
Abstract
Cereal grasses of the Triticeae tribe have been the major food source in temperate regions since the dawn of agriculture. Their large genomes are characterized by a high content of repetitive elements and large pericentromeric regions that are virtually devoid of meiotic recombination. Here we present a high-quality reference genome assembly for barley (Hordeum vulgare L.). We use chromosome conformation capture mapping to derive the linear order of sequences across the pericentromeric space and to investigate the spatial organization of chromatin in the nucleus at megabase resolution. The composition of genes and repetitive elements differs between distal and proximal regions. Gene family analyses reveal lineage-specific duplications of genes involved in the transport of nutrients to developing seeds and the mobilization of carbohydrates in grains. We demonstrate the importance of the barley reference sequence for breeding by inspecting the genomic partitioning of sequence variation in modern elite germplasm, highlighting regions vulnerable to genetic erosion.
Similar content being viewed by others

Main
Barley remains dated to the dawn of agriculture have been found at several archaeological sites1,2. In addition to indications that barley was an important food crop, recent excavations have fuelled speculation that beverages from fermented grains may have motivated early Neolithic hunter–gatherers to erect some of humankind’s oldest monuments3,4. Moreover, brewing beer may also have played a role in the eastward spread of the crop after its initial domestication in the Fertile Crescent5,6.
Since 2012, both genetic research and crop improvement in barley have benefited from a partly ordered draft sequence assembly7. This community resource has underpinned gene isolation8,9 and population genomic studies10. However, these and other efforts have also revealed limitations of the current draft assembly. The limitations are often direct consequences of two characteristic genomic features: the extreme abundance of repetitive elements, and the severely reduced frequency of meiotic recombination in pericentromeric regions11. These factors have limited the contiguity of whole-genome assemblies to kilobase-sized sequences originating from low-copy regions of the genome. Thus, a detailed investigation of the composition of the repetitive fraction of the genome—including expanded gene families—and of the distribution of targets of selection and crop improvement in (genetically defined) pericentromeric regions has been beyond reach.
Here we present a map-based reference sequence of the barley genome including the first comprehensively ordered assembly of the pericentromeric regions of a Triticeae genome. The resource highlights a conspicuous distinction between distal and proximal regions of chromosomes that is reflected by the intranuclear chromatin organization. Moreover, chromosomal compartments are differentiated by an exponential gradient of gene density and recombination rate, striking contrasts in the distribution of retrotransposon families, and distinct patterns of genetic diversity.
A chromosome-scale assembly of the barley genome
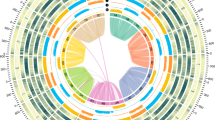
We adopted a hierarchical approach to generate a high-quality reference genome sequence of the barley cultivar Morex, a US spring six-row malting barley. First, a total of 87,075 bacterial artificial chromosomes (BACs) were sequenced, mainly using Illumina paired-end and mate-pair technology and assembled individually from 4.5 terabases of raw sequence data12,13,14 (Supplementary Note 1). In a second step, overlaps between adjacent clones15 were detected and validated by physical map information16, a genetic linkage17 and a highly contiguous optical map18 to construct super-scaffolds composed of merged assemblies of individual BACs (Table 1 and Extended Data Table 1). This increased the contiguity as measured by the N50 value (the scaffold size above which 50% of the total length of the sequence was included in the assembly) from 79 kb to 1.9 Mb. Scaffolds were assigned to chromosomes using a population sequencing (POPSEQ) genetic map17. Finally, we used three-dimensional proximity information obtained by chromosome conformation capture sequencing19,20,21 (Hi-C) to order and orient BAC-based super-scaffolds (Supplementary Note 2 and ref. 22). The final chromosome-scale assembly of the barley genome consists of 6,347 ordered super-scaffolds composed of merged assemblies of individual BACs, representing 4.79 Gb (~ 95%) of the genomic sequence content, of which 4.54 Gb have been assigned to precise chromosomal location in the Hi-C map (Table 1). Mapping of transcriptome data and reference protein sequences from other plant species to the assembly identified 83,105 putative gene loci including protein-coding genes, non-coding RNAs, pseudogenes and transcribed transposons (Fig. 1, Extended Data Fig. 1, Extended Data Table 2 and Supplementary Note 3). These loci were filtered further and divided into 39,734 high-confidence genes (with four different sub-categories) and 41,949 low-confidence genes on the basis of sequence homology to related species (Methods and Supplementary Note 3.4). Moreover, we predicted 19,908 long non-coding RNAs (Supplementary Note 3.7) and 792 microRNA precursor loci (Supplementary Note 3.8). The high co-linearity between the Hi-C-based pseudomolecules and linkage and cytogenetic maps22 as well as the conserved order of syntenic genes in pericentromeric regions compared with model grass Brachypodium distachyon (Extended Data Fig. 2a) corroborated the quality of the assembly. Extrapolating from a set of conserved eukaryotic core genes23, we estimate that the predicted gene models represent 98% of the cultivar Morex barley gene complement (Extended Data Fig. 2b).

a, The distribution of genomic features in 4 Mb windows is plotted along chromosome 1H. Analogous panels for the other chromosomes are found in Extended Data Fig. 5a. The left column in the legend refers to the background shading in the top panel; the right column indicates the colour code for lines in both panels. CDS, predicted coding sequences; cM, centimorgans. b, Enrichment of Gene Ontology (GO) terms in genomic compartments. Coloured rectangles indicate enrichment factors ranging from −2 (dark blue) to 2 (dark red). Numbers inside the rectangles indicate −log10-transformed P values.
Organization of chromatin
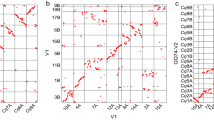
Barley has served as a model for traditional cytogenetics11; but relating chromosomal features to unique sequences has been challenging, requiring the cloning of repeat-free probes24. The reference sequence allowed us to employ the Hi-C data to interrogate the three-dimensional organization of chromatin in the nucleus. As in other eukaryotes20,25,26, the spatial proximity of genomic loci as measured by Hi-C link frequency is highly dependent on their distance in the linear genome (Fig. 2a). However, we observed an elevated link frequency at distances above 200 Mb and a pronounced anti-diagonal pattern in the intrachromosomal Hi-C contact matrices (Fig. 2b and Extended Data Fig. 3a), indicating an increased adjacency of regions on different chromosome arms. We interpret this pattern as reflective of the so-called Rabl configuration27 of interphase nuclei, where individual chromosomes fold back to juxtapose the long and short arms, with centromeres and telomeres of all chromosomes clustering at opposite poles of the nucleus (Fig. 2c and Supplementary Fig. 2.2). Fluorescence in situ hybridization (Fig. 2d) supported this hypothesis. Principal component analysis of the intrachromosomal proximity matrix showed that the first three principal components cumulatively explained ~70% of the variation and differentiated (1) distal from proximal regions, (2) interstitial from both distal and proximal regions and (3) the long arms from the short arms (Fig. 2f and Extended Data Fig. 4a). A linear model taking into account the genomic distance between two loci, as well as their relative distance from the centromere, accounted for 79% of the variation (Extended Data Fig. 4b) in the intrachromosomal proximity matrix at 1 Mb resolution.

a, Distance-dependent decay of contact probability. b, Intrachromosomal contact matrix. The intensity of pixels represents the normalized count of Hi-C links between 1 Mb windows on chromosome 1H on a logarithmic scale. c, Schematic model of the Rabl configuration of interphase chromosomes. Centromeres and telomeres are presented by red and green circles, respectively. d, Leaf interphase nucleus of barley. Chromatin was stained blue with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence in situ hybridization was performed with probes specific for centromeres (red) and telomeres (green). Scale bar, 5 μm. e, Interchromosomal contact matrix. The intensity of pixels represents the normalized count of Hi-C links between 1 Mb windows on chromosomes 1H (x axis) and 2H (y axis) on a logarithmic scale. A principal component analysis of the normalized contact matrix at 1 Mb resolution of chromosome 1H was conducted. f, The first and second eigenvectors are plotted against each other. Each point represents a 1 Mb window. Closer proximity to the centromere is indicated by a darker colour. Windows from the short and long arms are coloured blue and red, respectively.
