
Abstract
Constituting approximately 10% of flowering plant species, orchids (Orchidaceae) display unique flower morphologies, possess an extraordinary diversity in lifestyle, and have successfully colonized almost every habitat on Earth1,2,3. Here we report the draft genome sequence of Apostasia shenzhenica4, a representative of one of two genera that form a sister lineage to the rest of the Orchidaceae, providing a reference for inferring the genome content and structure of the most recent common ancestor of all extant orchids and improving our understanding of their origins and evolution. In addition, we present transcriptome data for representatives of Vanilloideae, Cypripedioideae and Orchidoideae, and novel third-generation genome data for two species of Epidendroideae, covering all five orchid subfamilies. A. shenzhenica shows clear evidence of a whole-genome duplication, which is shared by all orchids and occurred shortly before their divergence. Comparisons between A. shenzhenica and other orchids and angiosperms also permitted the reconstruction of an ancestral orchid gene toolkit. We identify new gene families, gene family expansions and contractions, and changes within MADS-box gene classes, which control a diverse suite of developmental processes, during orchid evolution. This study sheds new light on the genetic mechanisms underpinning key orchid innovations, including the development of the labellum and gynostemium, pollinia, and seeds without endosperm, as well as the evolution of epiphytism; reveals relationships between the Orchidaceae subfamilies; and helps clarify the evolutionary history of orchids within the angiosperms.
Similar content being viewed by others

Main
The Apostasioideae are a small subfamily of orchids that includes only two genera (Apostasia and Neuwiedia2,5), consisting of terrestrial species confined to the humid areas of southeast Asia, Japan, and northern Australia6. Although Apostasioideae share some synapomorphies with other orchids (for example, small seeds with a reduced embryo and a myco-heterotrophic protocorm stage), they possess several unique traits, the most conspicuous of which is their floral morphology7. Apostasia has a non-resupinate, solanum-type flower with anthers closely encircling the stigma (including postgenital fusion), a long ovary, and an actinomorphic perianth with an undifferentiated labellum. Three stamens (two of which are fertile) are basally fused to the style, forming a relatively simple gynostemium, and the anthers contain powdery pollen (grains not unified into pollinia). These characteristics (Extended Data Fig. 1a) differ from those of other Orchidaceae subfamilies, which have three sepals, three petals (of which one has specialized to form the labellum), and stamens and pistil fused into a more complex gynostemium (Extended Data Fig. 1b), but are similar to those of some species of Hypoxidaceae (a sister family to Orchidaceae, in the order Asparagales).
We sequenced the A. shenzhenica genome using a combination of different approaches; the total length of the final assembly was 349 Mb (see Methods and Supplementary Tables 1–4). We confidently annotated 21,841 protein-coding genes, of which 20,202 (92.50%) were supported by transcriptome data (Supplementary Fig. 1 and Supplementary Table 5). Using single-copy orthologues, we performed a BUSCO8 assessment that indicated that the completeness of the genome was 93.62%, suggesting that the A. shenzhenica genome assembly is of high quality (Supplementary Table 6). For comparative analyses, we also improved the quality of the previously published genome assemblies of the orchids Phalaenopsis equestris9 and Dendrobium catenatum10 (see Methods and Supplementary Tables 6 and 7).
We constructed a high-confidence phylogenetic tree and estimated the divergence times of 15 plant species using genes extracted from a total of 439 single-copy families (Fig. 1 and Extended Data Fig. 2). We undertook a computational analysis of gene family sizes (CAFÉ 2.211) to study gene family expansion and contraction during the evolution of orchids and related species (Fig. 1 and Supplementary Note 1.1). By comparing 12 plant species, we found 474 gene families (Extended Data Fig. 3) that appeared unique to orchids (Supplementary Note 1.2). Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis found these gene families to be specifically enriched in the terms ‘O-methyltransferase activity’, ‘cysteine-type peptidase activity’, ‘flavone and flavonol biosynthesis’ and ‘stilbenoid, diarylheptanoid and gingerol biosynthesis’ (Supplementary Note 1.2).

The phylogenetic tree shows the topology and divergence times for 15 plant species. As expected, as a member of the Apostasioideae, A. shenzhenica is sister to all other orchids. In general, the estimated orchid divergence times are in good agreement with recent broad scale orchid phylogenies2,3. Divergence times are indicated by light blue bars at the internodes; the range of these bars indicates the 95% confidence interval of the divergence time. Numbers at branches indicate the expansion and contraction of gene families (see Methods and Extended Data Fig. 2). MRCA, most recent common ancestor. The number in parentheses is the number of gene families in the MRCA as estimated by CAFÉ11.
Distributions of synonymous substitutions per synonymous site (KS) (see Supplementary Note 2.1) for paralogous A. shenzhenica genes showed a clear peak at KS ≈ 1 (Extended Data Fig. 4). Similar peaks at KS values of 0.7 to 1.1 were identified in 11 other orchids, covering all 5 orchid subfamilies (Supplementary Fig. 2). These peaks might reflect multiple independent whole-genome duplication (WGD) events across orchid sublineages or, more parsimoniously, a single WGD event shared by all (extant) orchids. Comparisons of orchid paralogue KS distributions with KS distributions of orthologues between orchid species, and between orchids and Asparagus officinalis (asparagus, Asparagaceae, a sister family to Orchidaceae in the order Asparagales) (Fig. 2a and Supplementary Note 2.1) indicated that the WGD signatures are not shared with non-orchid Asparagales. Absolute phylogenomic dating12 (Extended Data Fig. 5 and Supplementary Note 2.1) revealed that the WGDs and the earliest diversification of extant orchid lineages occurred relatively close together in time, supporting the possibility of a single WGD event in the most recent common ancestor of extant orchids.

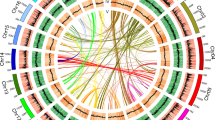
a, Distribution of KS for the one-to-one P. equestris–D. catenatum, A. shenzhenica–D. catenatum, A. shenzhenica–P. equestris and A. shenzhenica–A. officinalis orthologues (filled grey curves and left-hand y-axis). Distribution of KS for duplicated anchors found in co-linear regions of A. shenzhenica (green lines), D. catenatum (red lines) and P. equestris (blue lines). The filled grey curves and dashed coloured lines are actual data points from the distributions; the solid coloured lines are kernel density estimates (KDE) of the anchor-pair (duplicated genes found in co-linear regions) data scaled to match the corresponding dashed lines. All anchor-pair data are scaled up ×15 (right-hand y-axis) compared to the orthologue data. b, Syntenic dot plot of the self-comparison of A. shenzhenica. Only co-linear segments with at least 15 anchor pairs are shown. The sections on each scaffold with co-linear segments are shown in grey. The red bars below the dot plot illustrate the duplication depths (the number of connected co-linear segments overlapping at each position; see Methods). The co-linear regions in green indicate the four co-linear segments that have a common orthologous co-linear segment in A. trichopoda as shown in (c). c, Co-linear alignment of A. shenzhenica and A. trichopoda. The colours of genes in the alignment indicate gene orientation, with blue for forward strands and green for reverse strands. The grey links connect orthologues between A. shenzhenica and A. trichopoda. Scf86, scaffold00086 of the A. trichopoda genome (v1.0).
