
Abstract
Cervical cancer remains one of the leading causes of cancer-related deaths worldwide. Here we report the extensive molecular characterization of 228 primary cervical cancers, one of the largest comprehensive genomic studies of cervical cancer to date. We observed notable APOBEC mutagenesis patterns and identified SHKBP1, ERBB3, CASP8, HLA-A and TGFBR2 as novel significantly mutated genes in cervical cancer. We also discovered amplifications in immune targets CD274 (also known as PD-L1) and PDCD1LG2 (also known as PD-L2), and the BCAR4 long non-coding RNA, which has been associated with response to lapatinib. Integration of human papilloma virus (HPV) was observed in all HPV18-related samples and 76% of HPV16-related samples, and was associated with structural aberrations and increased target-gene expression. We identified a unique set of endometrial-like cervical cancers, comprised predominantly of HPV-negative tumours with relatively high frequencies of KRAS, ARID1A and PTEN mutations. Integrative clustering of 178 samples identified keratin-low squamous, keratin-high squamous and adenocarcinoma-rich subgroups. These molecular analyses reveal new potential therapeutic targets for cervical cancers.
Similar content being viewed by others

Main
Cervical cancer accounts for 528,000 new cases and 266,000 deaths worldwide each year, more than any other gynaecological tumour1. Ninety-five per cent of cases are caused by persistent infections with carcinogenic HPVs2. Effective prophylactic vaccines against the most important carcinogenic HPV types are available, but the number of people receiving the vaccine remains low. Although early cervical cancer can be treated with surgery or radiation, metastatic cervical cancer is incurable and new therapeutic approaches are needed3.
While most HPV infections are cleared within months, some persist and express viral oncogenes that inactivate p53 and RB, leading to increased genomic instability, accumulation of somatic mutations, and in some cases, integration of HPV into the host genome4. The association with cancer risk and histological subtypes varies substantially among carcinogenic HPV types, but the reasons for these differences are poorly understood. Furthermore, clinically relevant subgroups of cervical cancer patients have yet to be identified. Here we present a comprehensive study of invasive cervical cancer conducted as part of The Cancer Genome Atlas (TCGA) project, with a focus on identifying novel clinical and molecular associations as well as functionally altered signalling pathways that may drive tumorigenesis and serve as prognostic or therapeutic markers.
Samples and clinical data
Primary frozen tumour tissue and blood were obtained from women with cervical cancer who had not received prior chemotherapy or radiotherapy (Supplementary Information 1 and Supplementary Tables 1, 2). DNA, RNA and protein were processed as previously described5 (Supplementary Information 1, 3, 5 and 8). Mutations were called for 192 samples (the extended set), while all other platform (aside from protein) and integrated analyses were performed on a subset of 178 samples (the core set). Protein levels were measured on 155 samples (119 samples from both the core and extended sets plus 36 additional samples). The total number of non-overlapping samples in these three sets was 228 (Extended Data Fig. 1a). Of the 178 core-set samples, surgery was the primary treatment in 121 cases, median follow-up time was 17 months, and 145 patients were alive at the time of last follow-up. A committee of expert gynaecological pathologists reviewed most cases (Supplementary Information 1 and Extended Data Fig. 1b–g). The core set included 144 squamous cell carcinomas, 31 adenocarcinomas and 3 adenosquamous cancers.
Somatic genomic alterations
Whole-exome sequencing was performed on 192 extended-set tumour–blood pairs. All samples had at least 32 Mb of target exons covered with a median depth of 49× (range: 7–351×) for tumour samples and 47× (range: 9–341×) for normal samples. Collectively, the samples contained 43,324 somatic mutations, including 24,551 missense, 2,470 nonsense, 9,260 silent, 5,841 non-coding, 535 splice-site, 74 non-stop mutations, 475 frameshift insertions and deletions (indels) and 118 in-frame indels. Eleven tumours with outlier mutation frequencies (>600 per sample) were classified as ‘hypermutant’. The aggregate mutation density was 4.04 mutations per Mb across all tumours, and 2.53 when the hypermutant tumours were excluded.
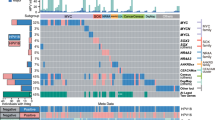
Fourteen genes that are significantly mutated (SMGs) with false-discovery rates (FDR) < 0.1 were found using the MutSig2CV6 algorithm (Supplementary Table 4). We identified SHKBP1, ERBB3, CASP8, HLA-A and TGFBR2 as novel SMGs in cervical cancer, and confirmed that PIK3CA, EP300, FBXW7, HLA-B, PTEN, NFE2L2, ARID1A, KRAS and MAPK1 are SMGs, as previously reported7,8 (Fig. 1, Extended Data Fig. 2a–g and Supplementary Fig. 6). Supplementary Table 4 shows the comparison of SMGs identified in the current TCGA set and a previously published dataset8. Mutations in 7 of the 14 SMGs in the TCGA set were present in at least one squamous cell carcinoma and one adenocarcinoma; however, mutations in HLA-A, HLA-B, NFE2L2, MAPK1, CASP8, SHKBP1 and TGFBR2 were found exclusively in squamous tumours.

a–d, Cervical carcinoma samples ordered by histology and mutation frequency (a), clinical and molecular platform features (b), SMGs (c), and select somatic copy number alterations (d) are presented. SMGs are ordered by the overall mutation frequency and colour-coded by mutation type. Novel SMGs identified in squamous cell carcinomas are labelled in turquoise text. The number of APOBEC signature mutations (red) and other mutations (blue) present in every SMG is plotted to the right of the SMG panel and the number of gene-level somatic copy number alterations across all genes is plotted as gain (red) and loss (blue) to the right of the somatic copy number alteration panel. CN, copy number; SCNAs, somatic copy number alterations; Adeno., adenocarcinomas; Adenosq., adenosquamous cancers; Squamous, squamous cell carcinomas.
PIK3CA had mostly activating helical-domain E542K and E545K mutations, with a marked relative decrease in mutations elsewhere in the gene (Extended Data Fig. 2g). This observation resembles findings in bladder cancer9 and HPV-positive head and neck squamous cell cancers (HNSCs)10, but it differs from observations in breast and most other cancers11. The underlying nucleotide substitution pattern in the E542K and E545K mutations is associated with mutagenesis by a subclass of APOBEC cytidine deaminases8,12,13,14,15, with 150 out of 192 exomes displaying significant (q < 0.05) enrichment (up to sixfold) for the APOBEC signature. Further, the APOBEC mutation load correlated strongly with the total number of mutations per sample (Extended Data Fig. 2h), suggesting that APOBEC mutagenesis is the predominant source of mutations in cervical cancers.
We found an average of 88 somatic copy number alterations per tumour, fewer than in HNSC, ovarian and serous endometrial carcinomas, but more than in endometrioid endometrial carcinomas10,16,17. GISTIC2.0 analysis (with a threshold of q < 0.25) revealed 26 focal amplifications and 37 focal deletions along with 23 recurrently altered whole arms (Extended Data Fig. 3c and Supplementary Table 7). Novel recurrent focal amplification events were identified (in genomic order) at 7p11.2 (EGFR, 17%), 9p24.1 (CD274, PDCD1LG2, 21%), 13q22.1 (KLF5, 18%) and 16p13.13 (BCAR4, 20%). Other previously reported amplification events occurred at 3q26.31 (TERC, MECOM, 78%), 3q28 (TP63, 77%), 8q24.21 (MYC, PVT1, 42%), 11q22.1 (YAP1, BIRC2, BIRC3, 17%), and 17q12 (ERBB2, 17%). Novel recurrent deletions were identified at 3p24.1 (TGFBR2, 36%) and 18q21.2 (SMAD4, 28%), in addition to previously identified deletions at 4q35.2 (FAT1, 36%) and 10q23.31 (PTEN, 31%). A cluster with high copy number alterations mostly contained squamous tumours with amplification events involving 11q22 (YAP1, BIRC2, BIRC3) and 7p11.2 (EGFR), whereas the cluster containing low copy number variations included most adenocarcinomas and was enriched for tumours with deletions in TGFBR2 and SMAD4, and gains in ERBB2 and KLF5 (Extended Data Fig. 3a, b). Notably, both groups had amplifications involving CD274 (PD-L1) and PDCD1LG2 (PD-L2) that correlated significantly (P < 0.0001) with expression of two key immune cytolytic effector genes, granzyme A and perforin18 (Extended Data Fig. 3d). This highlights the potential of immunotherapeutic strategies for a subset of cervical cancers.