Contacts between loci on different chromosomes followed a similar pattern (Fig. 2e and Extended Data Fig. 3b): a prominent cross pattern supporting a juxtaposition of long and short arms. In contrast to intrachromosomal matrices, contact probabilities between loci on, for instance, the short arm of one chromosome are equal for loci on both the short and the long arm on another chromosome having the same relative distance to the centromeres: that is, facing each other in the interphase nucleus. We also observed a higher contact frequency between telomere-near regions, as has been observed in Arabidopsis25.
To test whether pairs of homologous chromosomes are positioned closer to each other than to non-homologues, we performed diploid Hi-C28 on leaf tissue from F1 hybrids between the cultivars Morex and Barke, and assigned the resultant Hi-C links to the haplotypes of both inbred parents by mapping reads to a diploid reference. We did not observe any preferential interaction between homologues. Rather, contacts between the maternal and paternal copies of the same chromosome occurred as frequently as between non-homologues (Extended Data Fig. 4c).
We conclude that the frequency with which loci juxtapose in three-dimensional space is predominantly determined by their position in the linear genome. This is in sharp contrast to the organization of chromatin in human nuclei where two compartments corresponding to open and closed chromatin domains are evident at megabase resolution20, but is consistent with cytogenetic mapping of histone marks associated with heterochromatin in large, repeat-rich genomes29.
The genomic context of repetitive elements
Large plant genomes consist mainly of highly similar copies of repetitive elements such as long terminal repeat (LTR) retrotransposons and DNA transposons30,31. Our hierarchical sequencing strategy reduced the algorithmic complexity of assembling a highly repetitive genome from short reads. Instead of resolving complex repeat structures on the whole-genome level, we reconstructed the sequences of 100–150 kb BACs. This allowed us to disentangle nearly identical copies of highly abundant repetitive elements, as evidenced by the good representation of both mathematically defined repeats and retrotransposon families (Extended Data Fig. 2c, d). Homology-guided repeat annotation with a Triticeae-specific repeat library32 identified 3.7 Gb (80.8%) of the assembled sequence as derived from transposable elements (Table 1, Fig. 1a and Extended Data Table 3), most of which were present as truncated and degenerated copies, with only 10% of mobile elements intact and potentially active.
Median 20-mer frequencies were used to partition the seven barley chromosomes into three zones (Fig. 1 and Extended Data Fig. 5a), reminiscent of the three compartments of wheat chromosome 3B33. The distal zone 1 was characterized by an enrichment of low-copy regions, a high gene content and frequent meiotic recombination. Zone 2, occupying the interstitial regions of chromosomes, had the highest 20-mer frequencies and intermediate gene density. Surprisingly, the abundance of repetitive 20-mers decreased in the proximal zone 3, where older mobile elements with diverged, and thus unique, sequences predominated (Fig. 1). The three zones also differed in the composition of the gene space (Extended Data Table 2b and Supplementary Note 3). For example, genes involved in defence response and reproductive processes were preferentially found in distal regions, while proximal regions contained more genes related to housekeeping processes, such as photosynthesis and respiration, compared with other parts of the genome (Fig. 1b).
Transposable element groups exhibited pronounced variation in their insertion site preferences (Fig. 3a and Extended Data Fig. 5b). On a global scale, most miniature inverted-repeat transposable elements and long interspersed elements were found in gene-rich distal regions, as has been reported in other grass species34,35. By contrast, zone 3 was populated by Gypsy retrotransposons, while Copia elements favoured zones 1 and 2. These differences in the relative abundance of retrotransposon families were reflected by distinct distributions of functional domains. For example, sequences encoding the chromodomain (PF00385) are concentrated in the vicinity of the centromere and may be involved in the target specificity through incorporation in the integrase of Gypsy elements36 (Fig. 3a and Extended Data Fig. 5b).

a, Abundance of key genomic features, different transposon superfamilies and common Pfam domains across chromosome 1H. Analogous panels for the other chromosomes are found in Extended Data Fig. 5b. The colour scale of the heatmaps ranges from blue (0) to yellow (maximum across all chromosomes per track). Minimum and maximum values are indicated to the right of each track. MITEs, miniature inverted-repeat transposable elements; LINEs, long interspersed elements; fl, full-length; PR, protease; CH, chromodomain; RT, reverse transcriptase; NBS, NB-ARC; Pkin, protein kinase. b, Transposable elements up- and downstream of genes. Coding sequences of high-confidence genes were used as anchor points. Transposable element composition was determined 10 kb up- and downstream of each gene. The x axis indicates the position relative to the gene, while the y axis indicates how many genes had a transposable element of the respective superfamily at the respective position in their upstream/downstream region.
At a local scale, different types of elements also occupy different niches in the proximity of genes (Fig. 3b). Mariner transposons preferably reside within 1 kb up- or downstream of the coding regions of genes, while Harbinger and long interspersed elements are found further away. The observed distribution of different types of transposable elements around genes may reflect selective pressures, allowing only the smallest elements, namely Mariners, to be tolerated closest to genes. Intriguingly, Helitrons as well as elements of the Harbinger superfamily have a clear preference for promoter regions, while long interspersed elements have a preference for downstream regions (Fig. 3b). At greater distances from genes, large elements such as LTR retrotransposons and CACTA elements dominate.
Expansion of gene families
The barley reference sequence enabled us to disentangle complex gene duplications that may shed light on gene family expansion specific to barley or the Triticeae. A total of 29,944 genes belonged to families with multiple members (Fig. 4a and Supplementary Note 4.1). Gene families expanded in barley were tested for over-representation of Gene Ontology37 terms compared with sorghum, rice, Brachypodium and Arabidopsis. Among the most significant results were terms related to defence response and disease resistance (NBS-LRR and thionin genes), as well as thioredoxin genes (Supplementary Note 4.1).

a, OrthoMCL clustering of the barley high-confidence gene complement with B. distachyon, rice, sorghum and Arabidopsis thaliana genes. Numbers in the sections of the Venn diagram correspond to numbers of clusters (gene groups). The first number below the species name denotes the total number of proteins that were included into the OrthoMCL analysis for each species. The second number indicates the number of genes in clusters for a species. b, Phylogenetic tree of 68 full-length α-amylase protein sequences derived from amy genes identified in the genomes of barley, hexaploid wheat, B. distachyon, rice, sorghum and maize. Each wheat subgenome was considered separately to facilitate the comparison of gene copy numbers and duplication events across species. Note that for the amy4 subfamily, two to three genes per genome were identified in all genomes. These genes are located on distinct chromosomes and hence most probably did not originate from tandem gene duplications. While most species further contain only a single amy3 gene copy per genome, moderate copy number extension was observed in sorghum and rice where a potential tandem gene duplication resulted in two amy3 gene copies. Three genes of the amy2 subfamily were identified on chromosome 7H in barley and on chromosomes 7A, 7B, 7D in wheat. No similar copy number extension was observed in B. distachyon, Sorghum bicolor or Oryza sativa. In maize, two amy2 genes were identified. The amy1 subfamily shows the highest level of copy number extension. Tandem duplications are present in sorghum and rice. Two to three full-length genes were identified per genome in hexaploid wheat on group 6 chromosomes and five full-length amy1 genes on chromosome 6H and unanchored scaffolds in barley. Notably four of these barley genes share 99.8–100% sequence identity on protein and nucleotide level, indicating very recent duplication events. T. aestivum, Triticum aestivum; Z. mays, Zea mays. c, Expression of the SWEET11 gene subfamily in the developing barley grains. Left, expression profiles of SWEET11a and SWEET11b as determined by quantitative real-time PCR (qPCR) on total RNA isolated from micro-dissected developing grains. Right, localization of SWEET11a and SWEET11b expression in cross-section of immature seeds by RNA in situ hybridization. Hybridizations with sense probes are shown as negative controls in Extended Data Fig. 7a. Scale bars, 100 μm.