Intragenomic co-linearity and synteny analysis identified two WGD events in A. shenzhenica (Fig. 2b, Supplementary Fig. 3 and Supplementary Note 2.2). Co-linearity and synteny analyses between A. shenzhenica and Amborella trichopoda, and between A. shenzhenica and Vitis vinifera, also support at least two WGDs in A. shenzhenica (Supplementary Figs 4 and 5); for example, four paralogous segments in A. shenzhenica corresponded to one orthologous region in A. trichopoda (Fig. 2c). Detailed genome comparisons of A. shenzhenica with Ananas comosus (pineapple) and A. officinalis revealed a specific 4:4 co-linearity pattern (Extended Data Fig. 6 and Supplementary Figs 6–8) that is consistent with the two monocot WGDs proposed for A. comosus, indicating that all three species possess a similar evolutionary history with regard to WGDs (Supplementary Note 2.2). Together, these patterns of co-linearity suggest that the older of the two WGDs evident in A. shenzhenica is likely to be shared with A. comosus and A. officinalis (representing the τ WGD13,14 shared by most monocots), and corroborate the idea that the younger WGD represents an independent event, specific to the Orchidaceae lineage. Analyses of gene trees that contained at least one paralogue pair from co-linear regions from one of the three orchid genomes placed the younger A. shenzhenica WGD and the P. equestris and D. catenatum WGDs on the orchid stem branch, and also provided additional evidence for the monocot τ WGD13,14 (Fig. 3 and Supplementary Note 2.3). We therefore find strong support for a WGD event shared by all extant orchids, which is likely to be only slightly older than their earliest divergence and might be correlated with orchid diversification. In addition, as observed for many other plant lineages, this orchid-specific WGD might be associated with the Cretaceous/Palaeogene boundary15.

The numbers on the branches of the species tree indicate the number of gene families with one or more anchor pairs from at least one of the three orchids with genomes that coalesced on the respective branch (top), as well as the individual contributions of anchor pairs from the three orchids (bottom; A, A. shenzhenica; D, D. catenatum; P, P. equestris). The two WGD events identified are depicted by stars. Species with published genomes are in bold. All the duplication events have bootstrap values over 80% (see Methods; for results for bootstrap values over 50% see Supplementary Fig. 15).
Apostasia presents a number of characters that are plesiomorphic in orchids, such as an actinomorphic perianth with an undifferentiated labellum, a gynostemium with partially fused androecium and gynoecium, pollen that is not aggregated into pollinia, and underground roots for terrestrial growth1,5,6,7. The A. shenzhenica genome contains 36 putative functional MADS-box genes (Table 1, Supplementary Table 8 and Supplementary Fig. 9), 27 of which are type II MADS-box genes (Table 1). Two type II MADS-box classes appear to be reduced: A. shenzhenica seems to have fewer genes in the B-AP3 (two members) and E classes (three members) than P. equestris (four B-AP3 and six E-class members) and D. catenatum (four B-AP3 and five E-class members) (Fig. 4a). Previous studies have shown that expanded B-AP3 and E classes with members that have different expression patterns in floral organs are associated with the innovation of the unique labellum and gynostemium in orchids9,16,17, and that duplicated B-AP3 genes are responsible for the modularization of the perianth of orchid flowers18. We identified B-AP3 genes from the transcriptomes of species of each of the orchid subfamilies and the B-class MADS-box genes from the floral transcriptome data of Molineria capitulata, a member of Hypoxidaceae that possesses a flower with petaloid tepals and powdery pollen (similar to that found in Apostasia). We found one member in each of the two B-AP3 subclades for both A. shenzhenica and M. capitulata, but one or two members in each B-AP3 subclade for the other orchids (Extended Data Fig. 7). All these B-AP3 genes are highly expressed in flower buds (Extended Data Fig. 7). These similarities suggest that the lower gene numbers in MADS-box B-AP3 and E classes in Apostasia represent an ancestral state, responsible for producing the plesiomorphic flower with an undifferentiated labellum and partially fused gynostemium. The B-AP3 and E classes may have expanded independently only in the non-apostasioid orchids or, alternatively, in the common ancestor of all extant orchids, possibly as a result of the shared orchid WGD, with subsequent loss of paralogous genes in Apostasia causing reversion to the ancestral state. The B-AP3 gene tree topology and some evidence from co-linearity analysis of orchid B-AP3 genes (Supplementary Fig. 10) suggest the latter. We hypothesize that differential paralogue retention and subsequent sub- and neo-functionalization of B-AP3 and E-class members resulted in the derived labellum found in other orchids (Fig. 4b).

a, Phylogenetic analysis of MADS-box genes among A. shenzhenica, P. equestris, O. sativa and Arabidopsis. The B-AP3 and E-class, MIKC*, Mβ, and AGL12 and ANR1 subclades are marked by purple, orange, green and blue shading, respectively. b, A. shenzhenica, with fewer B-AP3 class and E class MADS-box genes, keeps an undifferentiated labellum and partially fused gynostemium, while P. equestris, with more B-AP3 class and E class MADS-box genes, develops the specialized labellum and column (in red). c, Loss of the P-subclade genes of MIKC* in P. equestris is likely to be related to the evolution of pollinia. d, The failed development of endosperm in orchids might be related to the missing type I Mβ MADS-box genes (Extended Data Fig. 9). e, A. shenzhenica, containing the AGL12 gene and expanded ANR1 genes, is a terrestrial orchid, while epiphytic orchids, such as P. equestris, have lost the AGL12 gene and some ANR1 genes.
The packaging of pollen grains into a compact unit known as the pollinium, specialized for transfer as a unit by pollinating vectors, was a key innovation in the evolutionary history of Orchidaceae and may have played a role in promoting the tremendous radiation of the group19. In seed plants, the P- and S-subclades of MIKC*-type genes are major regulators of male gametophytic development20,21. The P-subclade, however, is absent in all orchids except A. shenzhenica (Extended Data Fig. 8). Gene expression analysis showed that, in orchids and M. capitulata, MIKC*-type genes are expressed in the pollinia or pollen, suggesting they play roles in its development (Extended Data Fig. 9). Although most orchids have a pollinium, Apostasia has scattered pollen, similar to M. capitulata, Oryza sativa (rice), and Arabidopsis thaliana. Therefore, we propose that the loss of the P-subclade members of MIKC*-type genes is related to the evolution of the pollinium (Fig. 4a, c and Supplementary Note 3).
The reduction of seed volume and content to an absolute minimum is a pivotal aspect of Orchidaceae evolution: in all orchid species, endosperm is absent from the seed. Type I MADS-box genes are important for the initiation of endosperm development22, and transcripts of type I Mα and Mγ MADS-box genes were found in developing seeds of A. shenzhenica, P. equestris, and M. capitulata (Extended Data Fig. 10 and Supplementary Fig. 11). Notably, the three orchid genomes do not contain any type I Mβ MADS-box genes (Fig. 4a and Supplementary Fig. 12), which are found in Arabidopsis, Populus trichocarpa (poplar), O. sativa (Table 1), and in M. capitulata (Supplementary Fig. 13). The lack of endosperm in orchids might therefore be related to the missing type I Mβ MADS-box genes (Fig. 4d).
