysis of indicated repeat elements within the terminal 1,000 bp of all scaffolds (n?=?962). Expected values represent mean repeat frequency with 95% bootstrap confidence interval (n?=?1,000). e, Graphical representation of representative S. mediterranea (left, ~1.6?Mb) and D. melanogaster (right, ~1.7?Mb) MARVEL PacBio assembly graph segments. Thick lines, consensus sequence; thin lines, individual read alignments; colour-coding, alignment quality (blue, low; red, high; see spectra at bottom); black marks, repeats. The contig tour of the final haploid genome assembly is shown offset to the right and alternative regions are shown in red. f, Dot plot comparison between a representative alternative region and the corresponding main contig. Fwd, forward match; Rev, reverse match; Break, insertions or deletions over 99 bp. Break annotations (1–11, right) list repeat categories that cover more than 60% of the insertion/deletion sequence; ‘mixed’ indicates contributions of multiple repeat classes.
Overall, the combination of giant repeat elements, low-complexity regions and inbreeding-resistant heterozygosity provides an explanation for why previous short-read sequencing assemblies of S. mediterranea have proven so challenging. The long-range contiguity that we achieved in the S. mediterranea genome assembly, and similarly substantial improvements in the PacBio genome assembly of the flatworm Macrostomum lignano22 (Supplementary Table 2), further emphasize the improvements that a combination of long-read sequencing with the MARVEL assembler offers in the assembly of challenging genomes.
Comparative analysis of the planarian gene complement
We next annotated the S. mediterranea gene complement, relying on our planarian transcriptome resources18 (Supplementary Information S13). Our analysis showed a high divergence of S. mediterranea gene sequences (Supplementary Information S14), en par with Caenorhabditis elegans (Fig. 3a). By contrast, the low degree of sequence substitutions between the sexual and asexual S. mediterranea strains (Fig. 3a) and nearly identical mapping statistics of the two transcriptomes to the genome (Supplementary Information S7.1, Extended Data Fig. 1f) establish the utility of our assembly for both strains.

a, Protein sequence divergence amongst 51 single copy genes (Supplementary Table 3). Branch length shows substitutions per site. Red, flatworms; blue, lophotrochozoan outgroups. b, Whole genome alignments of S. mediterranea, M. lignano and H. sapiens against the indicated reference genomes. The distributions of the alignment score (top) and alignment span (bottom) of the top 10,000 chains of co-linear alignments are shown as box plots, with boxes indicating the first quartile, median and third quartile with whiskers extending up to 1.5 times the interquartile distance. Outliers are defined as more than 1.5 times the interquartile and are shown as dots. c, Presence (green) or absence (red) of highly conserved genes in the indicated species. The yellow box highlights S. mediterranea. Asterisks mark homologues secondarily identified by manual searches.
To evaluate the S. mediterranea genome structure, we performed whole-genome alignments (Supplementary Information S15) with the available parasitic flatworm genomes6 and a draft genome of the platyhelminth M. lignano22 (Fig. 3b). The highest alignment similarity was found between S. mediterranea and the parasitic flatworm Schistosoma mansoni, which is consistent with the platyhelminth phylogeny7. However, alignments were mostly limited to individual exons of specific genes, irrespective of the quality of the various assemblies (Extended Data Fig. 5a, b). In general, flatworm genome comparisons resulted in alignment chains that were much shorter and lower scoring than those obtained from comparisons across the tetrapod (human–frog) or vertebrate (human–zebrafish) clades (Fig. 3b). Together with more than 1,000 likely planarian-specific protein coding genes (Supplementary Information S16, Supplementary Table 5, Extended Data Fig. 6a–g), our data show a high degree of genome divergence between S. mediterranea and other flatworms.