In the following, we focused on a detailed analysis of gene families having particular importance for malting quality. Germinating barley grains possess high diastatic power: that is, the combined ability of a complex of enzymes to mobilize fermentable sugars from starch. Key diastatic enzymes include α-amylases. The genome of barley cultivar Morex contains 12 α-amylase (amy) family sequences (Supplementary Note 4.2 and Extended Data Table 4a), which can be classified into four subfamilies38. Gene duplication events have occurred in the subfamilies amy1 and amy2 (Fig. 4b), located on chromosomes 6H and 7H, respectively. The existence of these duplications had been speculated earlier, but could not be analysed further because of high sequence similarity between the copies. The reference assembly contained five full-length amy1 subfamily genes, four of which, here designated as amy1_1a–d, shared >99.8% identity at the nucleotide level including introns. Locus-specific PCR confirmed earlier suggestions39,40 of multiple, highly similar amy1_1 genes (Extended Data Fig. 6 and Supplementary Note 4.2). Given the relevance of α-amylase activity to the brewing process, the high variability of the amy1_1 multiple gene locus (Extended Data Fig. 6) observed in landraces and elite lines, including modern malting cultivars, is remarkable.
The accumulation of fermentable carbohydrates in the grain depends on the transfer of sugars from maternal tissue into the developing seeds. In contrast to the two routes of nutrient transfer in rice seeds—the nucellar projection and nucellar epidermis—delivery of assimilates into barley grains occurs predominantly via the nucellar projection41 and requires active transporters. The family of SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTER (SWEET) transmembrane proteins mediating sugar efflux42 consists of 23 members in barley (Extended Data Table 4b and Supplementary Note 4.3). There is a small extension of the sugar-transporting SWEET11, SWEET13, SWEET14 and SWEET15 subfamilies, with two or more genes for each subgroup compared with only a single orthologue in rice and Arabidopsis (Extended Data Table 4b). Duplication of SWEET11 was most likely followed by neofunctionalization as evidenced by divergent expression patterns. Both SWEET11a and SWEET11b were highly expressed in maternal seed tissue, but differed in the distribution of expression domains (Fig. 4c and Extended Data Fig. 7). Genes encoding a family of vacuolar processing enzymes, which are essential for programmed cell death in maternal tissue43 and starch accumulation in the grain (Supplementary Note 4.3 and V.R., unpublished observations) showed a similar expansion in barley (Extended Data Table 4c), pointing to the central role of the nucellar projection for grain filling in the Triticeae.
These examples of genes involved in sugar transport and metabolism illustrate that the high-quality reference genome sequence can serve as a springboard for the in-depth analysis of the evolutionary history of gene duplications, their relation to morphological and physiological innovations, and their impact on crop performance.
Molecular diversity and haplotype analysis
To explore how the new barley genome assembly could be exploited for genetics and breeding, we generated exome sequence data from 96 European elite barley lines, half with a spring growth habit, half with a winter one (Supplementary Table 5.1). We investigated the extent and partitioning of molecular variation within and between these groups using 71,285 single-nucleotide polymorphisms (SNPs). Plotting diversity values in 100 SNP windows both in linear order (Fig. 5a) and according to physical distance (Fig. 5b) revealed marked contrasts in the levels and distribution of diversity both within and between gene pools. In spring types, extensive regions on chromosomes 1H, 2H and 7H were virtually devoid of diversity, as was a large region on 5H in the winter gene pool. For these chromosomes, this results in a single gene-pool-specific haplotype across the extensive pericentromeric regions. Chromosomes 3H, 4H and 6H maintain higher diversity across these regions owing to the presence of multiple similarly extensive haplotypes. This is even more evident when diversity is plotted on a physical scale (Fig. 5b). We presume that the lack of observed variation in elite germplasm is a signature of intense selection during breeding for different end-use sectors (principally malting versus feed barley), and the virtual absence of allelic re-assortment during meiosis owing to restricted recombination in the pericentromeric regions.

Ninety-six elite barley cultivars, including 48 from the winter gene pool (blue line) and 48 from the spring gene pool (red line), were used. Diversity (unbiased heterozygosity, y axis) is plotted as the rolling average of 100 adjacent SNPs along each chromosome. For improved visualization, all chromosomes have been normalized to a standard length. a, Patterns of diversity on chromosomes 1H–7H (top to bottom). The distance between each SNP has been normalized (that is, this does not show genetic distance). The number of SNPs included on each chromosome is given at the bottom right of each plot. b, The same diversity values normalized according to physical distance. Extensive peri-centromeric regions of very low diversity in the spring gene pool are highlighted in green and low diversity in the winter gene pool in purple. Regions with similar levels of diversity in both gene pools are highlighted in orange. Coloured dots show the position of eight loci previously identified as being differentiated between the winter and spring gene pools.
Crosses between spring and winter barleys are rarely performed as they are considered to disrupt the gene-pool-specific gene complexes required for general performance (such as phenological adaptations) and end-use quality. Contrasting local patterns of diversity outside the pericentromeric regions therefore also most likely reflect the outcome of selection within alternative gene pools. We explored this further by comparing diversity in eight characterized genes whose variant alleles are important for conditioning barley’s seasonal growth habit (Supplementary Note 5). Of the eight genes, HvCEN is uniquely ‘locked’ in the pericentromeric region of chromosome 2H where alternative alleles at a single SNP confer both differences in days-to-heading44 and strong latitudinal differentiation10. The extensive pericentromeric haplotype in spring barleys (Fig. 5) may stem from selection for this single HvCEN SNP. While strong selection for other favourable alleles locked in the same region in spring barley cannot be ruled out, the virtual absence of recombination severely restricts exploitation of diversity across the entire region. Despite our focus here on life-history traits, strong selection for other traits mapping to pericentromeric regions45,46, including good malting quality in the spring gene pool on chromosomes 1H and 7H, would probably also reduce diversity in these regions. Interestingly, we are unaware of any phenotypic trait in the winter gene pool that would result in strong selection for a single pericentromeric haplotype on chromosome 5H.
We next explored patterns of linkage disequilibrium across the entire genome. As expected for two highly inbred and elite crop gene pools, we observed extensive linkage disequilibrium on all chromosomes in both spring and winter barleys (Extended Data Fig. 8). The number of discrete haplotype blocks in this germplasm set varied from 86 to 161 per chromosome (Extended Data Fig. 8). Surprisingly, the two-row spring gene pool, generally considered to be narrowest owing to intense selection for malting quality, exhibited a greater number of haplotype blocks than the winter lines for most chromosomes.
Discussion
To assemble a highly contiguous reference genome sequence for barley, we combined hierarchical shotgun sequencing, a strategy previously used for assembling large and complex plant genomes33,47, with novel technologies such as optical mapping18 and chromosome-scale scaffolding with Hi-C21. The latter technology was key to resolving the linear order of sequence scaffolds in pericentromeric regions. We anticipate the adoption of Hi-C-based genome mapping in other Triticeae species, such as bread and durum wheat and their wild relatives. Now that the quality of whole-genome shotgun assemblies is on a par with map-based assemblies48,49, we believe that the barley genome project will be one of the last such efforts to follow the laborious BAC-by-BAC approach.
The barley reference genome sequence constitutes an important community resource for cereal genetics and genomics. It will facilitate positional cloning, provide a better contextualization of population genomic datasets and enable comparative genomic analysis with other Triticeae in non-recombining regions that have been inaccessible to analysis of gene collinearity until now. The exciting methodological advances in sequence assembly and genome mapping have enabled even large and repeat-rich genomes to be unlocked48,50 and hold the promise of constructing reference-quality genome sequences, not only for a single cultivar, but also for representatives of major germplasm groups.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Sequencing and assembly of individual BAC clones
Barley genome sequencing relied exclusively on shotgun sequencing of 88,731 BAC clones using high-throughput next-generation sequencing-by-synthesis22. This comprised 15,661 so-called gene-bearing BAC clones, preselected mainly by overgo-probe hybridization for the presence of transcribed genes and fingerprinted for definition of a minimum tiling path of the barley gene space. These gene-space minimum tiling path BAC clones were sequenced as combinatorial pools by Illumina short-read technology and, after quality trimming of de-convoluted reads, were assembled using Velvet version 1.2.09 as previously described13. The remaining 73,070 BACs were selected from a minimum tiling path representing the physical map of the barley genome16. Minimum tiling path BAC clones assigned to different barley chromosomes were sequenced at one of four sequencing centres, relying on highly multiplexed paired-end and mate-pair sequencing libraries using either the Roche 454 Titanium or the Illumina MiSeq, HiSeq2000 and HiSeq2500 platforms (Supplementary Note 1 and ref. 51). In brief, sequencing reads were de-convoluted on the basis of the used BAC-specific barcode sequence tags and assembled with sequencing centre-specific assembly pipelines. BAC clones sequenced on the Roche 454 Titanium platform were assembled with MIRA51 according to previously described procedures52,53. Illumina HiSeq2000 paired-end sequencing data (2 × 100 nucleotides) of BAC clones were assembled either with CLC Assembly Cell version 4.0.6 beta (http://www.clcbio.com/products/clc-assembly-cell/) set to default parameters12, SOAPdenovo version 2.01 (ref. 54) or the ABySS assembler (version 1.5.1)55. Sequence contigs of the de novo BAC assemblies larger than 500 base pairs (bp) were scaffolded using mate-pair sequencing information either generated from BAC DNA-derived 8 kbp insert mate-pair sequencing libraries or from 2 kbp, 5 kbp or 10 kbp genomic DNA-derived mate-pair libraries. This was achieved by either using BWA mem version 0.7.4 (ref. 56) with default parameters for read mapping, followed by scaffolding individual BACs using SSPACE version 3.0 Standard57, or with SOAPaligner/soap2 version 2.21 and using SOAPdenovo54 scaffolder version 2.01.