Orchids are one of very few flowering plant lineages that have been able to successfully colonize epiphytic or lithophytic niches, clinging to trees or rocks and growing in dry conditions using crassulacean acid metabolism2,9,10. The roots of epiphytic orchids, such as Phalaenopsis and Dendrobium, are extremely specialized and differ from the roots of terrestrial orchids such as Apostasia. These aerial roots develop the velamen radicum, a spongy epidermis that traps the nutrient-rich flush during rainfall, representing an important adaptation of epiphytic orchids23,24,25. The Arabidopsis AGL12 gene is involved in root cell differentiation26. A. shenzhenica contains one AGL12 clade gene, as do Arabidopsis and rice. In addition, we found transcripts similar to AGL12 in M. capitulata. In both A. shenzhenica and M. capitulata, these genes are highly expressed in root tissue (Supplementary Fig. 14). Notably, we did not find similar genes in epiphytic orchids, suggesting that the loss of these gene(s) may be involved in losing the ability to develop true roots for terrestrial growth (Fig. 4e). Utricularia gibba, an asterid in the order Lamiales (only distantly related to the orchids) that lacks true roots, also lacks these AGL12 clade or similar genes27. The Arabidopsis ANR1 gene is a key gene involved in regulating lateral root development in response to external nitrate supply28. We found that the MADS-box gene subfamily ANR1 is probably reduced in P. equestris (two members) and D. catenatum (three members), compared with four members in A. shenzhenica (Fig. 4a): this is consistent with no development of lateral (aerial) roots in epiphytic orchids.
In conclusion, the genome sequence of A. shenzhenica, an orchid belonging to a small clade that is sister to the rest of Orchidaceae, provides a reference for studying orchid evolution, revealing clear evidence of an ancient WGD shared by all orchids, facilitating reconstruction of the ancestral orchid gene toolkit, and providing insights into many orchid-specific features such as the development of the labellum and gynostemium, pollinia, and seeds without endosperm, as well as the evolution of epiphytism.
Methods
No statistical methods were used to predetermine sample size.
Sample preparation and sequencing
For genome sequencing, we collected leaves, stems, and flowers from wild A. shenzhenica, a self-pollinating species found in southeast China4 that has a karyotype of 2N = 2X = 68 with uniform small chromosomes (Supplementary Fig. 16). We extracted genomic DNA using a modified cetyltrimethylammonium bromide (CTAB) protocol. Sequencing libraries with insert sizes ranging from 180 bp to 20 kb (Supplementary Table 1) were constructed using a library construction kit (Illumina). These libraries were then sequenced using an Illumina HiSeq 2000 platform. The 80.02-Gb raw reads generated were filtered according to sequencing quality, the presence of adaptor contamination, and duplication. Only high-quality reads were used for genome assembly.
Total RNA was extracted from this study’s samples using the RNAprep Pure Plant Kit and genomic DNA contamination was removed using RNase-Free DNase I (both from Tiangen). The integrity of RNA was evaluated on a 1.0% agarose gel stained with ethidium bromide (EB), and its quality and quantity were assessed using a NanoPhotometer spectrophotometer (IMPLEN) and an Agilent 2100 Bioanalyzer (Agilent Technologies). As the RNA integrity number (RIN) was greater than 7.0 for all samples, they were used in cDNA library construction and Illumina sequencing, which was completed by Beijing Novogene Bioinformatics Technology Co., Ltd. The cDNA library was constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) and 3 μg RNA per sample, following the manufacturer’s recommendations. The PCR products obtained were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. Library preparations were sequenced on an Illumina Hiseq 2000 platform, generating 100-bp paired-end reads.
Genome size estimation and preliminary assembly
The genome size of species in Apostasioideae is between 0.38 pg and 5.96 pg31, which is relatively small compared to that of other subfamilies (ranging from 0.38 pg to 55.4 pg)32. To estimate the genome size of A. shenzhenica, we used reads from paired-end libraries to determine the distribution of K-mer values. According to the Lander–Waterman theory33, genome size can be determined by the total number of K-mers divided by the peak value of the K-mer distribution. Given only one peak in the K-mer distribution, we found that A. shenzhenica has no heterozygosity (Supplementary Fig. 17). With the peak at the expected K-mer depth and the formula genome size = total K-mer/expected K-mer depth, the size of the haploid genome was estimated to be 471.0 Mb (haploid). We used ALLPATHS-LG software34 and obtained a preliminary assembly of A. shenzhenica with a scaffold N50 size of 1.196 Mb and corresponding contig N50 size of 30.1 Kb.
PacBio library construction and sequencing and filling gaps
The preliminary assembly of A. shenzhenica and the previous published genome assemblies of P. equestris9 and D. catenatum10 were improved using PacBio and 10X Genomics Linked-Reads.
Genomic DNA was isolated from the leaves of A. shenzhenica, P. equestris and D. catenatum. For a 20-kb insert size library, at least 20 μg of sheared DNA was required. SMRTbell template preparation involved DNA concentration, damage repair, end repair, ligation of hairpin adapters, and template purification, and used AMPure PB Magnetic Beads. Finally, the sequencing primer was annealed and sequencing polymerase was bound to SMRTbell template. The instructions specified as calculated by the RS Remote software were followed. We carried out 20-kb single-molecule real-time DNA sequencing by PacBio and sequenced the DNA library on the PacBio RS II platform, yielding about 5.44 Gb (A. shenzhenica), 10.54 Gb (P. equestris) and 11.06 Gb (D. catenatum) PacBio data (read quality ≥ 0.80, mean read length of A. shenzhenica ≥ 7 Kb, of P. equestris and D. catenatum ≥ 10 Kb) (Supplementary Table 2).
We used PBjelly software35 to fill gaps with PacBio data. The options were “<blasr>-minMatch 8 -sdpTupleSize 8 -minPctIdentity 75 -bestn 1 -nCandidates 10 -maxScore -500 -nproc 10 -noSplitSubreads</blasr>” for the protocol.xml file. Then, we used Pilon36 with default settings to correct assembled errors. For the input BAM file, we used BWA to align all the Illumina short reads to the assembly and SAMTOOLS to sort and index the BAM file.
10X Genomics library construction, sequencing, and extending scaffolds
DNA sample preparation, indexing, and barcoding were done using the GemCode Instrument from 10X Genomics. About 0.7 ng input DNA with 50 kb length was used for GEM reaction procedure during PCR, and 16-bp barcodes were introduced into droplets. Then, the droplets were fractured following the purifying of the intermediate DNA library. Next, we sheared DNA into 500 bp for constructing libraries, which were finally sequenced on the Illumina HiseqXTen37 (Supplementary Table 3).
We used BWA mem to align the 10X Genomics data to the filled gaps assembly using default settings. Then, we used fragScaff38 for scaffolding. The options were as follows: A. shenzhenica (stages1 “-m 3000 -q 30”; stages2 “-C 2”; stages3 “-j 1.25 -u 2”), D. catenatum (stages1 “-m 3000 -q 30”; stages2 “-C 1”; stages3 “-j 2 -u 2”) and P. equestris (stages1 “-m 3000 -q 30”; stages2 “-C 1”; stages3 “-j 2 -u 2”)39.
The total length of the final assembly for A. shenzhenica was 349 Mb with a scaffold N50 size of 3.029 Mb and corresponding contig N50 size of 80.1 Kb. (Supplementary Table 4). For the two previously published orchid genomes of P. equestris and D. catenatum, the scaffold N50 size as well as the completeness (see below) improved considerably: for P. equestris, the scaffold N50 size increased from 359.12 Kb9 to 1.217 Mb and the corresponding contig N50 size from 20.56 Kb9 to 45.79 Kb, while for D. catenatum the scaffold N50 size increased from 391.46 Kb10 to 1.055 Mb, and the corresponding contig N50 size from 33.1 Kb10 up to 51.7 Kb (Supplementary Table 7).