Structural rearrangements were identified by analysis of RNA sequencing (RNA-seq) (core set, n = 178) and whole-genome sequencing (WGS) data with low-pass (n = 50) and deep (n = 19) coverage. Both RNA-seq and WGS detected 22 putative structural rearrangements in 14 patients (Supplementary Table 8). In total, 26 recurrent fusions were found (Supplementary Table 9, with examples in Extended Data Fig. 4d). RNA-seq analysis revealed four samples with 16p13 ZC3H7A–BCAR4 gene fusions, whereby exon 1 of ZC3H7A was linked to the last exon of BCAR4. WGS revealed tandem duplication and copy number gain of BCAR4 on chromosome 16p13.13 (Extended Data Fig. 4c). BCAR4 is a metastasis-promoting long non-coding RNA that enhances cell proliferation in oestrogen-resistant breast cancer by activating the HER2/HER3 pathway. Lapatinib, an EGFR/HER2 inhibitor, counteracts BCAR4-driven tumour growth in vitro, and warrants evaluation as a possible therapeutic agent in BCAR4-positive cervical cancer19.
Integrated analysis of molecular subgroups
Integration of copy number, methylation, mRNA and microRNA (miRNA) data using iCluster20 highlighted the molecular heterogeneity of cervical carcinomas. Three clusters were identified that largely corresponded to mRNA clusters (Supplementary Fig. 9): a squamous cluster with high expression of keratin gene family members (keratin-high), another squamous cluster with lower expression of keratin genes (keratin-low), and an adenocarcinoma-rich cluster (adenocarcinoma). Keratin-high and keratin-low clusters included 133 out of 144 squamous cell carcinomas and the adenocarcinoma cluster contained 29 out of 31 adenocarcinomas (Fig. 2). KRAS (P = 9.7 × 10−5), ERBB3 (P = 2.6 × 10−3) and HLA-A (P = 0.03) mutations were significantly associated with clusters, whereby KRAS mutations were absent from the keratin-high cluster and HLA-A mutations were absent from the adenocarcinoma cluster (Fig. 2). Members of the SPRR and TMPRSS cornification gene families and the SMGs ARID1A (P = 0.02), NFE2L2 (P = 6.9 × 10−6) and PIK3CA (P = 0.01) were differentially expressed between keratin-low and keratin-high clusters (Extended Data Fig. 4b).

a, Integrative clustering of 178 core-set cervical cancer samples using mRNA, methylation, miRNA and copy number variation (CNV) data identifies two squamous-carcinoma-enriched groups (keratin-low and keratin-high) and one adenocarcinoma-enriched group, as shown in the feature bars (top). Features presented include histology, HPV clade, HPV integration status, UCEC-like status, APOBEC mutagenesis level, mRNA EMT score, tumour purity and three SMGs (KRAS, ERBB3 and HLA-A) that are significantly associated across the three clusters identified with iCluster (ERBB2 is presented for comparison purposes with its family member ERBB3). b, The cluster of clusters panel displays subtypes defined independently by mRNA, miRNA, methylation, reverse phase protein array (RPPA), CNV and PARADIGM data. C1–C6 indicate clusters. Black, sample is not represented in the cluster; red, sample is represented in the cluster; grey, data not available. c, The heatmaps show select mRNAs, miRNAs, proteins and CNVs that are either significantly associated with iCluster groups or have been identified as markers in other analyses. The heatmap colour scale bar represents the scale for the features presented in the heatmaps with a breakpoint of zero represented by white. APOBEC mut., APOBEC mutagenesis; inter., intermediate.
Unsupervised hierarchical clustering of variable DNA-methylation probes produced three groups (Extended Data Fig. 5a), including a small ‘CpG island hypermethylated’ (CIMP-high) cluster, a CIMP-intermediate cluster and a CIMP-low cluster that were associated with an epithelial–mesenchymal transition (EMT) mRNA score10,21 (Extended Data Fig. 5b). Most of the samples in the adenocarcinoma cluster were CIMP-high, whereas the other iCluster groups contained a mixture of CIMP-intermediate and CIMP-low samples (Fig. 2). Comparing all cervical carcinomas to 120 normal samples drawn from 12 TCGA projects, we identified 1,026 epigenetically silenced genes that were methylated to a greater extent in cancers than in normal tissues, including several zinc-finger (ZNF), protease (ADAM, ADAMTS), and collagen (COL) genes (Supplementary Tables 11 and 12).
Unsupervised clustering resulted in six miRNA clusters that were associated with the iCluster groups (P = 1.7 × 10−19) (Extended Data Fig. 6a). Samples from the adenocarcinoma cluster almost exclusively overlapped with miRNA cluster 5, and were characterized by high expression of miR-375 and low expression of miR-205-5p and miR-944 (Supplementary Table 31). Expression levels of tumour suppressors miR-99a-5p and miR-203a were significantly higher in samples from the keratin-high cluster than samples from the keratin-low cluster (Supplementary Table 31; P = 0.01 and P = 0.008, respectively). Among miRNAs with significant and functionally validated gene and protein anti-correlations22, one large subnetwork involved the miR-200 family and other miRNAs with expression patterns that anti-correlated with those of the EMT-related transcription factors ZEB1, ZEB2 and SNAI2, the Hippo and p73 transcriptional co-factor YAP1, the receptor tyrosine kinases (RTKs) ERBB2, ERBB3 and AXL, and the hormone receptor ESR1 (Extended Data Fig. 6b, Supplementary Figs 17, 18 and Supplementary Table 15).
Reverse phase protein array (RPPA) analysis of 155 samples with 192 antibodies (Extended Data Fig. 1a and Supplementary Table 17) identified three clusters significantly associated with the iCluster groups (P = 1.8 × 10−4) and EMT mRNA score (Fig. 3a, c, d and Supplementary Table 16). Samples from the EMT cluster were enriched in the keratin-low cluster, whereas PI3K–AKT and hormone cluster samples were enriched in the keratin-high and adenocarcinoma clusters, respectively, suggesting distinct pathway activation across integrated cervical cancer subtypes. Differential expression levels of phosphorylated (p)-MAPK, p-EGFR (Y1068), p-SRC (Y416), IGFBP2 and TIGAR between keratin-high and keratin-low clusters suggest diverse activation patterns of RTK, MAPK, PI3K and metabolic signalling pathways that may underlie the molecular diversity of cervical squamous cancers (Fig. 2).

a, Clustered heatmap of samples (columns) and 192 antibodies (rows) for 155 samples (112 overlap with the core set of 178; see Extended Data Fig. 1a). Clusters presented from left to right include hormone (dark blue), EMT (red) and PI3K–AKT (green). A subset of proteins differentially expressed between the clusters is highlighted. Tracks for clinical and molecular features are shown for features that were significantly associated with RPPA clusters (P < 0.05). Correlation between RPPA clusters and other categorical variables were detected by χ2 test, whereas correlations with continuous variables were analysed using the non-parametric Kruskal–Wallis test. In the heatmap, blue represents downregulated expression, red represents upregulated expression and white represents no change in expression. NA, data not available. b, Five-year Kaplan–Meier survival curves and log-rank test P value (P = 6.1 × 10−4) comparing overall survival (OS) across all RPPA clusters using 115 silhouette width core samples (silhouette core; see Supplementary Information 8). c, EMT mRNA score levels were calculated for all samples and compared across RPPA clusters. P = 0.001 (one-way ANOVA). d, Pathway scores for EMT, hormone-receptor and PI3K–AKT signalling pathways are presented for all RPPA clusters (x axis); Kruskal–Wallis test used to identify significant pathway score differences between the clusters.
The core members of each RPPA cluster with the highest silhouette width (>0.02, n = 115) were associated with five-year survival (Fig. 3b; P = 6.1 × 10−4), with the EMT group exhibiting worse outcome. Notably, this was the only platform where clusters associated with outcomes (Supplementary Figs 8, 9, 12 and 22; Supplementary Information 6). Samples in the EMT cluster exhibited high ‘reactive’ pathway scores11 (Supplementary Fig. 20), illustrating for the first time in cervical cancer the presence of a subset of stromal reactive tumours that have high expression of caveolin-1, MYH11 and RAB11, a subset which also appears in other diseases23 (Supplementary Table 16). YAP was the most significantly differentially expressed protein distinguishing samples from the EMT cluster from all others (Supplementary Table 18; P = 1.7 × 10−15) and YAP1 was significantly amplified in the samples from the EMT cluster compared to the hormone (P = 1.1 × 10−5) and PI3K–AKT cluster (P = 6.4 × 10−4) samples. Regulation of the EMT-related molecules YAP and ZEB1 (refs 24, 25, 26) may also be driven by significantly lower expression levels of miR-200a-3p in the samples from the EMT cluster compared to samples from the other RPPA clusters (Extended Data Figs 6b, 7a; P = 3.8 × 10−3). These results highlight potential roles for YAP and reactive stroma in EMT-regulated progression of cervical cancers.