Genome-wide three-dimensional chromatin conformation capture sequencing
To generate physical scaffolding information for the BAC sequence based genome assembly, as proposed in ref. 21, Hi-C and tethered conformation capture (TCC) sequencing data were generated from 7-day-old leaf tissue of greenhouse-grown barley plantlets by adapting previously published procedures (Supplementary Note 2). In brief, for Hi-C, freshly harvested leaves were cut into 2 cm pieces and vacuum infiltrated in nuclei isolation buffer supplemented with 2% formaldehyde. Crosslinking was stopped by adding glycine and additional vacuum infiltration. Fixed tissue was frozen in liquid nitrogen and ground to powder before re-suspending in nuclei isolation buffer to obtain a suspension of nuclei. About 107 purified nuclei were digested with 400 units of HindIII as described previously58. Digested chromatin was marked by incubating with biotin-14-dCTP and Klenow enzyme using a fill-in reaction20 resulting in blunt-ended repaired DNA strands. Biotin-14-dCTP from non-ligated DNA ends was removed owing to the exonuclease activity of T4 DNA polymerase, followed by phenol–chloroform extraction and washing of the precipitated DNA as described20. As an alternative to Hi-C, the TCC protocol was also adapted for barley. Nuclei were prepared from barley leaf tissue as described above for Hi-C, before biotinylating the isolated chromatin using EZlink Iodoacetyl-PEG2-Biotin. The samples were neutralized with SDS, and DNA was digested with HindIII, dialysed, followed by immobilization to low surface coverage using streptavidin-coated magnetic beads19. Open DNA ends were labelled with biotin-14-dCTP using Klenow enzyme, and blunt-ended, labelled DNA products were collected from the magnetic beads by reversing the formaldehyde crosslink using proteinase K19. Biotin-14-dCTP from non-ligated DNA ends was removed by using Exonuclease III19. Hi-C and TCC products were mechanically sheared to fragment sizes of 200–300 bp by applying ultrasound using a Covaris S220 device followed by size-fractionation using AMPure XP beads. DNA fragments in the range between 150 and 300 bp were blunt-end repaired and A-tailed before purification through biotin–streptavidin-mediated pull-down58. Illumina paired-end adapters were ligated to the Hi-C and TCC products, respectively, followed by PCR amplification, pooling of PCR products and purification with AMPure XP beads before quantification of Hi-C/TCC libraries by qPCR for Illumina HiSeq2500 PE100 sequencing20.
Nanochannel-based genome mapping
Long-range scaffolding of genome sequence assemblies was facilitated by BioNano genome maps generated by nanochannel electrophoresis of fluorescently labelled high-molecular mass DNA obtained from flow-sorted chromosomes59. High-molecular mass DNA was prepared from 3.5 × 106 purified chromosomes (whole genome) of barley cultivar Morex essentially following published procedures60,61. The purified chromosomes were embedded in agarose miniplugs to achieve approximate concentrations of 1 million chromosomes per 40 μl volume before being treated with proteinase K as described previously61. DNA was labelled at Nt.BspQI nicking sites (GCTCTTC) by incorporation of fluorescent-dUTP nucleotide analogues using Taq polymerase as described previously59. The labelled DNA was analysed on the Irys platform (BioNano Genomics) in 191 cycles in total, generating 243 Gb of data exceeding 150 kb. On the basis of the label positions on single DNA molecules, de novo assembly was performed by a pairwise comparison of all single molecules and graph building62. The parameter set for large genomes was used for assembly with the IrysView software. A P value threshold of 10−9 was used during the pairwise assembly, 10−10 for extension and refinement steps and 10−14 for merging contigs. A whole-genome map of 4.3 Gb was obtained (Extended Data Table 1).
Data integration for constructing pseudomolecules
The construction of pseudomolecules representing the seven barley chromosomes followed an iterative, mainly automated procedure which involved the integration of the following major datasets: (1) sequence assemblies of 87,075 unique, successfully sequenced and assembled BAC clones; (2) BAC assembly information from a genome-wide physical map of barley16; (3) 571,814 end-sequences of BAC clones7; (4) a dense linkage map assigning genetic positions to 791,177 contigs of a whole-genome shotgun assembly of barley cultivar Morex17; (5) Hi-C/TCC sequence information; and (6) the optical map of the genome of barley cultivar Morex. A schematic outline of the procedure is presented elsewhere22. In the first step, overlaps between individual BAC assemblies were searched with Megablast63 by either applying ‘stringent’ or ‘permissive’ alignment criteria22 and by combining with the high density genetic map information. On the basis of this initial analysis, a BAC overlap graph was constructed by use of the R package igraph64 considering the above-listed additional datasets in subsequent iterative steps. Building the overlap graph focused first on overlaps obtained under ‘stringent’ search criteria for BACs within individual physical map contigs (FP contigs) and then subsequently also between independent FP contigs. Subsequently, overlaps obtained under ‘permissive’ criteria were evaluated while checking for cumulative evidences provided by the additional datasets supporting the overlap information22. Ordering and orienting of the resultant sequence scaffolds were achieved by integrating the overlap graph with Hi-C /TCC data22. Before the construction of pseudomolecules, we (1) identified genes incomplete or missing in the non-redundant sequence, but represented by (a) BAC sequence that had been excluded from the construction of the non-redundant sequence, or by (b) Morex WGS contigs, and (2) performed a final scan for contaminant sequences. Then a single FASTA file containing a single entry for each barley chromosome (a ‘pseudomolecule’) and an additional entry combining all sequences not anchored to chromosomes was constructed22.
Three-dimensional chromatin conformation analysis
Mapping of Hi-C/TCC reads and assignment to restriction fragments were performed as described elsewhere22. Briefly, raw reads were trimmed with cutadapt65. Trimmed Hi-C reads were mapped to the barley pseudomolecule sequence with BWA mem (version 0.7.12)66. Duplicate removal and sorting were performed with NovoSort (http://www.novocraft.com/products/novosort/). Mapped reads were assigned to restriction fragments with BEDtools67, tabulated with custom AWK scripts and imported into R (https://www.r-project.org/). Raw counts of Hi-C links were aggregated in 1 Mb bins and normalized separately for intra- and interchromosomal contacts using HiCNorm68. Contact probability matrices were plotted using standard R functions69. Principal component analysis was performed with the R function prcomp() on the matrix of log-transformed normalized Hi-C link counts between 1 Mb fragments.
We fitted the linear model log10(nl) ~ log10(dist) + abs(cen_dist1 – cen_dist2) + arm1:arm2 + apos1:apos1 using the R function lm(). Here, nl is the normalized link count between two 1 Mb bins, dist is their distance in the linear genome, cen_dist1 and cen_dist2 are the relative distances from the centromere of both loci, arm1 and arm2 are the chromosome arm assignment of both loci, and apos1 and apos2 are the relative distances of both loci from the ends of the chromosome arm (that is, apos1 is close to zero if locus 1 is either near the centromere or the telomere, and close to one if locus 1 resides in interstitial regions). TCC reads of Morex × Barke F1 hybrids were mapped to a synthetic reference representing the parental genomes. An in silico Barke assembly was created by inserting SNPs discovered by aligning Barke WGS reads to the Morex reference assembly with BWA MEM66 and calling variants with SAMtools70. SNPs were then inserted into the Morex reference using the FastaAlternateReferenceMaker of GATK71. TCC reads of the hybrid were then mapped to the synthetic reference as described above. Only uniquely alignable read pairs were considered. Hi-C link counts were tabulated at the level of chromosomes.