Repeat prediction
A total of 146.65 Mb of repetitive elements occupying more than 42.05% of the A. shenzhenica genome were annotated using a combination of structural information and homology prediction10. Retrotransposable elements, known to be the dominant form of repeats in angiosperm genomes, constituted a large part of the A. shenzhenica genome and included the most abundant subtypes, such as LTR/Copia (4.97%), LTR/Gypsy (11.84%), LINE/L1 (2.78%) and LINE/RTE-BovB (9.32%), among others. In addition, the percentage of de novo predicted repeats was notably larger than that obtained for homologous repeats based on Repbase40, indicating that A. shenzhenica has multiple unique repeats compared with other sequenced plant species (Supplementary Table 9).
Gene and non-coding RNA prediction
MAKER41 was used to generate a consensus gene set based on de novo predictions from AUGUSTUS42 and GlimmerHMM43, homology annotation with the universal single-copy genes from CEGMA44 and the genes from Arabidopsis (TAIR10) and another four sequenced monocots (O. sativa, P. equestris, S. bicolor and Zea mays) using exonerate45, and RNA-seq prediction by Cufflinks46 and Tophat47. These results were integrated into a final set of protein-coding genes for annotation (Supplementary Table 5). Using the same annotation pipeline as for A. shenzhenica, 29,545 and 29,257 protein-coding genes were predicted for P. equestris and D. catenatum, respectively (Supplementary Table 7). A. shenzhenica was found to have a greater average gene length (here we considered the start and stop codons as the two boundaries for a gene) than most other sequenced plants, but this length was similar to that of P. equestris and D. catenatum (Supplementary Fig. 18 and Supplementary Table 10), in both of which this is due to a long average intron length9,10.
We then generated functional assignments of the A. shenzhenica genes with BLAST (version 2.2.28+) by aligning their protein-coding regions to sequences in public protein databases, including KEGG (59.3)48, SwissProt (release 2013_06)49, TrEMBL (release 2013_06)50 and NCBI non-redundant protein database (20150617), and InterProScan (v5.11-51.0)51 was also used to provide functional annotation (Supplementary Table 11). We were able to generate functional assignments for 84.2% of the A. shenzhenica genes from at least one of the public protein databases (Supplementary Table 11).
The tRNA genes were searched by tRNAscan-SE52. For rRNA identification, we downloaded the Arabidopsis rRNA sequences from NCBI and aligned them with the A. shenzhenica genome to identify possible rRNAs. Additionally, other types of non-coding RNAs, including miRNA and snRNA, were identified by using INFERNAL53 to search from the Rfam database. In the end, we identified 43 microRNAs, 203 transfer RNAs, 452 ribosomal RNAs and 93 small nuclear RNAs in the A. shenzhenica genome (Supplementary Table 12).
Transcriptome assembly
Before assembly, we got high-quality reads by removing adaptor sequences and filtered low-quality reads by using TRIMMOMATIC54 from raw reads with parameters: ILLUMINACLIP:path/adaptor:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:4:15 MINLEN:36. The resulting high-quality reads were de novo assembled and annotated with the TRINITY program55. The commands and parameters used for running TRINITY were as follows: Trinity –seqType fq –JM 200G –left sample_1.fq –right sample_2.fq –normalize_by_read_set –CPU 32 –output sample –min_kmer_cov 2. Protein sequences and coding sequences of transcripts were predicted using TransDecoder (http://transdecoder.github.io), a software tool that identifies likely coding sequences from transcript sequences and compares the translated coding sequences with the PFAM domain database55. For genes with more than one transcript, the longest one was used to calculate transcript abundance and coverage. Transcript abundance level was normalized using the fragments per kilobase per million mapped reads (FPKM) method, and FPKM values were computed as proposed by Mortazavi et al.56.
Transcriptomes of Agave deserti57 and Allium cepa58 were downloaded from Dryad (h5t68) and NCBI (PRJNA175446), respectively. We removed the redundant unigenes in A. cepa by CD-HIT-EST with 99% identity and used TransDecoder to predict proteins with default parameters.
We carried out BLASTP (E value <1 × 10−3) to search the best hits for the proteins predicted in the transcriptomes against a customized database, built with proteins from the genomes of A. shenzhenica, P. equestris9, D. catenatum10, and A. officinalis (GenBank accession number GCF_001876935.1) as well as public databases, such as NCBI Plant RefSeq (release 80), Ensembl (release 77), Ensembl Metazoa (release 24), Ensembl Fungi (release 24), and Ensembl Protists (release 24). Only plant-homologous proteins were retained in the transcriptomes to eliminate the effects of genes derived from commensal organisms, laboratory contaminants, and artefacts resulting from incorrect assembly (Supplementary Table 13).
Gene family identification
We downloaded genome and annotation data of A. trichopoda (http://amborella.huck.psu.edu, version1.0), A. comosus (GenBank accession number GCF_001540865.1), A. thaliana (TAIR 10), A. officinalis (GenBank accession number GCF_001876935.1), B. distachyon (purple false brome; Phytozome v9.0), M. acuminata (http://ensemblgenomes.org, release-21), O. sativa (Nipponbare, IRGSP-1.0), P. dactylifera (http://qatar-weill.cornell.edu/research/datepalmGenome), P. trichocarpa (http://ensemblgenomes.org, release-21), S. bicolor (sorghum; Phytozome v9.0), S. polyrhiza (common duckweed; http://www.spirodelagenome.org), and V. vinifera (Phytozome v9.0). We chose the longest transcript to represent each gene and removed gene models with open reading frames shorter than 150 bp. Gene family clustering was performed using OrthoMCL59 based on the set of 21,841 predicted genes of A. shenzhenica and the protein sets of the above ten other monocots, three dicots and the outgroup A. trichopoda. This analysis yielded 11,995 gene families in A. shenzhenica containing 18,268 predicted genes (83.6% of the total genes identified; orthologous genes in the 15 sequenced plant species are shown in Supplementary Fig. 19 and Supplementary Table 14) (see also Supplementary Note 1).
Phylogenetic tree construction and phylogenomic dating
We constructed a phylogenetic tree based on a concatenated sequence alignment of 439 single-copy gene families from A. shenzhenica and the 14 other plant species using MrBayes60 software with GTR+Γ model (Fig. 1). For the phylogenetic analysis incorporating ten additional transcriptome species (Extended Data Fig. 2), we first picked up the genes of A. shenzhenica, D. catenatum, and P. equestris in the single-copy gene families as seed genes, and then made a BLASTP alignment between the transcriptome unigenes and the seed sequences. For one single-copy family, if the three seed genes all had the identical best-hit to a unigene, this gene was identified as the orthologous gene to the gene family. With this method we found 132 single-copy gene families of the total 25 species, then constructed the phylogenetic tree based on a concatenated sequence alignment of them using PhyML61 with GTR+Γ model. Divergence times were estimated by PAML MCMCTREE62. The Markov chain Monte Carlo (MCMC) process was run for 1,500,000 iterations with a sample frequency of 150 after a burn-in of 500,000 iterations. Other parameters used the default settings of MCMCTREE. Two independent runs were performed to check convergence. The following constraints were used for time calibrations: (i) the O. sativa and B. distachyon divergence time (40–54 million years ago (Ma))63; (ii) the P. trichocarpa and A. thaliana divergence time (100–120 Ma)64; (iii) the monocot and eudicot divergence time with a lower boundary of 130 Ma65; and (iv) 200 Ma as the upper boundary for the earliest-diverging angiosperms66.