The mutual exclusivity modules in cancer (MEMo) algorithm27 uses somatic-mutation and copy number data to identify oncogenic networks with mutually exclusive genomic alterations. Because miR-200a and miR-200b (miR-200a/b) expression was negatively correlated with EMT mRNA scores (Extended Data Fig. 7b, d), we used MEMo to examine alterations in miR-200a/b and EMT gene networks and found a potential link between the TGFβ pathway and miR-200a/b alterations in regulating EMT28,29. Deletions and mutations affecting the receptor gene TGFBR2, the modulating genes CREBBP and EP300, and the transcription factor SMAD4 probably all affect growth-suppressive and pro-apoptotic functions driven by TGFβ (Fig. 4c) and were observed in 30% of squamous cell carcinomas (Fig. 4d). Tumours with both hypermethylation and downregulation of miR-200a/b (referred to as altered) were restricted to squamous cell carcinomas, were enriched in the keratin-low cluster (Fig. 4d and Extended Data Fig. 8; P = 0.001 for both miR-200a and miR-200b), showed significant upregulation of both ZEB1 and ZEB2 (Extended Data Fig. 9a–d), and were mutually exclusive with alterations in the TGFβ signalling pathway (Fig. 4d). Notably, samples with altered miR-200a/b exhibited higher EMT mRNA scores than unaltered samples, whereas no significant difference was found between samples with or without TGFβ-pathway alterations (Fig. 4d and Extended Data Fig. 7c, e). These findings highlight potential treatment approaches for this subgroup of cervical cancer patients, as targeting EMT may render tumours more sensitive to small-molecule inhibitors and cytotoxic chemotherapy21,30,31.

a, Multiple alterations affect RTK, AKT and MAPK signalling in both squamous cell carcinomas and adenocarcinomas. A schematic diagram of the pathways is shown for altered genes along with the percentage of alteration in squamous cell carcinomas and adenocarcinomas. Significant P values (P < 0.05, Student’s t-test) for alteration frequency differences between squamous cell carcinomas and adenocarcinomas are listed at the gene level, with significantly different genes marked with an asterisk. b, Distinct types of alterations (amplification, deletion, missense mutation and truncating mutation) affect genes (rows) in these pathways in each sample (columns). c, TGFβ signalling is frequently altered in cervical tumours. Alterations in this pathway are divided between those probably affecting TGFβ-tumour-suppressive functions and those affecting the TGFβ-driven EMT program. d, Samples with alterations targeting TGFβ-tumour-suppressive functions do not show significantly different EMT scores compared with all other samples; however, samples with low expression/high methylation of miR-200a/b have significantly higher EMT scores than all other samples. miR-down, samples met double threshold of methylated and downregulated as described in Methods. NS, not significant. Percentages in b and d, indicate per cent of the total histological subgroup population.
MEMo analysis also showed differences in therapeutically relevant alterations in RTK, PI3K and MAPK pathways across cervical cancers. MEMo identified mutual exclusivity modules involving alterations within both the PI3K and MAPK pathways (Supplementary Table 27; adjusted P = 0.06); however, there was a strong tendency for co-occurrence of ERBB2 and ERBB3 alterations within adenocarcinomas (P < 0.001, log odds ratio > 3), indicating that a subset of these tumours may exhibit aberrant HER3 signalling through interactions between mutant HER3 and activated HER2 and therefore could potentially benefit from HER2- and HER3-targeted therapies32 (Fig. 4a, b). Although not statistically significant, aberrations in PIK3CA also tended to co-occur with PTEN somatic mutations and deletions (P = 0.078, log odds ratio = 0.71), which is similar to endometrial tumours with few copy number alterations and suggests potential therapeutic benefit from PI3K-pathway-targeting agents17.
PARADIGM33,34, which integrates copy number, RNA-seq and pathway-interaction data, showed markedly different pathway activation profiles between squamous carcinomas and adenocarcinomas (Extended Data Fig. 10 and Supplementary Fig. 48). PARADIGM identified higher inferred activation of p53, p63, p73, AP-1, MYC, HIF1A, FGFR3 and MAPK signalling as key distinguishing features of squamous cell carcinomas, similar to other squamous cancers35. By contrast, adenocarcinomas exhibited higher inferred activation of ERα, FOXA1, FOXA2 and FGFR1 pathways (Extended Data Fig. 10, Supplementary Figs 25, 48 and Supplementary Table 18). Possible underlying mechanisms for ERα upregulation may stem from the expression of miR-193b-3p, a direct regulator of ESR1 that was significantly downregulated in adenocarcinomas compared to squamous carcinomas (Fig. 2, Extended Data Fig. 6 and Supplementary Table 14; P = 0.04), or from oestrogen signalling in stromal cells36.
Cross-cancer analysis
We next evaluated the relationship of cervical cancer subtypes with endometrial cancer, an adjacent cancer site with hormone-related carcinogenesis, and HNSC, a subset of which is caused by HPV. For this, hierarchical clustering of cervical, uterine corpus endometrial (UCEC)17, and HNSC10 mRNA-expression data was performed. Three major groups were observed, with cluster 1 including all UCEC samples and most cervical adenocarcinomas and characterized by overexpression of hormone-receptor genes ESR1 and PGR (Extended Data Fig. 4a). Cluster 2 included predominantly squamous cervical carcinomas and 23 out of 27 HPV-positive HNSC samples. Cluster 3 included few cervical cancers and the remaining HNSC cancers, which were mostly HPV-negative. This highlights the similarity of HPV-related squamous cancers at different anatomical sites.
Since a subset of cervical cancers clustered with endometrial samples, a gene-expression classifier was developed to predict whether carcinomas were cervical or endometrial (Supplementary Information 5). We classified 8 out of 178 (4.5%) cervical cancer samples as endometrial-like (UCEC-like) cancers, which were confirmed to be cervical cancers by study pathologists (Extended Data Fig. 1f, g). These tumours included 7 out of 9 HPV-negative cancers and 5 of the 8 were adenocarcinomas. Six UCEC-like cancers were in the adenocarcinoma cluster and two were in the keratin-low cluster. Despite their low number, the UCEC-like tumours accounted for 33%, 27% and 20% of mutations in ARID1A, KRAS and PTEN, respectively. They were associated with the RPPA hormone and miRNA C6 clusters, and all but one sample was CIMP-low and copy number-low (Supplementary Table 1).
HPV genotypes, variants and integration
Of the 178 core-set tumours, 169 (95%) were HPV-positive, 120 (67%) had alpha-9 (A9) types (103 HPV16), 45 (25%) had alpha-7 (A7) types (27 HPV18), and 9 (5%) were HPV-negative (Supplementary Table 3). HPV variants were predominantly European (137 out of 169, 81% A variants), and there was a significant association of non-European HPV16 variants with cervical adenocarcinomas (Supplementary Table 3; odds ratio = 5.3, P = 3 × 10–3). All HPV-positive cancers had detectable expression of HPV E6- and E7-oncogene mRNAs, which encode proteins that inhibit p53 and RB function, respectively37,38. Notably, HPV18 cancers had significantly higher ratios of unspliced to spliced transcripts encoding the active E6 oncoprotein than the HPV16 cancers (Extended Data Fig. 11a; P = 2 × 10–10), suggesting different functional implications of E6 and E7 in cancers associated with different HPV genotypes.
HPV A7 types were enriched in the keratin-low and adenocarcinoma clusters (P = 5 × 10–4). Most HPV clade A7 tumours were CIMP-low, and HPV-negative tumours formed a distinct subgroup within the CIMP-low cluster with a significantly lower mean promoter-methylation level than other samples in that cluster (Extended Data Fig. 5a; P = 5 × 10−3). Samples with the highest rate of gene silencing were HPV-positive adenocarcinomas, particularly those related to A9 types (t-test P < 0.001). Functional epigenetic module (Supplementary Information 13) analysis39, which integrates DNA-methylation and gene-expression data using protein–protein interaction networks, identified inverse correlations between methylation and gene expression in HPV-positive versus HPV-negative cervical cancers and HPV-positive (n = 36) versus HPV-negative (n = 243) HNSCs. The analysis revealed 12 statistically significant subnetworks for cervical cancer and 11 for HNSCs, with one common subnetwork centred around Forkhead Box A2 (FOXA2) (Supplementary Table 19 and Supplementary Fig. 32). miR-944, miR-767-5p and miR-105-5p were the most differentially expressed miRNAs between HPV-positive and HPV-negative samples (Supplementary Fig. 14e). miR-944 expression was also significantly higher, whereas miR-375 expression was significantly lower in HPV16-positive squamous cancers compared to HPV18-positive squamous cancers (Supplementary Fig. 14d). Notably, HPV-negative cancers had a significantly higher EMT mRNA score and a lower frequency of the APOBEC mutagenesis signature compared with HPV-positive tumours (Extended Data Fig. 11b and Supplementary Fig. 27; P = 0.02 and P = 0.004, respectively).
PARADIGM was used to evaluate molecular pathways differentially activated in squamous samples with A7- and A9-HPV infections. We observed higher inferred activation of p53 and p63 signalling and lower FOXA1 signalling in tumours infected with A9 types (Fig. 5a and Supplementary Fig. 23a). Higher SFN pathway activation was also observed for A9-positive tumours, which is consistent with the low methylation and high gene-expression patterns of SFN found in functional epigenetic module analysis (Fig. 5a and Supplementary Table 19). Notably, the SFN-encoded stratifin (also known as 14-3-3σ) adaptor protein has previously been associated with epithelial immortalization and squamous cell cancers40,41, altered p53-pathway activation42, and Wnt-mediated β-catenin signalling43.