Fluorescence in situ hybridization was performed with H. vulgare nuclei as described earlier72 using Arabidopsis-type telomere and barley centromere-specific [AGGGAG]5 repeat probes73.
Automated annotation of transcribed regions
Automated gene annotation of the barley reference sequence assembly was based on four datasets providing independent gene evidence information (Supplementary Note 3). This included (1) RNA sequencing (RNA-seq) data; (2) reference protein predictions from barley7, rice74, B. distachyon75 and S. bicolor76; (3) published barley full-length complementary DNA (fl-cDNA) sequences77; and (4) newly generated barley PacBio Iso-Seq data. Previously published7 and newly generated RNA-seq datasets were derived from a total of 16 different tissues, each with three biological replicates, including seven vegetative, six inflorescence, two developing grain and one germinating grain tissues. RNA-seq libraries were sequenced on Illumina HiSeq2000 in paired-end 2 × 100 nucleotides (PE100) mode (Supplementary Note 3). To support gene calling in general, and the identification of alternative splice forms in particular, enriched full-length transcript information was generated by the Iso-Seq method using the PacBio RS II system and DNA Sequencing Chemistry 4.0 version 2 (Supplementary Note 3). RNA-seq-based transcript structures, reference-based gene model predictions, structure information from Iso-Seq alignments as well as structure information from flcDNA sequence alignments were clustered into a consensus transcript set using Cuffcompare78 (Supplementary Note 3). Predicted transcript sequences were automatically extracted into a single FASTA file on the basis of respective coordinates in the genome assembly. Putative open reading frames and corresponding peptide sequences, including prediction of Pfam domains, were obtained by applying TransDecoder (https://transdecoder.github.io), which also resulted in reports about predicted alternative peptides per transcript (Supplementary Note 3). A single best translation per transcript was selected on the basis of BLASTP79 comparison of all predicted peptides to a comprehensive protein database containing high-confidence protein sequences from A. thaliana80, maize47, B. distachyon75, rice75 and S. bicolor76, followed by additional filtering procedures (Supplementary Note 3). Functional descriptions (‘human readable descriptions’) were generated for all potential genes using the AHRD pipeline (https://github.com/groupschoof/AHRD) on the basis of one representative protein sequence for each gene locus. Gene candidates were then classified into high- and low-confidence genes and further subdivided into nine classes, each supported by different levels of gene evidence (Supplementary Note 3). High-confidence protein-coding genes either showed significant sequence homology to a reference protein or were associated with a predicted function. Low-confidence genes were characterized by (1) having no or only weak sequence homology to reference proteins and no predicted function, (2) they were candidates for transposons or (3) they lacked an open reading frame of a minimal length (Supplementary Note 3). Completeness of gene-space representation was evaluated with the BUSCO pipeline23 (Extended Data Fig. 2b).
Feature distributions along the chromosomes
A sliding window approach with a window size of 4 Mb and a shift of 0.8 Mb was used to display the distribution of different genome components and other features such as GC content or recombination rate along the chromosomes. The resulting data were smoothed with the python function scipy.signal.gaussian (p1 = 40, p2 = 10 for Fig. 1a; p1 = 15, p2 = 3 for Fig. 2a). The boundaries of genomic compartments (Fig. 1) are given in Supplementary Table 4.4.
Annotation of the non-genic part of the genome
Transposable elements were detected and classified by homology search with Vmatch (http://www.vmatch.de) against the REdat_9.7_Triticeae section of the PGSB transposon library81. The following parameter settings were used: identity ≥70%, minimal hit length 75 bp, seed length 12 bp (exact commandline: -d -p -l 75 -identity 70 -seedlength 12 -exdrop 5). The Vmatch output was filtered for redundant hits by prioritizing higher-scoring matches and then either shortening (<90% coverage and ≥50 bp rest length) or removing lower-scoring overlaps.
The identification of full-length LTR retrotransposons with LTRharvest82 resulted in 143,957 non-overlapping candidate sequences using the following parameter settings: ‘overlaps best -seed 30 -minlenltr 100 -maxlenltr 2000 -mindistltr 3000 -maxdistltr 25000 -similar 85 -mintsd 4 -maxtsd 20 -motif tgca -motifmis 1 -vic 60 -xdrop 5 -mat 2 -mis -2 -ins -3 -del -3’. All candidates were annotated for PfamA domains with hmmer3 software83 and stringently filtered for false positives by several criteria, the main ones being the presence of at least one typical retrotransposon domain (for example, RT, RH, INT, GAG) and a tandem repeat content below 25%. This resulted in a final set of 24,952 high-confidence full-length LTR retrotransposons. Insertion ages of the LTR retrotransposons were calculated according to the method of ref. 84 by the divergence of 5′ and 3′ LTRs that had been identical at the time of transposition. We used a grass-specific mutation rate of 1 × 10−8. The average age of all full-length LTR elements was calculated in 4 Mb windows and plotted in Fig. 1a. The frequencies of 20-mers were determined using Tallymer85.
Phylogenetic analysis of Gypsy elements was performed on predicted protein sequences deposited at the TREP database32. Protein domains in predicted open reading frames were identified with Pfam86, SignalP87 and COILS88.
For the analysis of transposable element content in up- and downstream regions of genes, 10 kb immediately flanking the predicted coding sequences of all high-confidence genes were extracted from the genome assembly. The genomic segments were then used in BLASTN searches79 against the TREP database32. After an initial annotation, previously unclassified or poorly characterized transposable element families were re-analysed and new consensus sequences were constructed. Analysis of up- and downstream regions was then repeated with the updated TREP database. The transposable element family producing the longest BLASTN hit was determined for every 20th base position of each 10 kb segment, resulting in 500 data points for each up- and downstream region of the high-confidence genes.
Gene family analysis
Gene family clusters were defined from 39,734 barley high-confidence class genes and the annotated gene sets of Rice MSU7.0 (39,049 genes, http://rice.plantbiology.msu.edu/), B. distachyon version 3.1 (31,694 genes, https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Bdistachyon), S. bicolor version 3.1 (33,032 genes, https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Sbicolor) and A. thaliana TAIR10 (27,416 genes, https://www.arabidopsis.org/)) using OrthoMCL89 software version 2.0. Splice variants were removed from the datasets, keeping only the representative/longest protein sequence prediction, and datasets were filtered for internal stop codons and incompatible reading frames. In the first step, pairwise sequence similarities between all input protein sequences were calculated using BLASTP79 with an e-value cut-off of 10−5. Markov clustering of the resulting similarity matrix was used to define the orthologue cluster structure, using an inflation value (-I) of 1.5 (OrthoMCL default). Gene families with barley-specific gene duplications, compared with other plant species, were extracted from the ENSEMBL Compara pipeline90. Over- and under-representation of Gene Ontology terms between barley and other plant species (Supplementary Tables 4.1–4.3) and between genomic compartments (Supplementary Table 4.5) were analysed with a hypergeometric test using the functions GOstats and GSEABase from the Bioconductor R package91 against a universe of all genes with Gene Ontology annotations. REViGO92, which removes redundant and similar terms from long Gene Ontology lists by semantic clustering, was applied to visualize the enrichment results. Expansion of three barley gene families encoding α-amylases, the vacuolar processing enzyme VPE2 protein subfamily and the sugar transporters SWEET11 subfamily, with specific importance in barley grain filling/seed development or barley germination/malting, were analysed in greater detail using BLAST searches (versus genome and gene prediction) as well as GenomeThreader mappings to the barley genome assembly. Further details are provided in Supplementary Note 4. In situ hybridizations for SWEET genes were performed as described previously93.
Analysis of sequence and haplotype diversity
Ninety-six two-row spring (n = 48) and winter (n = 48) homozygous inbred elite barley lines (Supplementary Table 5.1) were subjected to exome capture using the barley Roche NimbleGen exome capture liquid array94 and sequenced on the Illumina HiSeq 2500 platform. An average of 2 × 21,876,780 paired-end Illumina reads per sample was generated. This corresponds to approximately 72× coverage of the 61 Mb exome capture space.