We therefore next investigated gene loss in planarians. Our analysis deliberately focused on highly conserved genes, such that the absence of sequence similarity alone provides a strong indication of loss (Supplementary Information S17). We identified 452 highly conserved genes that were lost in both S. mediterranea and other planarians (Fig. 3c), which compares to 284 and 757 such losses in D. melanogaster and C. elegans, respectively (Extended Data Fig. 5c). Gene loss in planarians is therefore broadly in the same range as in established invertebrate model organisms. However, the lost genes included 124 homologues of genes that are essential in humans or mice (Supplementary Table 6) and are generally key components of multiple cell biological core mechanisms (Fig. 3c). Specifically, planarians lack multiple highly conserved components of DNA double-stranded break (DSB) repair, including RAD52, XRCC4, NHEJ1 (also known as XLF), SMC5, SMC6 and the entire condensin II complex23. A possibly consequent reliance on mutagenic DSB repair pathways (for example, micro-homology-mediated end joining)24 could account for both the abundance of microsatellite repeats and the structural divergence of the S. mediterranea genome (Fig. 3b), but raises questions regarding the extraordinary resistance of planarians to DSB-inducing γ-irradiation4.
Planarians are also lacking recognizable homologues of key metabolic genes. Loss of the fatty acid synthase (FASN) gene is striking in the face of its essential role in de novo fatty acid synthesis in eukaryotes, and may indicate that planarians are particularly dependent on dietary lipids. The loss of the haem breakdown enzyme genes HMOX1 and BLVRB despite maintained haem biosynthesis capacity25 is similarly unusual for a free-living eukaryote (both are lost in C. elegans26). Remarkably, the above and multiple other genes were missing not only in planarians, but also in the parasite genomes6 and the transcriptome of the macrostomid M. lignano27 (Fig. 3c). Given their broad conservation in the lophotrochozoan sister clade, the broad absence of these genes in flatworms is likely to represent an ancestral loss. This complicates, for example, the interpretation of FASN loss in the parasitic lineages as a specific adaptation to parasitism6. Conversely, the absence of key metabolic genes as phylogenetic signal underscores the utility of free-living flatworms as model systems for the parasitic lineages and the development of anti-helminth reagents8.
A MAD1-MAD2-independent spindle check-point?
The apparent absence of MAD1 and MAD2 in planarians (Fig. 3c) raises the question of whether planarians have a functional SAC, and how essential cellular functions can be maintained in the absence of supposed core components. Both MAD1 and MAD2 (also known as MAD1L1 and MAD2L1) are near-universally conserved owing to their essential roles in the SAC, which guards against aneuploidy28 by inhibiting cell cycle progression as long as even a single chromosome remains unattached to the mitotic spindle14. Although MAD1 and MAD2 homologues are easily identifiable in all other flatworms examined (Extended Data Figs 7, 8), not even flatworm queries could identify significant homologues in S. mediterranea or the transcriptomes of five other planarian species. Therefore, planarians are likely to have lost MAD1, MAD2 and multiple other SAC components (Fig. 4a). The known M-phase arrest of planarian cells upon pharmacological interference with spindle function29 (Fig. 4b) is therefore remarkable, as it indicates the maintenance of a SAC-like response despite a lack of supposed SAC core components.

a, Cartoon illustration of SAC core components and function. Black and red denote components conserved or missing in S. mediterranea, respectively. KMN network: KNL1, MIS12 complex, NDC80 complex. b, Fractional abundance of mitotic cells under RNAi targeting the indicated SAC component genes, with (red) and without (cyan) nocodazole pre-treatment. Values are shown as mean with 95% confidence intervals (n?=?4 biological replicates, 10 pooled animals, 5 technical replicates with 5 or 6 images each). Cells treated with RNAi targeting CDC20 are shown as single replicates owing to rapid stem cell loss (Supplementary Information S18, Extended Data Fig. 9a, b). Significance assessed by two-way ANOVA, followed by Dunnett’s post-hoc test (****P?<?0.0001; NS, not significant), excluding RNAi targeting CDC20.