a, Cytoscape display of the largest interconnected regulatory network of PARADIGM integrated pathway level (IPL) features showing differential inferred activation between HPV A9 and A7 squamous carcinomas (n = 101 and n = 35, respectively). Node colour and intensity reflect the level of differential activation. Node size represents level of significance. SFN is within a subnetwork identified by functional epigenetic module analysis (Supplementary Information 13) as disrupted between HPV A9 and A7 squamous cell carcinomas, and is highlighted using a bold black outline. rRNA, ribosomal RNA. DST, DST isoform 3. b, Circos plot showing frequency (0–100%) of gains and losses for regions of each chromosome (outer circle). Lines within the inner circle indicate integration breakpoints from the HPV genome (L1, L2, E1, E2, E4, E5, E6 and E7 genes) to the human genome as defined in Methods, Supplementary Information 2, and Supplementary Table 3. Lines are colour coded by HPV clade.
Viral–cellular fusion transcripts indicating integration of HPV into the host genome were observed in 141 out of 169 (83%) HPV-positive cancers, including all HPV18-positive cancers. Of these 141 samples, 90 (64%) had a single HPV integration event, 35 had two events, and 16 had three or more events (totalling 220 unique integration events) (Supplementary Table 3). HPV integration events affected all chromosomes, including some previously described hotspots such as 3q28 and 8q24 (ref. 44) (Fig. 5b). Genomic loci affected by integration were characterized by increased somatic copy number alterations (P = 6.9 × 10−13 for HPV16 and P = 0.058 for HPV18) and increased gene expression (P = 1.6 × 10−11 for HPV16 and P = 0.011 for HPV18) (Extended Data Fig. 11c, d). In addition, 153 (70%) fusion transcripts included known or predicted genes, whereas the remainder included intergenic regions (Fig. 5b and Supplementary Table 3).
Conclusion
Through comprehensive molecular and integrative profiling, we identified novel genomic and proteomic characteristics that subclassify cervical cancers. Integrated clustering identified keratin-low squamous, keratin-high squamous, and adenocarcinoma-rich clusters defined by different HPV and molecular features (Extended Data Fig. 8). ERBB3, CASP8, HLA-A, SHKBP1 and TGFBR2 were identified as SMGs for the first time in cervical cancer, with ERBB3 (HER3) immediately applicable as a therapeutic target. For the first time in cancer, we report amplifications and fusion events involving the BCAR4 gene, which can be targeted indirectly by lapatinib. Further, we identified amplifications in CD274 and PDCD1LG2, two genes that encode well-known immunotherapy targets. A set of endometrial-like cervical cancers comprised predominantly of HPV-negative tumours and characterized by mutations in KRAS, ARID1A and PTEN was discovered, with PTEN and potentially ARID1A proteins serving as therapeutic targets. Importantly, over 70% of cervical cancers exhibited genomic alterations in either one or both of the PI3K–MAPK and TGFβ signalling pathways (Extended Data Fig. 9e), illustrating the potential clinical significance of therapeutic agents targeting members of these pathways. For the first time, we report distinct molecular pathways activated in cervical carcinomas caused by different HPV types, highlighting the biological diversity of HPV effects.
Together, these findings provide insight into the molecular subtypes of cervical cancers and rationales for developing clinical trials to treat populations of cervical cancer patients with distinct therapies.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Samples and data freeze
Each tissue source site provided documentation that their IRBs either: a) approved their participation specifically in the TCGA project, through an approved protocol, amendment, exemption, or waiver, and the documentation must include specific mention of TCGA; or b) provided documentation that the IRB does not consider participation in TCGA to constitute ‘human subjects research’, and therefore does not have purview.
The Core Data Freeze (core set) included 178 samples from cervical carcinoma batches 88, 114, 127, 148, 169, 179, 200, 217, 236, 256, 280, 297, 335 and 350 (Supplementary Table 1). This is a standard data freeze whereby the case list was ‘frozen’ and analyses used the one set even though other samples came through the pipeline. Samples in the core set had mRNA-seq, whole exome DNA-seq (WES), miRNA-seq, methylation, SNP6 copy number and clinical data available. Additional samples that had multicentre mutation calls and/or RPPA data included 67 samples from cervical carcinoma batches 88, 114, 127, 148, 169, 179, 200, 217, 236, 256, 280, 297, 335, 350, 361, 373, 380, 394 and 420 (Supplementary Table 2). Of these samples, 14 had mutations called and 60 had RPPA data available; however, RPPA data for 17 samples was excluded owing to low protein content within the samples (Supplementary Table 2). Mutations were called for 192 samples (extended set), while all other platform and integrated analyses (aside from protein) were performed on the subset of 178 core-set samples. Protein levels were measured on 155 samples, which included 119 samples from both the core and extended sets as well as 36 samples outside of these sets. The total number of non-overlapping samples across core, extended and RPPA datasets is 228 (Extended Data Fig. 1a).
HPV detection, variant calling and transcript analysis
HPV status was determined using consensus results from MassArray and RNA-seq (Supplementary Information 2). MassArray uses real-time competitive polymerase chain reaction and matrix-assisted laser desorption/ionization–time-of-flight mass spectroscopy with separation of products on a matrix-loaded silicon chip array, similar to the work described in ref. 45. Two approaches for pathogen detection from RNA-seq data were used. The first used the microbial detection pipeline at the British Columbia Cancer Agency’s Genome Sciences Centre (BC), which is based on BioBloom Tools (BBT, v1.2.4b1)46. The second used the PathSeq algorithm47 at the Broad Institute (BI) to perform computational subtraction of human reads followed by alignment of residual reads to a combined database of human reference genomes and microbial reference genomes including HPV. In 97% of samples, complete agreement between MassArray and both RNA-seq approaches was observed. The remaining discrepant samples were resolved by majority decision, assigning the genotype called by at least two of the methods. RNA-seq data in FASTA format was used to identify HPV variants (Supplementary Fig. 1). Unaligned reads were taken from the PathSeq analysis and aligned to HPV reference genomes using TopHat48 with default parameters49. The HPV variant lineages/sublineages were assigned based on the phylogenetic topology and confirmed visually using the SNP patterns50. HPV splice junctions from RNA-seq were determined using TopHat. Two transcript types were distinguished for HPV16 and HPV18: transcripts that included evidence of an unspliced sequence of E6, and transcripts spliced at the E6 splice donor site (position 226 for HPV16 and position 233 for HPV18) (Supplementary Fig. 2). Read counts for unspliced, spliced, as well as the ratio of unspliced/spliced transcripts were categorized into quartiles separately for HPV16 and HPV18.
HPV integration analysis
Using RNA-seq data, concordance of integration events based on alignments of contigs from de novo transcriptome assembly (BC) and read alignments (BI) was evaluated (Supplementary Fig. 3). We identified method-specific integration events by assigning all sites within a 500-kb sliding window to a single integration event located at the median coordinate of that assigned sites for that event. An integration event was labelled as ‘confident’ when the total read support for each of its supporting integration sites passed centre-specific read evidence thresholds. To take advantage of differences between the two integration methods (that is, contig and read), for the concordance analysis we used all method-specific integration events (both confident and non-confident events). We labelled an integration event as ‘concordant’ when both methods reported an integration event within 500 kb in the same patient’s sample. For some concordant events, both methods reported a confident event. An integration event was labelled as ‘discordant’ when only one centre reported a confident integration event within 500 kb (Supplementary Figs 4 and 5). For both intragenic and intergenic concordant events, we reported a range of coordinates that extends from the most proximal to the most distal supported integration site. We assessed gene-level expression relative to somatic copy number and structural-variant data for genes into which we had mapped viral–human junctions from RNA sequencing data and for genes that were associated with enhancers into which we had mapped RNA junctions.
DNA sequencing and mutation calling
Detailed methods for library hybrid capture, read alignments and somatic variant calling are documented in Supplementary Information 3. MutSig2CV6 was used to identify significantly mutated genes (SMGs) within the cervical cancer exome sequencing data. Mutations were analysed for the core set plus 14 samples for a total of 192 extended-set samples. Eleven samples were identified to exhibit greater than average mutations rates and were termed hypermutants (somatic mutations > 600). These 11 samples were excluded from the analysis for identifying SMGs. All three sample subsets (all samples, squamous carcinomas only, adenocarcinomas only) without hypermutants (Supplementary Table 4) were analysed using an FDR cut-off of 0.1. FDR values are shown in Supplementary Table 4. SMG analysis using the entire sample cohort in from ref. 8 was performed as described previously8.