Identification of WGD events in A. shenzhenica and phylogenomic analyses
KS-based age distributions were constructed as previously described67. In brief, the paranome was constructed by performing an all-against-all protein sequence similarity search using BLASTP with an E value cutoff of 1 × 10−10, after which gene families were built with the mclblastline pipeline (v10-201) (http://micans.org/mcl)68. Each gene family was aligned using MUSCLE (v3.8.31)69, and KS estimates for all pairwise comparisons within a gene family were obtained through maximum likelihood estimation using the CODEML program70 of the PAML package (v4.4c)62. Gene families were then subdivided into subfamilies for which KS estimates between members did not exceed a value of 5. To correct for the redundancy of KS values (a gene family of n members produces n(n−1)/2 pairwise KS estimates for n−1 retained duplication events), a phylogenetic tree was constructed for each subfamily using PhyML61 under default settings. For each duplication node in the resulting phylogenetic tree, all m KS estimates between the two child clades were added to the KS distribution with a weight of 1/m (where m is the number of KS estimates for a duplication event), so that the weights of all KS estimates for a single duplication event summed to one. The resulting age distribution of the A. shenzhenica paranome is shown in Extended Data Fig. 4a.
Absolute dating of the identified WGD event in A. shenzhenica was performed as previously described9,12. In brief, paralogous gene pairs located in duplicated segments (anchors) and duplicated pairs lying under the WGD peak (peak-based duplicates) were collected for phylogenetic dating. Anchors, assumed to correspond to the most recent WGD event, were detected using i-ADHoRe (v3.0)71,72. Their KS distribution is shown in Extended Data Fig. 4b. The identified anchors confirmed the presence of a WGD peak near a KS value of 1 (the long tail and additional peaks in the anchor pair distribution are most likely due to small saturation effects67 and the remnants of older WGD events in the monocot lineage, such as the τ WGD13,14). We selected anchor pairs and peak-based duplicates present under the WGD peak and with KS values between 0.6 and 1.4 (dashed lines in Extended Data Fig. 4a, b) for absolute dating. For each WGD paralogous pair, an orthogroup was created that included the two paralogues plus several orthologues from other plant species as identified by InParanoid (v4.1)73 using a broad taxonomic sampling: one representative orthologue from the order Cucurbitales, one from the Rosales, two from the Fabales, one from the Malpighiales, two from the Brassicales, one from the Malvales, one from the Solanales, two from the Poaceae (Poales), one from A. comosus14 (Bromeliaceae, Poales), one from either M. acuminata74 (Zingiberales) or P. dactylifera75 (Arecales), and one orthologue from the Alismatales, either from S. polyrhiza76 or Zostera marina77. In total, 85 orthogroups based on anchors and 230 orthogroups based on peak-based duplicates were collected. The node joining the two A. shenzhenica WGD paralogues was then dated using the BEAST v1.7 package78 under an uncorrelated relaxed clock model and an LG+G (four rate categories) evolutionary model. A starting tree with branch lengths satisfying all fossil prior constraints was created according to the consensus APGIV phylogeny79. Fossil calibrations were implemented using log-normal calibration priors on the following nodes: the node uniting the Malvidae based on the fossil Dressiantha bicarpellata80 with prior offset = 82.8, mean = 3.8528, and s.d. = 0.581; the node uniting the Fabidae based on the fossil Paleoclusia chevalieri82 with prior offset = 82.8, mean = 3.9314, and s.d. = 0.583; the node uniting the A. shenzhenica WGD paralogues with the other non-Alismatalean monocots based on fossil Liliacidites84 with prior offset = 93.0, mean = 3.5458, and s.d. = 0.585; and the root with prior offset = 124, mean = 4.0786, and s.d. = 0.586. The offsets of these calibrations represent hard minimum boundaries, and their means represent locations for their respective peak mass probabilities in accordance with some recent and most taxonomically complete dating studies available for these specific clades87. A run without data was performed to ensure proper placement of the marginal calibration prior distributions88. The MCMC for each orthogroup was run for 10 million generations with sampling every 1,000 generations, resulting in a sample size of 10,000. The resulting trace files of all orthogroups were evaluated manually using Tracer v1.578 with a burn-in of 1,000 samples to ensure proper convergence (minimum ESS for all statistics was at least 200). In total, 303 orthogroups were accepted, and all age estimates for the node uniting the WGD paralogous pairs were then grouped into one absolute age distribution (Extended Data Fig. 5; too few anchor pairs were available to evaluate them separately from the peak-based duplicates), for which KDE and a bootstrapping procedure were used to find the peak consensus WGD age estimate and its 90% confidence interval boundaries, respectively. More detailed methods are available in Vanneste et al.12.
To compare the relative timing of speciations and WGD event(s) in orchids based on KS distributions, we first identified 839 anchors from D. catenatum and 355 anchors from P. equestris using i-ADHoRe 3.0 and calculated their KS as described above. Identification of orthologues between A. shenzhenica and A. officinalis, A. shenzhenica and P. equestris, A. shenzhenica and D. catenatum, and P. equestris and D. catenatum was performed first by reciprocal BLASTP with E value <1 × 10−5 for proteins from the three orchids and asparagus, followed by sorting BLAST hits by bit-scores and E values. Reciprocal best hits in the four comparisons were selected as orthologues. In this way, we identified 9,142 orthologues between A. shenzhenica and A. officinalis, 10,699 orthologues between A.shenzhenica and P. equestris, 11,386 orthologues between A. shenzhenica and D. catenatum, and 13,139 orthologues between P. equestris and D. catenatum. For each pair of orthologues, ClustalW89 alignment was carried out to perform sequence alignment using the parameter for amino acids recommended by Hall90. PAL2NAL91 was then used to back-translate aligned protein sequences into codon sequences and to remove any gaps in the alignment. Estimates of KS values were obtained from CODEML in PAML using the Goldman-Yang model with codon frequencies estimated by the F3 × 4 model.
We performed pairwise co-linearity analysis within A. shenzhenica and between A. shenzhenica and A. officinalis, A. comosus, V. vinifera, and A. trichopoda. Homologous pairs of A. shenzhenica and the above species were identified by all-against-all BLASTP (E value <1 × 10−5), followed by the removal of weak matches by applying a c-score of 0.5 (indicating their BLASTP bit-scores were below 50% of the bit-scores of the best matches)92. Then, i-ADHoRe 3.0 was used to identify co-linear segments with parameters as described above except using ‘level_2_only = FALSE’, enabling the functionality to detect highly degenerated co-linear segments resulting from more ancient large-scale duplications (this is achieved by recursively building genomic profiles based on relatively recent co-linear segments). All co-linear dot plots were drawn by selecting co-linear segments according to a specified required number of anchor pairs (given in the figure legend of each of the dot plots). For the comparisons between A. shenzhenica and the chromosome-level assembled genomes (A. officinalis, A. comosus, and V. vinifera) we retained co-linear segments with at least ten anchor pairs (Extended Data Fig. 6 and Supplementary Figs 5, 7, 8). For the comparisons with fragmented genomes, like A. trichopoda, and the self-comparison of A. shenzhenica, we kept co-linear segments with five anchor pairs (Fig. 2b and Supplementary Figs 3, 4). The start and end boundaries of selected co-linear segments were used to define broader regions containing such segments on the chromosomes or scaffolds by further connecting co-linear segments if they overlapped with each other. Then, duplication depths, that is, the number of connected co-linear segments overlapping at each position of a broader region, were illustrated in the margins of the plots by mapping the connected co-linear segments over each other. The number of anchors required in the co-linear segments could affect the duplication depth in such a way that increasing the number of anchors required tends to remove co-linear segments originating from more ancient WGD(s) due to increased gene loss.