To explore the underlying mechanisms of the SAC-like response in S. mediterranea, we targeted remaining components of the SAC network (Fig. 4a) by RNA interference (RNAi) and quantified the fraction of M-phase arrested cells with or without the microtubule depolymerizing drug nocodazole (Fig. 4b, Supplementary Information S18). The marked increase in the proportion of M-phase cells and the subsequent loss of dividing cells under RNAi targeting CDC20 (Fig. 4b, Extended Data Fig. 9a) or the anaphase-promoting complex/cyclosome (APC/C) subunit gene CDC2330 indicate that APC/C inhibition remains rate limiting for progression from M-phase in planaria. The SAC-mediated regulation of CDC20 in human cells involves the recruitment of MAD1 and MAD2 to the kinetochore by two molecular complexes thought to act in parallel, the broadly conserved KNL1–BUB3–BUB1 (KBB) complex and the ROD–ZW10–ZWILCH (RZZ) complex, which has been studied less because of its absence in yeast31 (Fig. 4a). The lack of clear KNL1 and MIS12 homologues, and of a cell-cycle phenotype of RNAi targeting BUB3 (Fig. 4b), indicates that planarians have lost KBB complex function. However, we could identify clear RZZ complex homologues and, notably, knockdown of these homologues prevented nocodazole-mediated M-phase arrest without affecting basal stem cell numbers or proliferation (Fig. 4b, Extended Data Fig. 9b). Therefore, planarian RZZ components control APC/C–CDC20 either independently of MAD1 and MAD2 or in concert with homologues that have lost defining sequence features (Extended Data Figs 6, 7). Our results motivate the examination of putative MAD1 and MAD2-independent roles of the RZZ complex in other model systems and, together with the striking evolutionary plasticity of the SAC network in eukaryotes13, generally challenge our understanding of a core cell biological mechanism.
Discussion
We have described the highly contiguous genome sequence of the planarian model species S. mediterranea, which enables the genomic analysis of whole-body regeneration, stem cell pluripotency, lack of organismal ageing and other notable features of this model system. The resulting bird’s eye view of a ‘difficult’ genome using long-read sequencing and de novo assembly also highlights important challenges that remain to be overcome. In the case of S. mediterranea, these include an abundance of low-complexity microsatellite repeats, inbreeding-resistant heterozygosity and a new class of extraordinarily long LTR elements. However, the fact that the scaffold size of newly reported genome assemblies often remains substantially below the 3.85?Mb of the S. mediterranea assembly (Extended Data Table 1) indicates that similar challenges may be widespread. We therefore expect that the specific improvements of the MARVEL assembler towards heterozygous and/or compositionally biased sequencing data15 will be useful for enhancing assembly contiguity in de novo genome sequencing projects.
We have also found a high degree of structural rearrangement and the absence of a number of conserved genes in the S. mediterranea genome. However, D. melanogaster, C. elegans and other animals also show loss of ‘essential’ genes13,26,32, which raises a general conundrum: how can animals survive and compete while lacking core components of essential mechanisms? In cell biological terminology, a core mechanism signifies a chain of molecular interactions that explain a given process in multiple species, while essentiality indicates importance for organismal survival. The emergence of viable yeast strains upon deletion of essential genes33 or the competitiveness of hundreds of extant planarian species in a diversity of habitats worldwide34 both make it clear that essentiality is relative. The demonstration of SAC function in the likely absence of MAD1 and MAD2 suggests that our genetic and mechanistic understanding of SAC function is incomplete. Further studies on planarians and other ‘non-traditional’ model organisms are needed to understand the basis and mechanism of these cellular functions. Such a function-oriented, rather than gene-centric, view of biological mechanisms abstracts general function from individual molecules and is therefore likely to ultimately facilitate the reverse engineering of biology.
Data Availability
The S. mediterranea genome assembly is accessible at GenBank under accession number NNSW00000000 and can also be browsed at and downloaded from http://planmine.mpi-cbg.de. All DNA and RNA reads were deposited at the Sequence Read Archive under the bioproject accession PRJNA379262 and under the following SRA accession numbers: PacBio P4/C2 data, SRX2700681 and SRX2700682; PacBio P6/C4 data, SRX2700683; PacBio CCS data, SRX2700684; DNA shotgun, SRX2700686; DNA Chicago, SRX2700687; and RNA-seq, SRX2700685.
Code Availability
The current source code of the MARVEL assembler is available at https://github.com/schloi/MARVEL. The execution scripts used for S. mediterranea can be found in the smed subfolder of the examples folder.