Copy number analysis
DNA from each tumour or germline sample was hybridized to Affymetrix SNP 6.0 arrays using protocols at the Genome Analysis Platform of the Broad Institute as previously described51. Briefly, Birdseed was used to infer a preliminary copy number at each probe locus from raw .cel files52. For each tumour, genome-wide copy number estimates were refined using tangent normalization, in which tumour signal intensities are divided by signal intensities from the linear combination of all normal samples that are most similar to the tumour16. Individual copy number estimates then underwent segmentation using circular binary segmentation53, and segmented copy number profiles for tumour and matched control DNAs were analysed using Ziggurat Deconstruction54. Significance of copy number alterations were assessed from the segmented data using GISTIC2.0 (version 2.0.22)54. For the purpose of this analysis, an arm-level event was defined as any event spanning more than 50% of a chromosome arm. For copy number-based clustering, tumours were clustered based on copy number at regions using GISTIC analysis. Clustering was done in R on the basis of Euclidean distance using Ward’s method. Allelic and integer copy number, tumour purity and tumour ploidy were calculated using the ABSOLUTE algorithm55.
Detecting structural variants from RNA-seq and WGS data
Integrative analysis was performed to identify putative driver fusions using both WGS (low-pass and high-coverage) and RNA-seq data. RNA-seq data for 178 core-set samples were analysed using the TopHat-Fusion and BreakFusion, PRADA and MapSplice algorithms. To identify structural variations in WGS data, 50 low-pass WGS and 19 high-pass WGS samples were analysed. Detection of structural variations in low-pass WGS data was performed using two algorithms, BreakDancer56 and Meerkat57, with a requirement for at least two discordant read pairs supporting each event and at least one read covering the breakpoint junction. High-pass WGS data were analysed to detect somatic structural variations using two runs of BreakDancer and one run of SquareDancer (https://github.com/ding-lab/squaredancer). The gene fusion lists generated by all methods and platforms were integrated (see Supplementary Tables 8–10).
APOBEC mutagenesis analysis
Analysis is based on previous findings that APOBECs deaminate cytidines predominantly in a tCw motif and that the APOBEC mutagenesis signature is composed of approximately equal numbers of two kinds of changes in this motif: tCw→tTw and tCw→tGw mutations (flanking nucleotides are shown in small letters; w = A or T). Using mutation data from all 192 extended-set samples, we calculated on a per-sample basis the enrichment of the APOBEC mutation signature among all mutated cytosines in comparison to the fraction of cytosines that occur in the tCw motif among the ±20 nucleotides surrounding each mutated cytosine (APOBEC_enrich column in data files). The minimum estimate of the number of APOBEC-induced mutations in a sample (APOBEC_MutLoad_MinEstimate) was calculated using the formula: [tCw→G + tCw→T]×[(APOBEC_enrich−1)/APOBEC_enrich], which allows estimation of the number of APOBEC signature mutations in excess of what would be expected by random mutagenesis. APOBEC_MutLoad_MinEstimate was calculated only for samples passing the threshold of FDR < 0.05 for APOBEC enrichment ([BH_Fisher_p-value_tCw] < 0.05). Samples with a BH_Fisher_p-value_tCw > 0.05 were given a value of 0. The APOBEC_MutLoad_MinEstimate value shows high correlation (0.9–0.95) with all other parameters used to characterize the APOBEC mutagenesis pattern, such as APOBEC enrichment, and absolute and relative APOBEC mutation loads. For some analyses and figures, the APOBEC_MutLoad_MinEstimate parameter was converted into categorical values as follows: no, APOBEC_MutLoad_MinEstimate = 0; low, 0 < APOBEC_MutLoad_MinEstimate > median of non-zero values; high, APOBEC_MutLoad_MinEstimate > median of non-zero values. The median of non-zero values in the extended set = 33.
Methylation analysis
The Illumina Infinium HM450 array58 was used to evaluate DNA methylation in the core set of samples from cervical cancer patients. Unsupervised consensus clustering was performed with Euclidean distance and partitioning around medoids (PAM) using the most variable 1% of CpG-island promoter probes. Epigenetically silenced genes were identified as previously described59. A total of 120 normal samples were used for this analysis by selecting 10 samples at random from the 12 TCGA projects that included normal samples.
RNA-seq analysis
RNA was extracted, converted into mRNA libraries, and paired-end sequenced (paired 50 nucleotide reads) on Illumina HiSeq 2000 Genome Analyzers as previously described5. RNA reads were aligned to the hg19 genome assembly using Mapsplice version 12_0760. Gene expression was quantified for the transcript models corresponding to the TCGA GAF2.1 (https://gdc-api.nci.nih.gov/v0/data/a0bb9765-3f03-485b-839d-7dce4a9bcfeb) using RSEM4 (ref. 61) and normalized within a sample to a fixed upper quartile. To predict whether a cancer sample was from the cervix or the uterus, the data matrix of normalized gene-level RSEM values from 170 UCEC samples was merged with the data matrix from the core set (n = 178) of cervical cancers. This merged dataset was then randomly split into a training set (87 cervical carcinoma samples; 86 UCEC samples) and a test set (91 cervical carcinoma samples; 84 UCEC samples). A sample was predicted to be cervical carcinoma if the t-statistic versus UCEC was significant (P < 0.05), but was not significantly different from the cervical carcinoma mean (and vice versa for the UCEC prediction). A data matrix of RSEM values from 178 cervical carcinoma, 170 UCEC and 279 HNSC samples was used to identify expression patterns across the 3 cancer types. The gene expression matrix was further filtered to only include the top 25% most variable genes by mean absolute deviation (n = 4,039 genes).
EMT mRNA score analysis
The EMT score was computed as previously described10,21. Briefly, the EMT score was the value resulting from the difference between the average expression of mesenchymal (M) genes minus the average expression of epithelial (E) genes. All values for unavailable data (NA) were removed from the calculation. Two-sample t-test and ANOVA were applied to each comparison accordingly.
miRNA sequencing and analysis
MicroRNA-sequencing (miRNA-seq) data was generated for the core set of tumour samples using methods described previously11. We identified miRNAs that have been associated with EMT62,63,64,65,66 and then calculated Spearman correlations between the EMT scores and normalized expression (reads per million, RPM) for 5p and 3p mature strands for each of the miRNAs using MatrixEQTL and filtering by FDR < 0.05. An miRNA was considered to be epigenetically controlled if the BH-corrected P values were less than 0.01 for both (i) a Spearman correlation of miRNA abundance (RPM) to beta for probes in promoter regions associated with the miRNAs, and for (ii) a t-test of RPM between unmethylated (β < 0.1) and methylated (β > 0.3) samples (an epigenetically controlled pattern). We assessed potential miRNA targeting for all 178 samples and then separately for the 144 squamous samples by calculating miRNA–mRNA and miRNA–protein (RPPA) Spearman correlations with MatrixEQTL v2.1.1 using gene-level normalized abundance RNA-seq (RSEM) data and normalized RPPA data. Correlations were calculated with a P value threshold of 0.05, and then the anti-correlations were filtered at FDR < 0.05. We extracted miRNA–gene pairs that were functionally validated in publications reported by miRTarBase v4.5 (ref. 22). For miRNA–RPPA anti-correlations, all gene names that were associated with each antibody were used. Results were displayed with Cytoscape v2.8.3.
PARADIGM analysis
Integration of copy number, RNA-seq and pathway interaction data was performed on the core set of samples using PARADIGM33,34. Briefly, PARADIGM infers integrated pathway levels (IPLs) for genes, complexes and processes using pathway interactions, genomic and functional genomic data from each patient sample. One was added to all expression values, which were then log2-transformed and median-centred across samples for each gene. The log2-transformed, median-centred mRNA data were rank-transformed based on the global ranking across all samples and all genes and discretized (+1 for values with ranks in the highest tertile, −1 for values with ranks in the lowest tertile and 0 otherwise) before PARADIGM analysis.
Pathways were obtained in BioPax level 3 format, and included the NCIPID and BioCarta databases from http://pid.nci.nih.gov and the Reactome database from http://reactome.org. Gene identifiers were unified by UniProt ID and then converted to Human Genome Nomenclature Committee’s HUGO symbols using mappings provided by HGNC (http://www.genenames.org/). Altogether, 1,524 pathways were obtained. Interactions from all of these sources were then combined into a merged superimposed pathway (SuperPathway). Genes, complexes and abstract processes (for example, cell cycle and apoptosis) were retained and henceforth referred to collectively as pathway features. The resulting pathway structure contained a total of 19,504 features, representing 7,369 protein-coding genes, 9,354 complexes, 2,092 families, 82 RNAs, 15 miRNAs and 592 abstract processes.
The PARADIGM algorithm infers an IPL for each pathway element that reflects the log likelihood that contrasts the probability of activity against inactivity. An initial minimum variation filter (at least 1 sample with absolute activity >0.05) was applied, resulting in 15,502 concepts (5,898 protein-coding genes, 7,307 complexes, 1,916 families, 12 RNAs, 15 miRNAs and 354 abstract processes) with relative activities showing distinguishable variation across tumours.
iCluster analysis
Integrative clustering of RNA-seq, methylation, copy number and miRNA data was performed using the R package iCluster20. The core set of samples was used since all samples in this set had data available across these four platforms. RNA-seq, methylation, copy number and mature-strand miRNA datasets had 20,531, 395,552, 23,109 and 1,213 features, respectively. The 500 most variable features based on the standard deviation from each dataset were selected for the integrative clustering analyses. For analysis involving the RNA-seq and miRNA datasets, a log[x + 1] transformation was used in order to deal with skewness in the data67. Methylation data was logit transformed to make it closer to normal distribution. The CNV data included the regions determined from GISTIC2.0, with CNVs treated as a continuous measurement based on the segmentation mean value for the region.