The raw Illumina reads were mapped to the reference sequence with BWA-MEM version 0.7.10 (ref. 66), using a stringent mismatch setting of ≤2% mismatches per read. Variant calling was performed with the Genome Analysis Tool Kit (GATK)71 version 3.4.0, following the GATK Best Practices pipeline (https://www.broadinstitute.org/gatk/guide/best-practices.php). This included read de-duplication, indel realignment, base quality score recalibration and variant calling with the latest version of the HaplotypeCaller. The workflow was implemented in a BASH script. The Tablet assembly viewer95 was used for visual spot checks of mappings and SNPs calls.
Variant discovery resulted in 15,982,580 variants in total, of which 943,959 were multi-nucleotide polymorphisms or short insertions/deletions (indels), while the remainder represented SNPs. For subsequent genetic analysis, we first reduced the total variant dataset by applying rigorous filtering criteria to produce a highly robust subset of 72,563 SNPs distributed across all seven barley chromosomes. The filtering applied was as follows: (1) ≥8× coverage for ≥50% of the samples; (2) ≥95% of samples represented at each SNP locus; (3) ≥5% minor allele frequency at the level of the sample: that is, counting sample genotypes rather than individual reads; (4) a VCF SNP quality score ≥30; and (5) ≥98% of samples homozygous. These filters reduced false-positive variant calls by removing spurious variant calls resulting from systematic read mis-mapping. Of this filtered dataset, a subset of 3,500 randomly sampled markers from each chromosome was analysed with the Haploview software96. This subsampling was required as Haploview was unable to generate the required plots when larger data volumes were used as input. Haploview was run on defaults, using the GABRIEL blocks method. The genotype calls were also imported into the genotype visualization software Flapjack97 to produce chromosome-scale images of haplotype diversity within the spring and winter pools. Diversity statistics were calculated in GenAlEx version 6.502 (ref. 98) and rolling averages based on 100 adjacent SNPs were plotted in Microsoft Excel 2010.
Data availability
The genome assembly for barley has been deposited in the Plant Genomics and Phenomics Research Data Repository under digital object identifier http://dx.doi.org/10.5447/IPK/2016/34. Accession numbers for all deposited datasets are listed in Supplementary Note 1. The barley genome assembly has been deposited on the IPK Barley Blast Server (http://webblast.ipk-gatersleben.de/barley_ibsc/). All other data are available from the corresponding authors upon reasonable request.
References
van Zeist, W. & Bakker-Heeres, J. A. H. Archaeological studies in the Levant 1. Neolithic sites in the Damascus basin: Aswad, Ghoraifé, Ramad. Palaeohistoria 24, 165–256 (1985)
Riehl, S., Zeidi, M. & Conard, N. J. Emergence of agriculture in the foothills of the Zagros Mountains of Iran. Science 341, 65–67 (2013)
Dietrich, O., Heun, M., Notroff, J., Schmidt, K. & Zarnkow, M. The role of cult and feasting in the emergence of Neolithic communities. New evidence from Göbekli Tepe, south-eastern Turkey. Antiquity 86, 674–695 (2012)
Hayden, B., Canuel, N. & Shanse, J. What was brewing in the Natufian? An archaeological assessment of brewing technology in the Epipaleolithic. J. Archaeol. Method Theory 20, 102–150 (2013)
Wang, J. et al. Revealing a 5,000-y-old beer recipe in China. Proc. Natl Acad. Sci. USA 113, 6444–6448 (2016)
Zohary, D ., Hopf, M. & Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin (Oxford Univ. Press, 2012)
International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012)
Yang, P. et al. PROTEIN DISULFIDE ISOMERASE LIKE 5-1 is a susceptibility factor to plant viruses. Proc. Natl Acad. Sci. USA 111, 2104–2109 (2014)
Pourkheirandish, M. et al. Evolution of the grain dispersal system in barley. Cell 162, 527–539 (2015)
Russell, J. et al. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 48, 1024–1030 (2016)
Künzel, G., Korzun, L. & Meister, A. Cytologically integrated physical restriction fragment length polymorphism maps for the barley genome based on translocation breakpoints. Genetics 154, 397–412 (2000)
Beier, S. et al. Multiplex sequencing of bacterial artificial chromosomes for assembling complex plant genomes. Plant Biotechnol. J. 14, 1511–1522 (2016)
Muñoz-Amatriaín, M. et al. Sequencing of 15 622 gene-bearing BACs clarifies the gene-dense regions of the barley genome. Plant J. 84, 216–227 (2015)
Ounit, R., Wanamaker, S., Close, T. J. & Lonardi, S. CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics 16, 236 (2015)
Colmsee, C. et al. BARLEX - the Barley Draft Genome Explorer. Mol. Plant 8, 964–966 (2015)
Ariyadasa, R. et al. A sequence-ready physical map of barley anchored genetically by two million single-nucleotide polymorphisms. Plant Physiol. 164, 412–423 (2014)
Mascher, M. et al. Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant J. 76, 718–727 (2013)
Lam, E. T. et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30, 771–776 (2012)
Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F. & Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–98 (2011)
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009)
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013)
Beier, S. et al. Construction of a map-based reference genome sequence for barley, Hordeum vulgare L. Sci. Data 4, 170044 (2017)
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015)
Fuchs, J., Houben, A., Brandes, A. & Schubert, I. Chromosome ‘painting’ in plants - a feasible technique? Chromosoma 104, 315–320 (1996)
Grob, S., Schmid, M. W. & Grossniklaus, U. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol. Cell 55, 678–693 (2014)
Duan, Z. et al. A three-dimensional model of the yeast genome. Nature 465, 363–367 (2010)
Tiang, C. L., He, Y. & Pawlowski, W. P. Chromosome organization and dynamics during interphase, mitosis, and meiosis in plants. Plant Physiol. 158, 26–34 (2012)
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014)
Houben, A. et al. Methylation of histone H3 in euchromatin of plant chromosomes depends on basic nuclear DNA content. Plant J. 33, 967–973 (2003)
Flavell, R. B., Bennett, M. D., Smith, J. B. & Smith, D. B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 12, 257–269 (1974)
SanMiguel, P. et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 274, 765–768 (1996)
Wicker, T., Matthews, D. E. & Keller, B. TREP: a database for Triticeae repetitive elements. Trends Plant Sci. 7, 561–562 (2002)
Choulet, F. et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 345, 1249721 (2014)
Bureau, T. E. & Wessler, S. R. Stowaway: a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell 6, 907–916 (1994)
Bureau, T. E. & Wessler, S. R. Mobile inverted-repeat elements of the Tourist family are associated with the genes of many cereal grasses. Proc. Natl Acad. Sci. USA 91, 1411–1415 (1994)
Malik, H. S. & Eickbush, T. H. Modular evolution of the integrase domain in the Ty3/Gypsy class of LTR retrotransposons. J. Virol. 73, 5186–5190 (1999)
Harris, M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004)
Huang, N., Sutliff, T. D., Litts, J. C. & Rodriguez, R. L. Classification and characterization of the rice α-amylase multigene family. Plant Mol. Biol. 14, 655–668 (1990)
Muthukrishnan, S., Gill, B. S., Swegle, M. & Chandra, G. R. Structural genes for α-amylases are located on barley chromosomes 1 and 6. J. Biol. Chem. 259, 13637–13639 (1984)
Khursheed, B. & Rogers, J. C. Barley α-amylase genes. Quantitative comparison of steady-state mRNA levels from individual members of the two different families expressed in aleurone cells. J. Biol. Chem. 263, 18953–18960 (1988)
Melkus, G. et al. Dynamic 13C/1H NMR imaging uncovers sugar allocation in the living seed. Plant Biotechnol. J. 9, 1022–1037 (2011)
Chen, L. Q. et al. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211 (2012)
Tran, V., Weier, D., Radchuk, R., Thiel, J. & Radchuk, V. Caspase-like activities accompany programmed cell death events in developing barley grains. PLoS ONE 9, e109426 (2014)
Comadran, J. et al. Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat. Genet. 44, 1388–1392 (2012)
Schmalenbach, I., Léon, J. & Pillen, K. Identification and verification of QTLs for agronomic traits using wild barley introgression lines. Theor. Appl. Genet. 118, 483–497 (2009)
Han, F. et al. Dissection of a malting quality QTL region on chromosome 1 (7H) of barley. Mol. Breed. 14, 339–347 (2004)
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009)
Zimin, A. V. et al. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the mega-reads algorithm. Preprint at http://biorxiv.org/content/early/2016/07/26/066100 (2016)
Pendleton, M. et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015)