To identify the duplication events that resulted in the 1,488 anchor pairs in A. shenzhenica, the 839 anchor pairs in D. catenatum, and the 355 anchor pairs in P. equestris, we performed phylogenomic analyses employing protein-coding genes from 20 species, including 12 orchids across all five subfamilies of Orchidaceae (the three orchids with genomes (A. shenzhenica, D. catenatum and P. equestris) plus nine orchid transcriptomes (Supplementary Table 13)), four non-orchid Asparagales (A. officinalis (genome), M. capitulata (Supplementary Table 13), A. deserti57 and A. cepa58), three commelinid monocots (Elaeis guineensis, P. dactylifera, and A. comosus), and A. trichopoda. OrthoMCL (v2.0.9)59 was used with default parameters to identify gene families based on sequence similarities resulting from an all-against-all BLASTP with E value <1 × 10−5. Then, 1,101 of the 2,582 anchor pairs with KS values greater than five were removed. If the remaining anchors fell into different gene families, indicating incorrect assignment of gene families by OrthoMCL, we merged the corresponding gene families. In this way, we obtained 32,217 multi-gene gene families. Next, phylogenetic trees were constructed for the subset of 777 gene families with no more than 300 genes that had at least one pair of anchors and one gene from A. trichopoda. Multiple sequence alignments were produced by MUSCLE (v3.8.31) using default parameters. These were trimmed by trimAl (v1.4)93 to remove low-quality regions based on a heuristic approach (-automated1) that depends on a distribution of residue similarities inferred from the alignments for each gene family. RAxML (v8.2.0)94 was then used with the GTR+Γ model to estimate a maximum likelihood tree starting with 200 rapid bootstraps followed by maximum likelihood optimizations on every fifth bootstrap tree. Gene trees were rooted based on genes from A. trichopoda if these formed a monophyletic group in the tree; otherwise, mid-point rooting was applied. The timing of the duplication event for each anchor pair relative to the lineage divergence events was then inferred using the following approach (Supplementary Fig. 20): we first mapped internodes from a gene tree to the species phylogeny according to the common ancestor of the genes in the gene tree. Each internode of the gene tree was then defined as either a duplication node, a speciation node, or a ‘dubious’ node. A duplication node is a node that shares at least one pair of paralogues, a speciation node is a node that has no paralogues and is consistent with divergence in the species phylogeny, and a ‘dubious’ node is a node that has no paralogues and is inconsistent with divergence in the species phylogeny. Then, if a pair of anchors coalesced to a duplication node, we traced back its parental node(s) until we reached a speciation node in the gene tree. In this way, we circumscribed the duplication event as between these two nodes with the duplication node as the lower bound and the speciation node as the upper bound on the species tree. If the two nodes were directly connected by a single branch on the species tree, the duplication was thus considered to have occurred on the branch. To reduce biased estimations, we used the bootstrap value on the branch leading to the common ancestral node of an anchor pair as support for a duplication event. In total, 628 anchor pairs in 493 gene families coalesced as duplication events on the species phylogeny, and duplication events from 318 anchor pairs in 262 gene families (or from 448 anchor pairs in 367 gene families) had bootstrap values greater than or equal to 80% (or 50%).
Evolution and expression analysis of orchid MADS box genes
We identified candidates of MADS-box genes by searching the InterProScan51 result of all the predicted A. shenzhenica proteins. The candidates of MADS-box genes were further determined by SMART95, which identified MADS-box domains comprised by 60 amino acids. The protein-sequence set of the MADS-box gene candidates was BLAST against the assembled A. shenzhenica transcriptomes with the TBLASTN program. The matched transcript sequences were then assembled with the candidates of MADS-box genes using Sequencher v5.1 (Gene Codes Corp.) and the exon structure of the final MADS-box genes was manually edited (Supplementary Data 1). In the end, we aligned all the identified MADS-box genes using the ClustalW program89. An unrooted neighbour-joining phylogenetic tree was constructed in MEGA596 with default parameters.
Transcriptomic analysis of other orchids
In addition, 53 more transcriptomes derived from 9 more taxa and 8 tissues (flower bud, anther, pollinium, shoot, stem, leaf, aerial root and root) (Supplementary Table 13) were sampled to investigate the roles of the genes that may be important for the evolution of orchid traits. The gene expression levels were indicated by FPKM on the longest assembled transcript.
Data availability
Genome sequences and whole-genome assembly of A. shenzhenica and whole transcriptomes have been submitted to the National Center for Biotechnology Information (NCBI) database under BioProject PRJNA310678; the remaining transcriptomes used in this study can be found in the previously available BioProjects PRJNA288388, PRJNA304321, and PRJNA348403; the raw data and the updated whole-genome assembly of P. equestris have been submitted to NCBI under BioProject PRJNA389183; and the raw data and the updated whole-genome assembly of D. catenatum have been renewed under the already existing BioProject PRJNA262478. All other data are available from the corresponding authors upon reasonable request.
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Accession codes
References
Roberts, D. L. & Dixon, K. W. Orchids. Curr. Biol. 18, R325–R329 (2008)
Givnish, T. J. et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. R. Soc. B 282, 1553 (2015)
Givnish, T. J. et al. Orchid historical biogeography, diversification, Antarctica and the paradox of orchid dispersal. J. Biogeogr. 43, 1905–1916 (2016)
Chen, L. J. & Liu, Z. J. Apostasia shenzhenica: a new species of Apostasioideae (Orchidaceae) from China. Plant Science Journal 29, 38–41 (2011)
Kocyan, A., Qiu, Y.-L., Endress, P. K. & Conti, E. A phylogenetic analysis of Apostasioideae (Orchidaceae) based on ITS, trnL-F and matK sequences. Plant Syst. Evol. 247, 203–213 (2004)
Dressler, R. L. Phylogeny and Classification of the Orchid Family (Discorides, 1993)
Kocyan, A. & Endress, P. K. Floral structure and development of Apostasia and Neuwiedia (Apostasioideae) and their relationships to other Orchidaceae. Int. J. Plant Sci. 162, 847–867 (2001)
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015)
Cai, J. et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 47, 65–72 (2015)
Zhang, G.-Q. et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 19029 (2016)
De Bie, T ., Cristianini, N ., Demuth, J. P & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006)
Vanneste, K., Baele, G., Maere, S. & Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous-Paleogene boundary. Genome Res. 24, 1334–1347 (2014)
Jiao, Y., Li, J., Tang, H. & Paterson, A. H. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell 26, 2792–2802 (2014)
Ming, R. et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 47, 1435–1442 (2015)
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017)
Tsai, W. C., Kuoh, C. S., Chuang, M. H., Chen, W. H. & Chen, H. H. Four DEF-like MADS box genes displayed distinct floral morphogenetic roles in Phalaenopsis orchid. Plant Cell Physiol. 45, 831–844 (2004)
Pan, Z. J. et al. Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA-like genes. New Phytol. 202, 1024–1042 (2014)
Mondragón-Palomino, M. & Theissen, G. Why are orchid flowers so diverse? Reduction of evolutionary constraints by paralogues of class B floral homeotic genes. Ann. Bot. 104, 583–594 (2009)
Johnson, S. D. & Edwards, T. J. The structure and function of orchid pollinaria. Plant Syst. Evol. 222, 243–269 (2000)
Liu, Y. et al. Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 25, 1288–1303 (2013)