MEMo analysis
High DNA-methylation levels upstream of miR-200a and miR-200b corresponded to transcriptional downregulation of the miRNAs (Extended Data Fig. 9a). For a sample to be called altered for either miR-200a or miR-200b (or both), we required both high DNA-methylation level upstream of the miRNA (β > 0.3) and low miRNA expression (log2[RPM] < 9.3 for miR-200a and log2[RPM] < 9 for miR-200b). Binary calls were given to altered and unaltered samples based on this double threshold (1 = altered, 0 = unaltered).
The mutual exclusivity modules in cancer (MEMo) algorithm27 was run on all core-set samples. MEMo was first run on 27 regions of recurrent copy number gain, 36 regions of copy number loss and 22 recurrently mutated genes. In order to include alterations for miR-200a and miR-200b in the MEMo analysis, a custom network was designed where each miRNA was connected to its known and validated targets (see above). Second, this network was merged with the comprehensive pathway network used by MEMo to search for modules of altered genes that include at least one of the miRNAs. Extracted modules were tested for mutual exclusivity using statistical framework of MEMo (Supplementary Table 27). A Student’s t-test was performed for comparison of the EMT mRNA scores between groups.
Data availability
The primary and processed data used in analyses can be downloaded by registered users from https://gdc-portal.nci.nih.gov/ and the TCGA publication page (https://tcga-data.nci.nih.gov/docs/publications/cesc_2016/).
Change history
16 March 2017
The Creative Commons licence information was added to the HTML and PDF.
References
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386 (2015)
Schiffman, M. et al. Human papillomavirus testing in the prevention of cervical cancer. J. Natl. Cancer Inst. 103, 368–383 (2011)
Uyar, D. & Rader, J. Genomics of cervical cancer and the role of human papillomavirus pathobiology. Clin. Chem. 60, 144–146 (2014)
Moody, C. A. & Laimins, L. A. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10, 550–560 (2010)
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012)
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013)
Chung, T. K. H. et al. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. Int. J. Cancer 137, 776–783 (2015)
Ojesina, A. I. et al. Landscape of genomic alterations in cervical carcinomas. Nature 506, 371–375 (2014)
Cancer Genome Atlas Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322 (2014)
Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015)
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012)
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013)
Burns, M. B., Temiz, N. A. & Harris, R. S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 (2013)
Henderson, S., Chakravarthy, A., Su, X., Boshoff, C. & Fenton, T. R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Reports 7, 1833–1841 (2014)
Roberts, S. A. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976 (2013)
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011)
The Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013)
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015)
Godinho, M. F. E. et al. BCAR4 induces antioestrogen resistance but sensitises breast cancer to lapatinib. Br. J. Cancer 107, 947–955 (2012)
Shen, R., Olshen, A. B. & Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 25, 2906–2912 (2009)
Byers, L. A. et al. An epithelial–mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 19, 279–290 (2013)
Hsu, S.-D. et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 42, D78–D85 (2014)
Akbani, R. et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 5, 3887 (2014)
Seton-Rogers, S. Oncogenes: all eyes on YAP1. Nat. Rev. Cancer 14, 514–515 (2014)
Shao, D. D. et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184 (2014)
Vandewalle, C., Van Roy, F. & Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 66, 773–787 (2009)
Ciriello, G., Cerami, E., Sander, C. & Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 22, 398–406 (2012)
Gregory, P. A. et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601 (2008)
Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012)
Haslehurst, A. M. et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 12, 91 (2012)
Taube, J. H. et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl Acad. Sci. USA 107, 15449–15454 (2010)
Jaiswal, B. S. et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617 (2013)
Sedgewick, A. J., Benz, S. C., Rabizadeh, S., Soon-Shiong, P. & Vaske, C. J. Learning subgroup-specific regulatory interactions and regulator independence with PARADIGM. Bioinformatics 29, i62–i70 (2013)
Vaske, C. J. et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26, i237–i245 (2010)
Hoadley, K. A. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014)
den Boon, J. A. et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: role of stromal estrogen receptor signaling. Proc. Natl Acad. Sci. USA 112, E3255–E3264 (2015)
Roman, A. & Munger, K. The papillomavirus E7 proteins. Virology 445, 138–168 (2013)
Vande Pol, S. B. & Klingelhutz, A. J. Papillomavirus E6 oncoproteins. Virology 445, 115–137 (2013)
Jiao, Y., Widschwendter, M. & Teschendorff, A. E. A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 30, 2360–2366 (2014)
Dellambra, E. et al. Downregulation of 14-3-3σ prevents clonal evolution and leads to immortalization of primary human keratinocytes. J. Cell Biol. 149, 1117–1130 (2000)
Moreira, J. M. A., Gromov, P. & Celis, J. E. Expression of the tumor suppressor protein 14-3-3σ is down-regulated in invasive transitional cell carcinomas of the urinary bladder undergoing epithelial-to-mesenchymal transition. Mol. Cell. Proteomics 3, 410–419 (2004)
Hermeking, H. et al. 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1, 3–11 (1997)
Chang, T.-C. et al. 14-3-3σ regulates β-catenin-mediated mouse embryonic stem cell proliferation by sequestering GSK-3β. PLoS One 7, e40193 (2012)
Wentzensen, N., Vinokurova, S. & von Knebel Doeberitz, M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 64, 3878–3884 (2004)
Tang, A. L. et al. UM-SCC-104: a new human papillomavirus-16-positive cancer stem cell-containing head and neck squamous cell carcinoma cell line. Head Neck 34, 1480–1491 (2012)
Chu, J. et al. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics 30, 3402–3404 (2014)
Kostic, A. D. et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 29, 393–396 (2011)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25, 1105–1111 (2009)
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009)
Schiffman, M. et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 70, 3159–3169 (2010)
McCarroll, S. A. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 40, 1166–1174 (2008)
Korn, J. M. et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 40, 1253–1260 (2008)
Olshen, A. B., Venkatraman, E. S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004)
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011)
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012)
Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009)
Yang, L. et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153, 919–929 (2013)
Bibikova, M. et al. High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295 (2011)
Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 (2014)
Wang, K. et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178 (2010)
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011)
Carstens, J. L., Lovisa, S. & Kalluri, R. Microenvironment-dependent cues trigger miRNA-regulated feedback loop to facilitate the EMT/MET switch. J. Clin. Invest. 124, 1458–1460 (2014)
Ceppi, P. & Peter, M. E. MicroRNAs regulate both epithelial-to-mesenchymal transition and cancer stem cells. Oncogene 33, 269–278 (2014)
Díaz-Martín, J. et al. A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. J. Pathol. 232, 319–329 (2014)
Kiesslich, T., Pichler, M. & Neureiter, D. Epigenetic control of epithelial–mesenchymal-transition in human cancer. Mol. Clin. Oncol. 1, 3–11 (2013)
Tam, W. L. & Weinberg, R. A. The epigenetics of epithelial–mesenchymal plasticity in cancer. Nat. Med. 19, 1438–1449 (2013)
Zwiener, I., Frisch, B. & Binder, H. Transforming RNA-seq data to improve the performance of prognostic gene signatures. PLoS One 9, e85150 (2014)
Acknowledgements
We would like to acknowledge the late H. Salvesen (the University of Bergen), who provided critical clinical and translational insight, and we dedicate this manuscript to her memory. We also acknowledge L. Gaffney (The Broad Institute) for her work in preparing some of the figures. In addition, this study was supported by National Institutes of Health (NIH) grants U54 HG003273, U54 HG003067, U54 HG003079, U24 CA143799, U24 CA143835, U24 CA143840, U24 CA143843, U24 CA143845, U24 CA143848, U24 CA143858, U24 CA143866, U24 CA143867, U24 CA143882, U24 CA143883, U24 CA144025 and P30 CA016672.