Accession codes
Primary accessions
BioProject
NCBI Reference Sequence
Sequence Read Archive
References
Rink, J. C. Stem cell systems and regeneration in planaria. Dev. Genes Evol. 223, 67–84 (2013)
Saló, E. & Agata, K. Planarian regeneration: a classic topic claiming new attention. Int. J. Dev. Biol. 56, 3–4 (2012)
Reddien, P. W. & Sánchez Alvarado, A. Fundamentals of planarian regeneration. Annu. Rev. Cell Dev. Biol. 20, 725–757 (2004)
Wagner, D. E., Wang, I. E. & Reddien, P. W. Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 332, 811–816 (2011)
Onal, P. et al. Gene expression of pluripotency determinants is conserved between mammalian and planarian stem cells. EMBO J. 31, 2755–2769 (2012)
Tsai, I. J. et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature 496, 57–63 (2013)
Laumer, C. E., Hejnol, A. & Giribet, G. Nuclear genomic signals of the ‘microturbellarian’ roots of platyhelminth evolutionary innovation. eLife 4, e05503 (2015)
Collins, J. J., III & Newmark, P. A. It’s no fluke: the planarian as a model for understanding schistosomes. PLoS Pathog. 9, e1003396 (2013)
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008)
Robb, S. M. C., Gotting, K., Ross, E. & Sánchez Alvarado, A. SmedGD 2.0: The Schmidtea mediterranea genome database. Genesis 53, 535–546 (2015)
Nishimura, O. et al. Unusually large number of mutations in asexually reproducing clonal planarian Dugesia japonica. PLoS One 10, e0143525 (2015)
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009)
van Hooff, J. J., Tromer, E., van Wijk, L. M., Snel, B. & Kops, G. J. Evolutionary dynamics of the kinetochore network in eukaryotes as revealed by comparative genomics. EMBO Rep. 18, 1559–1571 (2017)
Musacchio, A. & Salmon, E. D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 (2007)
Nowoshilow, S. et al. The axolotl genome and the evolution of key tissue formation regulators. Nature https://doi.org/10.1038/nature25458 (2018)
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017)
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016)
Brandl, H. et al. PlanMine—a mineable resource of planarian biology and biodiversity. Nucleic Acids Res. 44, D764–D773 (2016)
Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002)
Macas, J. & Neumann, P. Ogre elements—a distinct group of plant Ty3/gypsy-like retrotransposons. Gene 390, 108–116 (2007)
Guo, L., Zhang, S., Rubinstein, B., Ross, E. & Alvarado, A. S. Widespread maintenance of genome heterozygosity in Schmidtea?mediterranea. Nat. Ecol. Evol. 1, 0019 (2016)
Wasik, K. et al. Genome and transcriptome of the regeneration-competent flatworm, Macrostomum lignano. Proc. Natl Acad. Sci. USA 112, 12462–12467 (2015)
Lai, A. G., Kosaka, N., Abnave, P., Sahu, S. & Aboobaker, A. A. The abrogation of condensin function provides independent evidence for defining the self-renewing population of pluripotent stem cells. Dev. Biol. 433, 218–226 (2018)
Ceccaldi, R., Rondinelli, B. & D’Andrea, A. D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64 (2016)
Stubenhaus, B. M. et al. Light-induced depigmentation in planarians models the pathophysiology of acute porphyrias. eLife 5, e14175 (2016)
Rao, A. U., Carta, L. K., Lesuisse, E. & Hamza, I. Lack of heme synthesis in a free-living eukaryote. Proc. Natl Acad. Sci. USA 102, 4270–4275 (2005)
Grudniewska, M. et al. Transcriptional signatures of somatic neoblasts and germline cells in Macrostomum lignano. eLife 5, e20607 (2016)
Santaguida, S. & Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 16, 473–485 (2015)
McWhinnie, M. A. & Gleason, M. M. Histological changes in regenerating pieces of Dugesia dorotocephala treated with colchicine. Biol. Bull. 112, 371–376 (1957)
Kang, H. & Sánchez Alvarado, A. Flow cytometry methods for the study of cell-cycle parameters of planarian stem cells. Dev. Dyn. 238, 1111–1117 (2009)
Silió, V., McAinsh, A. D. & Millar, J. B. KNL1-Bubs and RZZ provide two separable pathways for checkpoint activation at human kinetochores. Dev. Cell 35, 600–613 (2015)
Sekelsky, J. DNA repair in Drosophila: mutagens, models, and missing genes. Genetics 205, 471–490 (2017)