Kwantes, M., Liebsch, D. & Verelst, W. How MIKC* MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol. Biol. Evol. 29, 293–302 (2012)
Masiero, S., Colombo, L., Grini, P. E., Schnittger, A. & Kater, M. M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23, 865–872 (2011)
Zotz, G. & Winkler, U. Aerial roots of epiphytic orchids: the velamen radicum and its role in water and nutrient uptake. Oecologia 171, 733–741 (2013)
Chomicki, G. et al. The velamen protects photosynthetic orchid roots against UV-B damage, and a large dated phylogeny implies multiple gains and losses of this function during the Cenozoic. New Phytol. 205, 1330–1341 (2015)
Gravendeel, B., Smithson, A., Slik, F. J. W. & Schuiteman, A. Epiphytism and pollinator specialization: drivers for orchid diversity? Phil. Trans. R. Soc. Lond. B 359, 1523–1535 (2004)
Tapia-López, R. et al. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 146, 1182–1192 (2008)
Ibarra-Laclette, E. et al. Architecture and evolution of a minute plant genome. Nature 498, 94–98 (2013)
Zhang, H. & Forde, B. G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 279, 407–409 (1998)
Leseberg, C. H., Li, A., Kang, H., Duvall, M. & Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 378, 84–94 (2006)
Arora, R. et al. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8, 242 (2007)
Jersáková, J. et al. Genome size variation in Orchidaceae subfamily Apostasioideae: filling the phylogenetic gap. Bot. J. Linn. Soc. 172, 95–105 (2013)
Leitch, I. J. et al. Genome size diversity in orchids: consequences and evolution. Ann. Bot. 104, 469–481 (2009)
Lander, E. S. & Waterman, M. S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2, 231–239 (1988)
Butler, J. et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008)
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7, e47768 (2012)
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014)
Zheng, G. X. et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 34, 303–311 (2016)
Adey, A. et al. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 24, 2041–2049 (2014)
Mostovoy, Y. et al. A hybrid approach for de novo human genome sequence assembly and phasing. Nat. Methods 13, 587–590 (2016)
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015)
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011)
Keller, O., Kollmar, M., Stanke, M. & Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763 (2011)
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004)
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007)
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005)
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protocols 7, 562–578 (2012)
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013)
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999)
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000)
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003)
Zdobnov, E. M. & Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001)
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997)
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009)
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014)
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protocols 8, 1494–1512 (2013)
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008)
Gross, S. M. et al. De novo transcriptome assembly of drought tolerant CAM plants, Agave deserti and Agave tequilana. BMC Genomics 14, 563 (2013)
Duangjit, J., Bohanec, B., Chan, A. P., Town, C. D. & Havey, M. J. Transcriptome sequencing to produce SNP-based genetic maps of onion. Theor. Appl. Genet. 126, 2093–2101 (2013)
Li, L., Stoeckert, C. J., Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003)
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001)
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010)
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007)
International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010)
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006)
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007)
Magallón, S., Hilu, K. W. & Quandt, D. Land plant evolutionary timeline: gene effects are secondary to fossil constraints in relaxed clock estimation of age and substitution rates. Am. J. Bot. 100, 556–573 (2013)
Vanneste, K., Van de Peer, Y. & Maere, S. Inference of genome duplications from age distributions revisited. Mol. Biol. Evol. 30, 177–190 (2013)
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002)
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
Goldman, N. & Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11, 725–736 (1994)
Proost, S. et al. i-ADHoRe 3.0—fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 40, e11 (2012)
Fostier, J. et al. A greedy, graph-based algorithm for the alignment of multiple homologous gene lists. Bioinformatics 27, 749–756 (2011)
Ostlund, G. et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 38, D196–D203 (2010)
D’Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012)
Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat. Biotechnol. 29, 521–527 (2011)
Wang, W. et al. The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat. Commun. 5, 3311 (2014)
Olsen, J. L. et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335 (2016)
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012)
The Angiosperm Phylogeny Group. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016)
Gandolfo, M., Nixon, K. & Crepet, W. A new fossil flower from the Turonian of New Jersey: Dressiantha bicarpellata gen. et sp. nov. (Capparales). Am. J. Bot. 85, 964–974 (1998)
Beilstein, M. A., Nagalingum, N. S., Clements, M. D., Manchester, S. R. & Mathews, S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 107, 18724–18728 (2010)
Crepet, W. & Nixon, K. Fossil Clusiaceae from the late Cretaceous (Turonian) of New Jersey and implications regarding the history of bee pollination. Am. J. Bot. 85, 1122–1133 (1998)
Xi, Z. et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl Acad. Sci. USA 109, 17519–17524 (2012)
Ramírez, S. R., Gravendeel, B., Singer, R. B., Marshall, C. R. & Pierce, N. E. Dating the origin of the Orchidaceae from a fossil orchid with its pollinator. Nature 448, 1042–1045 (2007)
Janssen, T. & Bremer, K. The age of major monocot groups inferred from 800+rbcL sequences. Bot. J. Linn. Soc. 146, 385–398 (2004)
Smith, S. A., Beaulieu, J. M. & Donoghue, M. J. An uncorrelated relaxed-clock analysis suggests an earlier origin for flowering plants. Proc. Natl Acad. Sci. USA 107, 5897–5902 (2010)
Clarke, J. T., Warnock, R. C. & Donoghue, P. C. Establishing a time-scale for plant evolution. New Phytol. 192, 266–301 (2011)
Heled, J. & Drummond, A. J. Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Syst. Biol. 61, 138–149 (2012)
Oliver, T., Schmidt, B., Nathan, D., Clemens, R. & Maskell, D. Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics 21, 3431–3432 (2005)
Hall, B. G. Phylogenetic Trees Made Easy (Sinauer, 2004)
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006)
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007)
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009)
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014)
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 (2015)
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011)
Acknowledgements
We acknowledge support from The Funds for Environmental Project of Shenzhen, China (no. 2013-02); The 948 programme of the State Forestry Administration P. R. China (no. 2011–4–53); The Funds for Forestry Science and Technology Innovation Project of Guangdong, China (no. 2016KJCX025; no. 2013KJCX014-05); Fundamental Research Project of Shenzhen, China (no. JCYJ20170307170746099; no. JCYJ20150403150235943); and the teamwork projects funded by Guangdong Natural Science Foundation (no. 2017A030312004) awarded to Z.-J.L. Y.V.d.P. acknowledges the Multidisciplinary Research Partnership ‘Bioinformatics: from nucleotides to networks’ Project (no. 01MR0310W) of Ghent University and the European Union Seventh Framework Programme (FP7/2007-2013) under European Research Council Advanced Grant Agreement 322739–DOUBLEUP.