Author information
Authors and Affiliations
Consortia
Contributions
The Cancer Genome Atlas research network contributed collectively to this work. Biospecimens were collected at the tissue source sites (TSSs) and processed by the biospecimen core resource (BCR). Data was generated by the genome sequencing and genome data analysis centres, with analyses performed by members across the network. Data were stored and released through the data coordinating centre (DCC). The NCI project coordinator was I. Felau and the overall analysis coordinator and data coordinator was C. P. Vellano. Special thanks also go out to TCGA network members who made substantial contributions to this work: C. P. Vellano (analysis coordinator, data coordinator, co-manuscript coordinator, RPPA analysis), N. Wentzensen (co-manuscript coordinator, HPV-analysis subgroup co-leader), A. I. Ojesina (co-manuscript coordinator, HPV-analysis subgroup co-leader, somatic-alteration analysis), A. G. Robertson (miRNA analysis, HPV analysis), M. D. McLellan (mutation calling), L. Danilova (methylation analysis), B. A. Murray (copy number and ABSOLUTE analysis), Z. Ju (RPPA analysis), J. T. Auman (mRNA-sequencing analysis, fusion analysis), P. Chalise (iCluster analysis), C. Yau (PARADIGM pathway analysis), G. Ciriello (MEMo pathway analysis), D. A. Gordenin (APOBEC analysis), R. Zuna (pathologist), H. Zhang (mutation analysis, Firehose), A. Pantazi (structural-variant-analysis subgroup leader, low-pass sequencing), M. H. Bailey (mutation analysis), L. Diao (EMT analysis), D. Koestler (methylation data processing, functional epigenetic module analysis), K. Mungall (HPV analysis), L. Lim (HPV analysis), R. Bowlby (miRNA analysis), S. Sadeghi (HPV analysis), D. Brooks (miRNA analysis), C. Sekhar Pedamallu (HPV analysis), K. Chen (fusion analysis), H. Zhao (fusion analysis), Z. Chong (fusion analysis), E. Martinez-Ledesma (fusion analysis), R. G. Verhaak (fusion analysis), K. M. Leraas (BCR), T. M. Lichtenberg (BCR), D. G. Tiezzi (immune-response gene analysis), M. C. Ryan (splicing analysis), S. M. Reynolds (regulome explorer analysis), G. B. Mills (project co-chair) and J. S. Rader (project co-chair).
Corresponding authors
Ethics declarations
Competing interests
The author declare no competing financial interests.
Additional information
The Cancer Genome Atlas Research Network (participants are arranged by institution)
Extended data figures and tables
Extended Data Figure 1 Sample sets and histological patterns of cervical cancer.
a, Summary of sample numbers and degree of overlap between the core, extended and RPPA datasets. b, Example of a large-cell non-keratinizing squamous cell carcinoma. Tongues of highly atypical polygonal neoplastic squamous cells infiltrate through a fibrotic stroma. The cells show abundant eosinophilic cytoplasm with pleomorphic nuclei and prominent mitotic figures. Although the tumour cells contain abundant cytokeratin filaments, this tumour has traditionally been termed non-keratinizing because of the absence of characteristic keratin pearls. c, An example of a large-cell keratinizing squamous cell carcinoma. Nests of atypical squamous cells infiltrate through a fibrotic stroma. In addition, this tumour shows highly eosinophilic keratin pearls with small, inky dark nuclei that imperfectly mimic the normal keratinization that is found in the epidermis. This differentiation pattern is aberrant in the cervix in which the squamous epithelium is normally a non-keratinizing squamous mucosa. d, An example of an endocervical adenocarcinoma (well differentiated). Closely set, atypical glands with enlarged nuclei and scattered mitotic figures infiltrate through the connective tissue of the cervix. The tall columnar tumour cells show basally placed, crowded, enlarged nuclei that show frequent mitotic figures. Compared with normal endocervical cells, the tumour cells show relative loss of intra-cytoplasmic mucin and are frequently called mucin-depleted, although most, but not all endocervical adenocarcinomas show varying amounts of intracytoplasmic mucin at least focally. e, Adenosquamous carcinoma of cervix. This tumour shows both nests of non-keratinizing squamous cell carcinoma and glands composed of tall columnar adenocarcinoma reflecting the origin of most cervical cancers in the transformation zone of the cervix in which both squamous and glandular cells normally differentiate. Despite this biphasic differentiation potential, adenosquamous carcinomas are relatively uncommon in the cervix. f, UCEC-like HPV-negative endocervical adenocarcinoma from a radical hysterectomy specimen. The endometrium in the uterus was benign. g, UCEC-like HPV-positive endocervical adenocarcinoma from a radical hysterectomy specimen. The endometrium in the uterus was benign. All samples were stained with haematoxylin and eosin (20×). Scale bar, 100 μm.
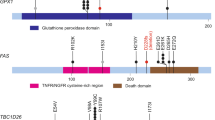
Extended Data Figure 2 SMGs and the role of APOBEC in cervical cancer mutagenesis.
a–f, High-confidence somatic mutations in SMGs among 192 exome-sequenced samples in the extended case set are shown. Domains are labelled according to Gencode 19, corresponding to Ensembl 74. Mutations at canonical intronic splice acceptor (e−1 and e−2) are labelled based on proximity to the nearest coding exon. Panels display somatic mutations detected in novel cervical cancer SMGs, with HLA-B included for comparison with its family member HLA-A. Each axis is the protein-coding portion of a gene and each highlighted section represents the UniProt functional domain. Vertical lines indicate the boundaries of multiple annotation sources within common domain annotations as outlined in Supplementary Table 5. Horizontal lines distinguish overlapping domains. Circles represent a single mutation and are coloured based on mutation type. Mutations present in squamous cell carcinomas are black, whereas those present in adenocarcinomas are pink. g, PIK3CA mutations and recurrence are shown in a stacked circle plot, as above. Additionally, lolliplot sticks are coloured red if the mutation type coincides with patterns of APOBEC mutagenesis. h, The minimal estimated number of APOBEC-induced mutations (APOBEC_MutLoad_MinEstimate column in Supplementary Table 1) strongly correlates with total number of mutations in a sample, as well as with the number of single-nucleotide variants (SNVs) in G:C pairs that are the exclusive substrate for mutagenesis by APOBEC cytidine deaminases. Although correlation with mutagenesis in A:T base pairs, which cannot be mutated by APOBEC enzymes, is statistically significant (two-tailed P = 0.047), it is very weak. Pearson correlation and R2 were calculated for all 192 exome-sequenced samples, including samples with zero values. Only samples with non-zero values of APOBEC_MutLoad_MinEstimate are presented.
Extended Data Figure 3 Copy number alterations in cervical cancer.
a, A log2-centred heatmap of somatic copy number alterations across 178 core-set cervical tumours. The x axis includes samples that have been ordered based on the cluster assignment. The y axis is based on genomic position, from 1p to Xq. Features associated with copy number clusters are annotated with asterisks; *P < 0.05; **P < 0.01. b, GISTIC2.0 amplification and deletion plots within copy number clusters. Chromosomal locations for peaks of significantly recurrent focal amplifications (red) and deletions (blue) are plotted by −log10 q value for the high (CN High) and low (CN Low) copy number clusters. Peaks are annotated with cytoband and candidate driver genes. The total number of genes in the peak region is indicated in parentheses. Peaks with more than 30 genes in the peak region are excluded. Any genes annotated have a significant positive correlation with mRNA expression. c, Chromosomal locations for peaks of significantly recurrent focal amplifications (red) and deletions (blue) are plotted by −log10 q value for all core set samples. Peaks are annotated with cytoband and candidate driver genes. The total number of genes in the peak region is indicated in parentheses. Peaks consisting of more than 30 genes in the peak region are excluded. Annotated genes have a significant positive correlation with mRNA expression. d, Cytolytic activity (CYT) associations with PD-L1 and/or PD-L2 amplification. Each bar represents a single tumour and the height of that bar represents the z score of the cytolytic activity of that tumour compared to the rest of the cohort. Bars are coloured according to their PD-L1 and/or PD-L2 amplification status and sorted from the highest to the lowest z score.
Extended Data Figure 4 Gene-expression patterns and fusion genes found in cervical cancer.
a, Hierarchical clustering (uncentred correlation with centroid linkage as the clustering method) was performed on 4,039 expressed and highly variable genes across samples from 178 cervical, 170 endometrial and 279 head and neck cancer patients. Normalized gene-level RSEM values were median-centred before clustering and relative increased expression values are indicated in red and relative decreased expression values are indicated in blue. Samples from patients with cervical (CESC, light blue), endometrial (UCEC, purple) and head and neck (HNSC, orange) cancer are categorized by different colours as indicated. Also included are indications of HPV status, histology of cervical and endometrial cancers, and tissue site for head and neck cancer samples. Select genes are noted to the right of their locations on the heatmap. b, Box plots of the three differentially expressed SMGs and top six significantly differentially expressed non-SMGs across the iCluster groups using Kruskal–Wallis test. All genes are significantly different between the keratin-low and keratin-high clusters. Significant P values across keratin-low and keratin-high clusters are presented. c, A schematic of BCAR4 tandem duplication in one case (C5-A3HF), detected by analysis of somatic copy number (top) and structural variation (middle). Split reads and genomic breakpoints indicating the tandem duplication are shown. At the RNA level (bottom) the last exon of BCAR4 forms a fusion gene with the first exon of ZC3H7A (red bars indicate the location of mRNA breakpoints; NR_024049 shown as BCAR4 representative transcript). d, Schematic of recurrent fusions (CPSF6–C9orf3, ARL8B–ITPR1 and MYH9–TXN2) or fusions with known occurrences in other cancer types (FGFR3–TACC3), detected by at least two RNA-seq fusion callers in 178 samples. Red bars indicate the mRNA breakpoints.