Rancati, G. et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135, 879–893 (2008)
Schockaert, E. R. et al. Global diversity of free living flatworms (Platyhelminthes, ‘Turbellaria’) in freshwater. Hydrobiologia 595, 41–48 (2008)
Wurtzel, O. et al. A generic and cell-type-specific wound response precedes regeneration in planarians. Dev. Cell 35, 632–645 (2015)
Acknowledgements
We thank J.-H. Lee for multiple sequence alignments; T. Boothe and M. V. Farré for karyotyping; A. Hejnol, A. Desai and J. Mansfeld for critical reading of the manuscript; and DoveTail Genomics staff for graphical support. We thank the following MPI-CBG facilities for their support: DNA sequencing, Scientific computing and Light microscopy. We thank V. Benes and the EMBL GeneCore and A. Dahl and the Deep Sequencing Group (SFB 655/BIOTEC) for RNA sequencing, and S. von Kannen, H. Andreas, S. Clausing and N. Gscheidel for technical support. This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement number 649024) and the Max Planck Society. G.R.Y. was supported by the Francis Crick Institute under award FC001162 (J. P. Stoye). The Crick Institute receives its core funding from Cancer Research UK, the UK Medical Research Council and the Wellcome Trust.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.C.R., E.M., S.S.; methodology: M.A.G., M.P., A.R., G.R.Y., S.W., H.B., I.H., M.H., J.C.R; formal analysis: M.A.G., M.P., A.R., G.R.Y., H.B., I.H., S.W., M.H.; investigation: M.A.G., M.P., A.R., M.H., J.C.R; writing (original draft): J.C.R.; writing (review and editing): J.C.R., E.M., M.H., S.P.; visualization: J.C.R., M.H., S.P., I.H., H.B., S.W., G.R.Y., A.R., M.P., M.A.G.; funding acquisition: J.C.R., E.M., S.S., A.D. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 S. mediterranea sequencing and assembly quality control.
a, S. mediterranea genomic DNA preparation. The established protocol (top) yields a black solution owing to co-purification of porphyrin pigments. The improved protocol (bottom) removes contaminants including the pigment and therefore results in clear preparations. b, The improved protocol consistently yields high-molecular-weight DNA, as shown by the pulse field gel electrophoresis of two independent preparations (right-hand two lanes) and DNA size markers (left-hand two lanes). c, Overview of all PacBio sequencing runs for the S. mediterranea assembly. d, Sequencing statistics of a representative PacBio RS II SMRT cell (P6/C4 chemistry). Total output: 1,053.4?Mb; reads of insert: 976.4?Mb; maximal read length: 52,441 bp. e, Connectivity matrix plot illustrating Chicago library read-pair distances after HiRise scaffolding. Colour coding identifies individual contigs contributing to the scaffold dd_Smed_g4_1. f, Mapping characteristics of S. mediterranea transcriptomes against the genome assembly with more than 60% query coverage and more than 60% sequence identity as cut-off criteria. Left, the dd_Smes_v1.PCFL transcriptome of the sequenced strain. Right, the dd_Smed_v6.PCFL transcriptome of the asexual strain. The pie charts show the absolute number and relative proportions of transcripts mapping with the indicated characteristics. g, Further analysis of the 538 non-mapping S. mediterranea transcripts from e (Supplementary Information S7). Missing gene, transcripts that map uniquely to the SmedSxl v4.0 assembly10 and have annotated orthologues in at least five other planarian species in PlanMine18. Putative contaminant, Top RefSeq BLAST hit in a likely contaminant species. Unknown, all remaining transcripts. The fact that only 46 out of 31,966 S. mediterranea transcripts are classified as genuinely missing indicates that the S. mediterranea assembly is largely complete. In contrast, 1,229 transcripts that uniquely mapped to the S. mediterranea genome and had orthologues in at least five other planarian species failed to map to the previously published SmedSxl v4.0 assembly10. Substantial gaps in the previous assembly also mean that the number of missing genes in the S. mediterranea assembly may be slightly higher, as some may have been classified as unknown.