Author information
Authors and Affiliations
Contributions
Z.-J.L. managed the project; Z.-J.L., G.-Q.Z., Y.V.d.P., K.-W.L., W.-C.T., Y.-B.L. and C.-M.Y. planned and coordinated the project; Z.-J.L., K.-W.L., W.-C.T., Y.V.d.P., R.L. and Z.L. wrote the manuscript; Z.-J.L., L.-J.C., S.-C.N., M.W., G.-H.L., X.-J.X., H.-X.H., J.-Y.W., S.-J.Z. and L.P. collected and grew the plant material; Q.X., Z.-J.L., W.-C.T., K.-W.L., L.-J.C., X.-Y.W. and M.L. prepared samples; G.-Q.Z., Z.-J.L. and Y.-Q.Z. sequenced and processed the raw data; Z.-W.W., Z.-J.L., G.-Q.Z., K.Y., S.F., N.M., S.S., M.O.-T., M.Y. and C.-M.Y. annotated the genome; Z.-J.L., Z.-W.W., W.-C.T. and G.-Q.Z. analysed gene families; Z.L., R.L., Y.V.d.P. and Y.-C.L. conducted whole genome duplication analysis; W.-C.T., Y.-Y.H., Z.-J.L., C.-M.Y., S.-B.C., W.-L.W., Y.-Y.C., C.-Y.S. and K.-W.L. conducted the MADS-box gene analysis; Z.-J.L., G.-Q.Z., Y.-Q.Z., K.-W.L., S.-C.N, J.-Y.W., Q.X., S.S., M.O.-T. and C.-M.Y. conducted transcriptome sequencing and analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks V. Albert, J. Leebens-Mack, J. C. Pires and S. Ramírez for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 The morphology of orchid flowers.
a, Illustration of an Apostasia flower. b, Illustration of a Phalaenopsis flower.
Extended Data Figure 2 Phylogenetic tree showing the topology and divergence times for 15 genomes (A. trichopoda, P. trichocarpa, A. thaliana, V. vinifera, Spirodela polyrhiza, O. sativa, Brachypodium distachyon, Sorghum bicolor, A. comosus, Musa acuminata, Phoenix dactylifera, A. officinalis, A. shenzhenica, P. equestris and D. catenatum) and 10 transcriptomes (Apostasia odorata, Cypripedium margaritaceum, Galeola faberi, Habenaria delavayi, Hemipilia forrestii, Lecanorchis nigricans, M. capitulata, Neuwiedia malipoensis, Paphiopedilum malipoense, Vanilla shenzhenica).
The unigenes of the transcriptomes of the 10 ‘transcriptome’ species were aligned to the 439 single-copy gene families of the 15 ‘genome’ species. One hundred and thirty-two single-copy gene families for the 25 species could be identified, and were used to construct a phylogenetic tree based on the PhyML software61 with the GTR+Γ model, while divergence times (indicated by light blue bars at the internodes) were predicted by MCMCTREE62. The range of the bars indicates the 95% confidence interval of the divergence times.
Extended Data Figure 3 Venn diagram showing unique and shared gene families among members of Orchidaceae, dicots, and Poaceae, and M. acuminata and P. dactylifera.
Numbers represent the number of gene families. Comparison of the 4 groups revealed 474 gene families unique to Orchidaceae and which exist in all 3 Orchidaceae species. If we consider lineage-specific gene families for each group (that is, gene families present in one or a few but not all species in a group), then there are 4,958 unique gene families for Orchidaceae, 7,503 for Poales, 4,494 for the dicots, and 1,560 for the group of M. acuminata and P. dactylifera.
Extended Data Figure 4 A. shenzhenica KS-based age distributions.
a, Distribution of KS for the whole A. shenzhenica paranome. b, Distribution of KS for duplicated anchors found in co-linear regions as identified by i-ADHoRe. A WGD event is identified in both distributions with its peak centred on a KS value of 1. The dashed lines indicate the KS boundaries used to extract duplicate pairs for absolute phylogenomic dating of the WGD event (see Methods and Extended Data Fig. 5).
Extended Data Figure 5 Absolute age of the A. shenzhenica WGD event.
Absolute age distribution obtained by phylogenomic dating of A. shenzhenica paralogues. The solid black line represents the KDE of the dated paralogues, and the vertical dashed black line represents its peak at 74 Ma, which was used as the consensus WGD age estimate. The grey lines represent density estimates from 2,500 bootstrap replicates and the vertical black dotted lines represent the corresponding 90% confidence interval for the WGD age estimate, 72–78 Ma (see Methods). The histogram shows the raw distribution of dated paralogues.
Extended Data Figure 6 Co-linearity and synteny between A. shenzhenica and A. comosus.
Only co-linear segments with at least 20 anchor pairs are shown. The sections on each scaffold with co-linear segments between A. shenzhenica and A. comosus are shown in grey. The red bars below the dot plot illustrate the duplication depths (the number of connected co-linear segments overlapping at each scaffold/chromosomal position; see Methods). Only connected co-linear segments with at least ten anchor pairs were used to calculate the duplication depths. The co-linear regions in green highlight the four co-linear segments in A. shenzhenica that correspond to a specific set of four co-linear segments in A. comosus, which originated from one of the seven ancestral pre-τ-WGD chromosomes in monocots (known as Anc6)14. The phylogenetic tree above the dot plot indicates how Anc6 evolved into (segments of) the current four chromosomes in A. comosus (the pair of paired LG18 and LG04, and LG13 and LG23; see Figure 2 in Ming et al.14) through two rounds of WGDs. Names of very small A. shenzhenica scaffolds are omitted for clarity. A part of the alignment of the co-linear segments between A. shenzhenica and A. comosus is shown below. The colours of genes in the alignment indicate anchor pairs with genes of the same colour being homologous. The grey links connect anchor pairs between the two closest segments.
Extended Data Figure 7 Phylogenetic and expression analysis of orchid B-AP3 genes.
Ash, A. shenzhenica; Dca, D. catenatum; Hf, H. forrestii; Mc, M. capitulata; Peq, P. equestris; Pm, P. malipoense; Vs, V. shenzhenica. Expressions of B-class genes derived from H. forrestii are not shown, because only a flower sample was collected from H. forrestii. The expression levels (FPKM value) are represented by the colour bar.
Extended Data Figure 8 Phylogenetic tree of MIKC*-type genes.
The red boxes indicate MADS-box genes from A. shenzhenica. Ash, A. shenzhenica; Dca, D. catenatum; Hf, H. forrestii; Mc, M. capitulata; Peq, P. equestris; Pm, P. malipoense; Vs, V. shenzhenica. MIKC* sequences of the other species were retrieved from GenBank based on Liu et al.20.
Extended Data Figure 9 Expression patterns of MIKC* MADS-box genes.
Ash, A. shenzhenica; Dca, D. catenatum; Mc, M. capitulata; Peq, P. equestris. The expression levels (FPKM value) are represented by the colour bar.
Extended Data Figure 10 Expression of type I Mγ MADS-box genes in M. capitulata, A. shenzhenica and P. equestris.
As, A. shenzhenica; Mc, M. capitulata; Pe, P. equestris. The expression levels (FPKM value) are represented by the colour bar.
Supplementary information
Supplementary Information
This file contains Supplementary Notes 1-3, Supplementary Figures 1-27, Supplementary Tables 1-23, Supplementary Data and additional references. (PDF 7375 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, GQ., Liu, KW., Li, Z. et al. The Apostasia genome and the evolution of orchids. Nature 549, 379–383 (2017). https://doi.org/10.1038/nature23897
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature23897
This article is cited by
-
The structure and diversity of bacteria and fungi in the roots and rhizosphere soil of three different species of Geodorum
BMC Genomics (2024)
-
Root growth in Orchid Dendrobium cv. Sonia requires shade avoidance response of phytochromes along with regulation of auxin pathway genes
Plant Physiology Reports (2024)
-
Probing of plant transcriptomes reveals the hidden genetic diversity of the family Secoviridae
Archives of Virology (2024)
-
Genome-wide analysis of the NAC gene family and functional verification of the DcNAC043s in Dendrobium catenatum
Plant Growth Regulation (2024)
-
Genome of the early spider-orchid Ophrys sphegodes provides insights into sexual deception and pollinator adaptation
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