Hirsch, C. et al. Draft assembly of elite inbred line PH207 provides insights into genomic and transcriptome diversity in maize. Plant Cell 28, 2700–2714 (2016)
Chevreux, B., Wetter, T. & Suhai, S. Genome sequence assembly using trace signals and additional sequence information. In Computer Science and Biology: Proc. 99th German Conference on Bioinformatics (eds Hofestädt, R. et al. 45–56 (GCB, 1999)
Steuernagel, B. et al. De novo 454 sequencing of barcoded BAC pools for comprehensive gene survey and genome analysis in the complex genome of barley. BMC Genomics 10, 547 (2009)
Taudien, S. et al. Sequencing of BAC pools by different next generation sequencing platforms and strategies. BMC Res. Notes 4, 411 (2011)
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012)
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011)
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012)
Stanˇková, H. et al. BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotechnol. J. 14, 1523–1531 (2016)
Lysák, M. A. et al. Flow karyotyping and sorting of mitotic chromosomes of barley (Hordeum vulgare L.). Chromosome Res. 7, 431–444 (1999)
Šimková, H., Cˇ íhalíková, J., Vrána, J., Lysák, M. & Doležel, J. Preparation of HMW DNA from plant nuclei and chromosomes isolated from root tips. Biol. Plant. 46, 369–373 (2003)
Cao, H. et al. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. Gigascience 3, 34 (2014)
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214 (2000)
Csardi, G. & Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 1695 (2006)
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011)
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013)
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010)
Hu, M. et al. HiCNorm: removing biases in Hi-C data via Poisson regression. Bioinformatics 28, 3131–3133 (2012)
R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2015)
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011)
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011)
Aliyeva-Schnorr, L. et al. Cytogenetic mapping with centromeric bacterial artificial chromosomes contigs shows that this recombination-poor region comprises more than half of barley chromosome 3H. Plant J. 84, 385–394 (2015)
Hudakova, S. et al. Sequence organization of barley centromeres. Nucleic Acids Res. 29, 5029–5035 (2001)
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005)
International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010)
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009)
Matsumoto, T. et al. Comprehensive sequence analysis of 24,783 barley full-length cDNAs derived from 12 clone libraries. Plant Physiol. 156, 20–28 (2011)
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012)
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990)
Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000)
Spannagl, M. et al. PGSB PlantsDB: updates to the database framework for comparative plant genome research. Nucleic Acids Res. 44 (D1), D1141–D1147 (2016)
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008)
Eddy, S. R. Accelerated profile HMM searches. PLOS Comput. Biol. 7, e1002195 (2011)
SanMiguel, P., Gaut, B. S., Tikhonov, A., Nakajima, Y. & Bennetzen, J. L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 20, 43–45 (1998)
Gremme, G., Steinbiss, S. & Kurtz, S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinformatics 10, 645–656 (2013)
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 32, D138–D141 (2004)
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011)
Lupas, A., Van Dyke, M. & Stock, J. Predicting coiled coils from protein sequences. Science 252, 1162–1164 (1991)
Li, L., Stoeckert, C. J. Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003)
Bolser, D., Staines, D. M., Pritchard, E. & Kersey, P. Ensembl Plants: integrating tools for visualizing, mining, and analyzing plant genomics data. Methods Mol. Biol. 1374, 115–140 (2016)
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004)
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of Gene Ontology terms. PLoS ONE 6, e21800 (2011)
Radchuk, V., Weier, D., Radchuk, R., Weschke, W. & Weber, H. Development of maternal seed tissue in barley is mediated by regulated cell expansion and cell disintegration and coordinated with endosperm growth. J. Exp. Bot. 62, 1217–1227 (2011)
Mascher, M. et al. Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant J. 76, 494–505 (2013)
Milne, I. et al. Tablet—next generation sequence assembly visualization. Bioinformatics 26, 401–402 (2010)
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005)
Milne, I. et al. Flapjack—graphical genotype visualization. Bioinformatics 26, 3133–3134 (2010)
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539 (2012)
The International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788 (2014)
Acknowledgements
This work was performed in the frame of the International Barley Genome Sequencing Consortium and was supported by German Ministry of Education and Research grants 0314000 and 0315954 to K.F.X.M., M.P., U.S. and N.S., and 031A536 to U.S. and K.F.X.M.; Leibniz ‘Pakt f. Forschung und Innovation’ grant ‘sequencing barley chromosome 3H’ to N.S. and U.S.; Scottish Government/UK Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/100663X/1 to R.W., P.E.H. and J.R.; BBSRC grants BB/I008357/1 to M.D.C. and M.C., and BB/I008071/1 to P.K.; Finland grant 266430 and a BioNano grant to A.H.S.; Carlsberg Foundation grant 2012_01_0461 to the Carlsberg Research Laboratory; Grains Research and Development Corporation (GRDC) grant DAW00233 to C.L. and P.L.; Department of Agricultural and Food, Government of Western Australia grant 681 to C.L.; National Natural Science Foundation of China (NSFC) grant 31129005 to C.L. and G. Zhang; NSFC grant 31330055 to G. Zhang.; Czech Ministry of Education, Youth and Sports grant LO1204 to J.D.; US National Science Foundation (NSF) grant DBI 0321756 to T.J.C. and S.L.; US Department of Agriculture–Cooperative State Research, Education, and Extension Service–National Institute of Food and Agriculture (USDA–CSREES–NIFA) grants 2009-65300-05645 and 2011-68002-30029 to T.J.C., S.L. and G.J.M.; NSF Advances in Biological Informatics grant DBI-1062301 to T.J.C. and S.L.; University of California grant CA-R-BPS-5306-H to T.J.C. and S.L.; NSF grant DBI 0321756 to S.L. BBSRC National Capability in Genomics (BB/J010375/1) and BBSRC Institute Strategic Programme funding for Bioinformatics (BB/J004669/1) to M.D.C., S.A. and M.C.; winter and spring barley accessions were a subset of genotypes selected from BBSRC and Agriculture and Horticulture Development Board projects AGOUEB and IMPROMALT (RD-2012-3776). We acknowledge (1) the technical assistance of S. König, M. Knauft, U. Beier, A. Kusserow, K. Trnka, I. Walde, S. Driesslein and C. Voss; (2) D. Stengel, A. Fiebig, T. Münch, D. Schüler, D. Arend, M. Lange and P. Rapazote-Flores for data management and submission; (3) K. Lipfert for artwork; (4) H. Berges, A. Bellec and S. Vautrin (CNRGV) for management and distribution of BAC libraries; (5) A. Graner and D. Marshall for scientific discussions.
Author information
Authors and Affiliations
Contributions
Project coordination: M.S., I.B., C. Li, R.W. (co-leader), N.S. (leader); BAC sequencing and assembly (1H, 3H, 4H): S.B., A. Himmelbach, S.T., M.F., M.G., M.M., U.S. (co-leader), M.P. (co-leader), N.S. (leader); BAC sequencing and assembly (2H, unassigned): D.S., D.H., S.A. (co-leader), M.D.C. (co-leader), M.C. (co-leader), R.W. (leader); BAC sequencing and assembly (5H, 7H): X.Z., R.A.B., Q.Z., C.T., J.K.M., B.C., G. Zhou, F.D., Y.H., S.Y., S. Cao, S. Wang, X.L., M.I.B., P.L., G. Zhang (co-leader), C. Li (leader); BAC sequencing and assembly (6H): S.B., S. Wang, C. Lin, H. Li, U.S., M.H. (co-leader), I.B. (leader); BAC sequencing (gene-bearing): M.M.-A., R.O., S. Wanamaker, S.L. (co-leader), T.J.C. (leader); optical mapping: A. Hastie, H.Š., J.T., H.S., J.V., S. Chan, M.M., N.S., J.D., A.H.S. (leader); data integration: M.M. (leader), S.B., C.C., D.B., L.L., T.S., J.A.P., P.K., N.S., U.S. (co-leader); transcriptome sequencing and analysis: P.E.H., M.B., J.R., H. Liu, S.T., M.F., M.G., M.P., R.W. (leader); annotation of transcribed regions: S.O.T., G.H., R.A.B., L.L., G.J.M., K.F.X.M. (co-leader), M.S. (leader); repetitive DNA analysis: T.W. (co-leader), J.T., K.F.X.M., A.H.S., H.G. (leader); gene family analysis: Q.Z., M.S., V.R., C.D., G.H., A.C., D.B., P.W., L.B., N.S., P.K., C. Li (co-leader), I.B. (leader); chromosome conformation capture: A. Himmelbach, S.G., L.A.-S., A. Houben, M.M. (co-leader), N.S. (leader); resequencing and diversity analysis: J.R., M.B., P.E.H., L.R., L.C., R.W. (leader); writing: M.M. (co-leader), M.S., A.H.S., G.J.M., R.W., N.S. (leader). All authors read and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A. Hastie is an employee of Bionano Genomics.