Extended Data Figure 2 Assembly validation by high stringency transcript back-mapping.
a, Quality control of the S. mediterranea assembly by means of high stringency back-mapping of 1,509 high confidence (HC)-cDNAs. HC-cDNAs were defined as having BLAST hits with more than 90% query and subject coverage in seven other planarian transcriptomes in PlanMine18. HC-cDNAs were mapped to the S. mediterranea assembly using more than 90% query coverage and sequence identity as cut-off criteria. The pie chart shows the absolute number and relative proportions of HC-cDNAs mapping with the indicated characteristics. b, Further analysis of the ten HC-cDNAs classified as non-mapping from a by intersection with the mapping results of Extended Data Fig. 1g. Of these, two were designated as ‘false negative’, as both mapped to the S. mediterranea genome with more than 90% query coverage and sequence identity using BLAT. c, UCSC genome browser screenshot (75-kb window) of the genomic mapping location of one of the two ‘unknown’ HC-cDNAs as a single example of a mapping failure due to an actual assembly error. The example documents inversion of the 5′-end of the cDNA within a low-confidence stretch at a contig end (lack of coverage in the Quiver track). The inversion is supported by inverted RNA-seq read mapping and inversion of the cDNA sequence shown in the respective tracks. Below, colour-coded Miropeats similarity plots of respective regions. d, e, Examples of genomic mapping loci of HC-cDNA transcripts from the multi-mapping category in a, browser screen shots as described in c. d, Example of a likely legitimate (biological) gene duplication in a gap-free high-confidence region. e, Micro tandem duplication surrounding a scaffolding gap in a repeat-rich region. f, Multi-mapping HC-cDNAs map preferentially to contig ends. The histogram plots the distance to the closest gap or contig end for the 67 multi-mappers and a corresponding number of unique mappers (a). g, Estimated size of the duplicated regions of multi-mapping HC-cDNAs. This analysis identifies a small fraction of small-scale duplications at assembly gaps in the S. mediterranea assembly, which can be easily identified with the help of the various quality control tracks in the PlanMine genome browser.
Extended Data Figure 3 Repeats in the S. mediterranea assembly.
a, Estimation of the abundance of solo and full-length LTR elements in the S. mediterranea assembly. Elements SLF-8 and SLF-14 show a large number of solo LTRs compared to full-length copies, indicating a large number of excision events by homologous recombination. Of the Burro elements, Burro-1 was the most abundant, with 124 full-length copies, followed by Burro-3 and Burro-2 with 25 and 23 full-length copies, respectively. b, Comparison of lengths of indicated repeat consensus classes in H. sapiens, D. melanogaster and C. elegans. For S. mediterranea, we used a custom library generated in this study. Dark colours indicate predominant lengths of specific repeat classes. Red, repeat consensuses more than 15?kb in length. c, Expression analysis of gypsy LTR elements in S. mediterranea RNA-seq data using TETranscripts. The three most transcriptionally active elements were Burro-1, Burro-2 and SLF-8. d, LTR expression analysis by whole-mount in situ hybridization and single-cell expression data35. Top, SLF-9-derived transcript. Bottom, Burro-1-derived transcript. Both are broadly transcribed in many S. mediterranea cell types (CIW4 strain, n?=?1 biological replicate, 10 animals). Scale bar, 250?μm. e, Kimura distance plot of S. mediterranea LTR elements. Substitution levels varied by element, but also within element groups. Burro-1, Burro-2, Burro-3 and SLF-8 all contain elements spread over a large range of substitution levels, possibly indicative of continued activity over large time scales. The remaining elements are characterized by more defined peaks in expansion, with the highest average divergences being seen in the smallest elements characterized (SLF-10, SLF-11, SLF-12), making these amongst the oldest within the genome. Notably, both SLF-8 and SLF-9 have representative elements with particularly low substitution rates, potentially indicating a recent or ongoing expansion.