Extended Data Figure 5 Unsupervised clusters of DNA methylation data.
a, Heatmap showing β values of 178 core-set samples ordered by CIMP clusters. Samples are presented in columns and the CpG island promoter CpG loci are presented in rows. An annotation panel on the right of the heatmap indicates CpG loci that are differentially methylated within a particular feature (see Supplementary Table 13). All features (marked with an asterisk) are significantly associated with DNA-methylation clusters (Fisher’s Exact test P < 0.01), except APOBEC mutagenesis level, UCEC-like status and HPV integration status. b, Box plots of the EMT mRNA score and tumour purity by CIMP clusters. *P < 0.05; **P < 0.01 (Student’s t-test).
Extended Data Figure 6 miRNA clusters and miRNA-gene/protein anti-correlations in cervical cancer.
a, Unsupervised clustering for miRNA profiles across 178 core-set tumour samples. Top to bottom: a normalized abundance heatmap for the fifty 5p or 3p strands that were highly ranked as differentially abundant by a SAMseq multiclass analysis; a silhouette width profile calculated from the consensus membership matrix; covariates with associated P values; and a summary table of the number of samples in each cluster. The scale bar shows row-scaled log10[RPM + 1] normalized abundances. Blue triangles mark selected cancer-associated miRs that were both differentially abundant across the subtypes and abundant in at least one subtype. b, Subnetworks of potential targeting relationships for a subset of miRNAs, as significance-thresholded (FDR < 0.05) miRNA–mRNA and miRNA–protein anti-correlations that are supported by functionally validated publications. For genes (nodes), colour distinguishes those that are only present in mRNA data (grey) from those that are present in both mRNA and RPPA data (green). Edges represent anti-correlations, and colour distinguishes anti-correlations between miRNA and mRNA (blue); and miRNA and unphosphorylated protein (green). In the n = 178 core-set cohort, no correlations satisfying FDR < 0.05 were reported between miRNA and phosphorylated protein.
Extended Data Figure 7 EMT-associated miRNAs and their relationship to miRNA clusters and TGFβR2 somatic alterations.
a, Normalized miR-200a-3p abundance (RPM) across RPPA clusters for all 112 (top) and 92 squamous (bottom) samples of the core set for which RPPA data are available. P values presented are from two-sided Kolmogorov–Smirnov tests for RPPA-based EMT cluster versus non-EMT cluster samples. For n = 112 samples, median miR-200a-3p RPM = 296.4 within the EMT cluster (n = 29) and 410.0 (n = 83) in non-EMT cluster samples. For squamous samples, median miR-200a-3p RPM = 296.4 (n = 29) within the EMT cluster and 393.4 (n = 63) in non-EMT cluster samples. EK-A2R7, which is in the hormone RPPA cluster, has an RPM value of 4,267 and is not shown. Results are not presented for adenocarcinoma samples separately owing to limited sample numbers (n = 18 from the core set with RPPA data available). b, Negative and positive Spearman correlation coefficients (FDR < 0.05) between EMT mRNA score and normalized abundance (RPM) for miRNA mature strands (n = 178). miRNAs that have been reported as associated with EMT (see Methods) are highlighted by blue bars. c, Normalized abundance heatmap of miRNAs most strongly negatively and positively correlated with EMT mRNA scores, with samples grouped by miRNA cluster and sorted by EMT score within each cluster. Somatic mutations (MUT) and deletions (HOMDEL) are shown for TGFBR2, CREBBP, EP300 and SMAD4. Methylation and concomitant downregulated expression alterations (ALT) as defined in Methods for miR-200a/b are also shown. miRNAs in blue represent those highlighted by blue bars in b. d, e, Same as b, c, for the n = 144 squamous tumour samples.
Extended Data Figure 8 Distinguishing features of cervical cancer integrated molecular subtypes.
a, Integrative clustering of 178 core-set samples from patients with cervical cancer using mRNA, methylation, miRNA and copy number data identified three iCluster groups: keratin-low, keratin-high and adenocarcinoma-rich (adenocarcinoma). Relative frequencies of various cervical cancer classifications defined by histology, HPV clade, CNVs, methylation, miRNA and RPPA are plotted. The key for each feature is shown at the bottom. For each category, the statistically significantly enriched features in each cluster are highlighted with asterisks and include the name of the enriched feature. Each category was significantly associated with the clusters (χ2 test; P < 0.05). The width of each plot is scaled according to the number of samples within each cluster. b, The frequencies of somatic alterations and additional novel features that distinguish the clusters, specifically those that do not occur in all three clusters, are plotted. The ‘somatic mutations’ panel shows the presence or absence of mutations for 7 of the identified SMGs. The ‘copy number alterations’ panel shows select copy number alterations (high-level amplifications and focal deletions) that are differentially present across the iCluster groups. The ‘additional features’ panel highlights miscellaneous features that also distinguish the clusters, including the presence of miR-200a/b alterations, UCEC-like samples and BCAR4-fusion events. The key for each feature is shown on the right.
Extended Data Figure 9 miR-200a/b associations with EMT-regulating genes and somatic alterations within RTK, PI3K, MAPK and TGFβR2 pathways in cervical cancer.
a, Expression levels for miR-200a and miR-200b compared to DNA-methylation level at their promoter. Samples were called altered if the miRNAs were concurrently hypermethylated (β > 0.3) and downregulated (red). b, mRNA expression levels for ZEB2, a target of both miR-200a and miR-200b, in subsets of miR-200a/b altered samples. ZEB2 is upregulated in samples with concurrent hypermethylation and downregulation of the miRNAs. c, mRNA expression levels of both ZEB1 and ZEB2 in miR-200a/b hypermethylated/downregulated (altered) and all other (WT) samples. d, Correlations of miR-200a and miR-200b expression with multiple genes involved in EMT signalling across squamous cell carcinomas and adenocarcinomas. e, Extent of genetic alterations and miRNA downregulation in the RTK, PI3K, MAPK and TGFβ pathways across all cervical tumours.
Extended Data Figure 10 Pathway biomarkers differentiating squamous cell carcinomas and adenocarcinomas.
a, Cytoscape display of the largest interconnected regulatory network of PARADIGM pathway features that are differentially activated between squamous cell carcinomas and adenocarcinomas connected through hubs with ≥10 downstream targets. Hubs with ≥10 downstream targets are labelled. Genes showing mRNA–miRNA expression anti-correlation with strong supporting evidence are highlighted with a thicker black outline and are labelled. Top differentially expressed genes relating to immune function are also labelled. Node size is proportional to significance of differential activation. b, Zoom-in display of the p63 sub-network neighbourhood. First neighbours (upstream or downstream) of four p63 complexes (bold text) are displayed in this view.
Extended Data Figure 11 HPV integration and molecular characteristics in cervical cancer.
a, E6 unspliced/spliced ratio for HPV16 and HPV18 intragenic, enhancer and intergenic sites. HPV16, median = 0.44 (n = 102); HPV18, median = 0.93 (n = 40). The P value is from a two-sided Kolmogorov–Smirnov test. b, Distribution of RNA-seq-based EMT score for HPV-negative (HPV−) and HPV-positive (HPV+) samples (n = 178). The P value was calculated as in a. c, Distributions of somatic copy number alterations and mRNA abundance ranks (left) and distribution functions for somatic copy number alterations and mRNA abundance ranks with 500 random samples shown close to the diagonals (grey) (right) for genomic loci with integrated HPV16. d, Distributions as in c for genomic loci with integrated HPV18. Benjamini–Hochberg-corrected P values for the somatic copy number alteration and mRNA abundance ranks are medians of the P values from Kolmogorov–Smirnov tests for all random samples.
Supplementary information
Supplementary
This file contains Supplementary Methods, Results and associated references for each core and integrated analysis. It also contains Supplementary Figures 1-48. (PDF 26248 kb)
Supplementary Tables
This zipped file contains Supplementary Tables 1-31 and a Supplementary Table guide. (ZIP 4107 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 543, 378–384 (2017). https://doi.org/10.1038/nature21386
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature21386
This article is cited by
-
Phylogenetic analysis and antigenic epitope prediction for E6 and E7 of Alpha-papillomavirus 9 in Taizhou, China
BMC Genomics (2024)
-
High-risk human papillomavirus genotyping in cervical cancers in Tanzania
Infectious Agents and Cancer (2024)
-
RGS20 promotes non-small cell lung carcinoma proliferation via autophagy activation and inhibition of the PKA-Hippo signaling pathway
Cancer Cell International (2024)
-
HPV16-miRNAs exert oncogenic effects through enhancers in human cervical cancer
Cancer Cell International (2024)
-
LncRNA WWTR1-AS1 upregulates Notch3 through miR-136 to increase cancer cell stemness in cervical squamous cell carcinoma
BMC Women's Health (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