Additional information
Reviewer Information Nature thanks M. Bevan, B. Keller and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Gene annotation pipeline.
a, Gene annotation pipeline combined gene evidence information from four data sources. Open reading frames were then predicted for 83,105 gene candidates. b, Gene candidates were classified into high-confidence (HC) and low-confidence (LC) genes on the basis of homology to reference proteins and alignment to library of repeat elements. Additional filtering procedures were applied before defining the final gene sets. Arrows between boxes with counts of high-confidence and low-confidence genes in each step indicate re-classifications (high-confidence to low-confidence, or low-confidence to high-confidence).
Extended Data Figure 2 Assembly validation.
a, Conserved gene order between barley (y axis) and B. distachyon (x axis). b, Completeness of the gene annotation as assessed by BUSCO. c, Representation of repetitive k-mers in reads and assemblies. d, Representation of full-length LTR retrotransposons in sequence assemblies of plant genomes with different sizes (represented by black points). The map-based reference sequence of barley reported in the present paper is shown in blue. Red dots correspond to shotgun assemblies of the barley genome7 and wheat chromosome 3B99.
Extended Data Figure 3 Hi-C contact matrices.
a, Intrachromosomal contacts. b, Interchromosomal contacts. Darker red indicates a higher contact probability.
Extended Data Figure 4 Global patterns in Hi-C contact matrices.
a, Principal component analysis of intrachromosomal Hi-C contact matrices. The eigenvectors of the first three principal components are plotted. Centromere positions are marked with a red line. b, Proportion of variance explained by linear models incorporating position informational in the linear genome fitted to the Hi-C contact matrices. c, Hi-C link counts in Morex × Barke F1 hybrids within the same chromosome, between homologous chromosomes and between non-homologous chromosomes.
Extended Data Figure 6 Experimental strategy to distinguish individual amy1_1 copies by PCR from genomic DNA through polymorphisms in the extended promoter regions of amy1_1 full-length copies.
a, Experimental strategy, primers CD52_amy1fw and CD53_amy1rc bind in the extended promotor region of all full-length amy1_1 copies (expected amplicon sizes are 225 bp for amy1_1a, 299 bp for amy1_1b and amy1_1d and 336 bp for amy1_1c). Forward primers CD54_fw1a, CD55_fw1b and CD56_fw1c are designed to specifically amplify copies amy1_1a, amy1_1b and amy1_1c, respectively when used with reverse primer CD58_amy1rc, which binds in the coding region of all amy1_1 copies. Expected amplicon sizes are 1,024 bp (amy1_1a), 1,026 bp (amy1_1b) and 757 bp (amy1_1c). Primer pair (CD55_fw1b–CD58_amy1rc) further binds to copy amy1_1d: here, sequences of the expected amplicons contain sufficient polymorphisms to distinguish these copies from each other. Positions of selected sequence polymorphisms and deleted regions suitable to distinguish single copies are indicated as black vertical bars and gaps, respectively. Numbering was done in respect of copy amy1_1b. b, PCR amplification of amy1_1 promoter regions in six barley cultivars and landraces. As expected, a PCR for cultivar Morex, using universal primers CD52_amy1fw and CD53_amy1rc, resulted in three amplicons of the expected sizes 225, 299 and 336 bp (compare a), which was confirmed by Sanger sequencing. Further primers CD52_amy1fw and CD53_amy1rc were used to amplify the amy1_1 extended promoter region in various barley cultivars. These experiments indicate polymorphic variation in, or even absence of, single promoters of amy1_1 in the different cultivars. The cultivars analysed differ in row type (six-rowed: cultivars Morex, Masan Naked 1, Akashinriki, Etincel; two-rowed: cultivars Barke, Bowman), growth habit (spring barley: cultivars Morex, Barke, Bowman, Masan Naked 1, Akashinriki; winter barley: cultivar Etincel) and geographic origin (North America: cultivars Morex, Bowman; Europe: cultivars Barke, Etincel; Asia: cultivars Masan Naked 1, Akashinriki). The cultivars Masan Naked 1 and Akashinriki depict landraces used for food, Bowman was classified as non-malting barley, while Morex, Barke and Etincel represent modern malting barley. c, Copy-specific PCR amplification of amy1_1 extended promoter regions. PCR amplification and Sanger sequencing identified three amy1_1 copies in barley cultivar Morex: amy1_1a (CD54_fw1a–CD58_amy1rc), amy1_1b (CD55_fw1b–CD58_amy1rc) and amy1_1c (CD56_fw1c–CD58_amy1rc). Additionally, sequencing revealed two polymorphic sites in PCR amplicon amy1_1b (CD55_fw1b–CD58_amy1rc) at positions 721 bp (T/C) and 1175 bp (C/T) (see a), indicating the presence of one or two additional amy1_1b-like copies in the genome of the analysed individual. The presence of copy amy1-1d could not be confirmed. The reason for that might have been sequence deviations in the cultivar Morex accession used for BAC library construction versus that used for the presented experiments, or differences in PCR efficiency for amplification of copies amy1_1b and amy1_1d.
Extended Data Figure 7 SWEET gene expression.
a, Control experiment for mRNA in situ hybridizations shown in Fig. 3c. In situ hybridization with sense probes for SWEET11a (top) and SWEET11b (bottom). Scale bars, 100 μm. b, Expression of SWEET11a and SWEET11b. Results of qPCR in different plant organs and in the developing grains at 7 days after flowering (DAF).
Extended Data Figure 8 Haplotype blocks in sets of 48 samples each of elite two-row spring barley lines (top half of each chromosome’s figure) and winter barley lines (bottom half), separately for each chromosome.
We restricted the number of SNPs per chromosome by randomly choosing 3,500 to fit with the maximum permitted by the software. The red and green plots in the centre of each chromosome figure represent whole-canvas dumps produced with the Flapjack software97. Markers are arranged in columns in linear order along the chromosome; red pixels represent reference alleles, while green pixels represent alternative alleles. Each row represents a barley cultivar; these have been sorted top to bottom by year of introduction (ascending). The Flapjack plots are framed by cropped linkage disequilibrium plots generated with the HaploView software96. Colour intensity conveys the extent of linkage between pairs of markers (red, highest). Approximate centromere positions are indicated by semi-opaque grey squares. The triangles with the thin black outline represent haplotype blocks as computed by HaploView. In some regions, extensive stretches exist where no blocks were detected (for example, chr2H, spring lines in top half, near centromere). These generally present highly monomorphic regions where there is no evidence for multiple haplotypes, and consequently blocks were not called.
Supplementary information
Supplementary Information
This file contains Supplementary Notes 1-5 and Supplementary References – see contents page for details. (PDF 3453 kb)
Supplementary Data
This zipped file contains Supplementary Tables 4.1-4.5, 4.7 and 5.1. (ZIP 25593 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mascher, M., Gundlach, H., Himmelbach, A. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017). https://doi.org/10.1038/nature22043
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature22043
This article is cited by
-
Evolution of Einkorn wheat centromeres is driven by the mutualistic interplay of two LTR retrotransposons
Mobile DNA (2024)
-
Transcriptional changes during crown-root development and emergence in barley (Hordeum vulgare L.)
BMC Plant Biology (2024)
-
A guide to barley mutants
Hereditas (2024)
-
Loss of Lateral suppressor gene is associated with evolution of root nodule symbiosis in Leguminosae
Genome Biology (2024)
-
Genomic resources for a historical collection of cultivated two-row European spring barley genotypes
Scientific Data (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