Extended Data Figure 4 AT-rich microsatellites in the S. mediterranea genome.
a, Features of AT-rich microsatellites. Top, inter-repeat spacing of repeats more than 99 bp in length. Bottom, repeat length. AT-rich microsatellites with an average length of 127 bp occur every approximately 3,500 bp. b, Genomic distribution of repeats more than 99 bp in length. c, Increased probability of read alignment termination within microsatellite repeats. Individual size bins were analysed separately for microsatellite repeats (red) or non-repetitive regions (cyan). Although accounting for only 4.2% of the assembly size, microsatellite repeats significantly limit assembly contiguity owing to an increased probability of read alignment loss. d, Genome-wide coverage ratios of insertion or deletion sequences more than 99 bp in length and excluding AT repeats. e, Read length variation analysis across AT-rich repeat regions (AAT) in regular PacBio sequencing data compared to circular consensus sequencing (CCS) coverage of the same region. CCS reads sample the same genomic region multiple times. The lack of a clear difference in the length variation of specific AT repeats between repetitive sequencing of the same DNA molecule (CCS) and that of sequencing reads representing different DNA molecules (regular PacBio data, P6/C4) indicates that repeat length variations are mainly technical in nature. Rather than repeat length polymorphisms, the most likely cause of the detrimental effect of the repeats is the increased ambiguity in low complexity sequence alignments (Supplementary Information S11.4). Unique (UQ) regions were included as controls. CCS_UQ: CCS subread length variation versus the consensus length of all subreads in binned unique regions (n?=?3,300). CCS_AAT: CCS subread length variation versus the consensus length of all subreads in binned AT repeat regions (n?=?4,825). P6_UQ: Length variation of individual reads in the regular PacBio sequencing data (P6/C4) versus the consensus length of the region in the S. mediterranea assembly in binned unique regions (n?=?3,310). P6_AAT: Length variation of individual reads in the regular PacBio sequencing data (P6/C4) versus the consensus length of the region in the S. mediterranea assembly in binned AT repeat regions (n?=?5,085). Dots, outliers; central horizontal line, median; box ranges, from first quartile to third quartile; whiskers, interquartile range (midspread): 75th and 25th percentile.
Extended Data Figure 5 Comparative genomics.
a, Contig and scaffold N50 statistics of the genomes used for the comparative genome alignments in Fig. 3b. The basal vertebrate lamprey genome assembly is more fragmented (similar or lower N50 values) than most other platyhelminth genomes. Nevertheless, the human-to-lamprey genome alignment has equivalent or even higher alignment chain scores and span lengths than any of the platyhelminth genome alignment comparisons, indicating that the true extent of sequence divergence and loss of conserved gene order in platyhelminths is likely to be greater than estimated. b, Example of a top-scoring alignment chain. The UCSC genome browser screenshot of the S. mediterranea genome shows that alignments predominantly overlap exons of the two transcripts shown at the top. This example is one of the few cases of apparent gene order conservation between S. mediterranea and S. mansoni. Blocks in the alignment chains represent local alignments, connecting single lines represent deletions in the query genome and double lines represent regions with sequence in both S. mediterranea and the query genome that do not align. c, Comparative loss analysis of highly conserved genes across the 26 indicated species. Red, conserved gene fraction, defined as the proportion of orthogroups containing at least 9 out of the 14 non-flatworm species and the query species. Blue, lost fraction of highly conserved genes, defined as the proportion of orthogroups containing at least 9 out of the 14 non-flatworm species, but not the query species (Supplementary Information S17). Absolute numbers of highly conserved genes are shown at the top, with slight fluctuations caused by species-specific sequence duplications.
Extended Data Figure 6 Planarian-specific genes.
a, Conservation of 1,165 flatworm-specific genes (Supplementary Information S16.1) amongst flatworm species. Only 61 sequences had sequence homologues in the indicated flatworm species (other denotes Taenia solium, Echinococcus multilocularis, Echinococcus granulosus, Hymenolepis microstoma), indicating that this gene set mostly represents planarian-specific genes. b, c, Characteristics of planarian-specific genes. b, Distribution of exon numbers compared to a control gene set (HC-cDNAs; Extended Data Fig. 2a), indicating enrichment of single-exon genes. c, Number of predicted domains (InterProScan), indicating that only a minority of genes contain predicted domains. d, Identity of detected domains (Pfam and SUPERFAMILY). Unintegrated signatures, recurring sequence motifs that are not grouped into InterPro entries. These might represent so far uncurated or weakly supported motifs that do not pass InterPro’s integration standards. e, Differential expression of 626 planarian-specific genes in published S. mediterranea RNA-seq data sets of different regeneration phases (left), stem cells or progeny populations (middle) or specific developmental stages (right). Red lines indicate differential expression relative to the control of each series (white indicates no change). Genes were ordered using rank by sum. The high proportion of differential expression indicates the widespread contribution of lineage-specific genes to planarian biology. f, g, Specific examples of non-conserved genes. Top, SMART domain representation. Bottom, Differential expression under the indicated conditions.
Extended Data Figure 7 Sequence conservation of MAD1 protein in non-planarian flatworms.
a, COBALT multiple protein sequence alignment of the MAD1 homologues of the indicated species (including all the non-planarian flatworm species from Fig. 3c). b, Heat map of BLOSUM62 sequence similarity matrix generated from alignment in a, demonstrating significant sequence conservation of MAD1 homologues even in flatworms.
Extended Data Figure 8 Sequence conservation of MAD2 protein in non-planarian flatworms.
a, COBALT multiple protein sequence alignment of the MAD2 homologues of the indicated species (including all the non-planarian flatworm species from Fig. 3c). b, Heat map of BLOSUM62 sequence similarity matrix generated from alignment in a), demonstrating significant sequence conservation of MAD2 homologues even in flatworms.
Extended Data Figure 9 Effect of RNAi targeting CDC20 and SAC components on the planarian stem cell compartment.
a, Fluorescent whole-mount in situ hybridization of the planarian head region. Stem cells (neoblasts) were visualized using a smedwi-1 probe (red), early and late progeny by pooled prog-1 and agat-1 probes (green). Blue, nuclear counterstaining by DAPI. Top, RNAi control against EGFP; bottom, RNAi targeting CDC20, which results in a markedly decreased number of smedwi-1 and prog-1/agat-1 positive cells after three rounds of RNAi feeding. This indicates the loss of neoblasts and a concomitant reduction in progenitor numbers (n?=?1 biological replicate, 10 animals). Scale bar, 200?μm. b, Effect of indicated RNAi treatments on planarian stem cell abundance. Representative images of cell macerates, stained with DAPI (nuclei, blue), anti-H3ser10P (mitotic cells, magenta) and smedwi-1 in situ hybridization (stem cells, yellow). Numbers indicate the mean fraction?±?s.d. of smedwi-1 positive cells of total cells quantified by nuclear counting using DAPI (n?=?1, 10 pooled animals, 5 technical replicates with 5 images each). Scale bar, 50?μm.
Supplementary information
Supplementary Information
This file contains Supplementary Data S1-S19, Supplementary Table legends 1-7 and Supplementary references. (PDF 1284 kb)
Supplementary Table
This file contains Supplementary Table 1. (XLSX 49 kb)
Supplementary Table
This file contains Supplementary Table 2. (XLSX 11 kb)
Supplementary Table
This file contains Supplementary Table 3. (XLS 29 kb)
Supplementary Table
This file contains Supplementary Table 4. (XLS 32 kb)
Supplementary Table
This file contains Supplementary Table 5. (XLS 177 kb)
Supplementary Table
This file contains Supplementary Table 6. (XLS 126 kb)
Supplementary Table
This file contains Supplementary Table 7. (XLSX 8 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Grohme, M., Schloissnig, S., Rozanski, A. et al. The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms. Nature 554, 56–61 (2018). https://doi.org/10.1038/nature25473
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25473
This article is cited by
-
Why study sleep in flatworms?
Journal of Comparative Physiology B (2024)
-
Nuclear genome annotation of wheel animals and thorny-headed worms: inferences about the last common ancestor of Syndermata (Rotifera s.l.)
Hydrobiologia (2024)
-
CRISPR/Cas9-based depletion of 16S ribosomal RNA improves library complexity of single-cell RNA-sequencing in planarians
BMC Genomics (2023)
-
The BAF chromatin remodeling complex licenses planarian stem cells access to ectodermal and mesodermal cell fates
BMC Biology (2023)
-
The planarian flatworm Schmidtea mediterranea
Nature Methods (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
