
Abstract
Elucidation of the evolutionary history and interrelatedness of Plasmodium species that infect humans has been hampered by a lack of genetic information for three human-infective species: P. malariae and two P. ovale species (P. o. curtisi and P. o. wallikeri)1. These species are prevalent across most regions in which malaria is endemic2,3 and are often undetectable by light microscopy4, rendering their study in human populations difficult5. The exact evolutionary relationship of these species to the other human-infective species has been contested6,7. Using a new reference genome for P. malariae and a manually curated draft P. o. curtisi genome, we are now able to accurately place these species within the Plasmodium phylogeny. Sequencing of a P. malariae relative that infects chimpanzees reveals similar signatures of selection in the P. malariae lineage to another Plasmodium lineage shown to be capable of colonization of both human and chimpanzee hosts. Molecular dating suggests that these host adaptations occurred over similar evolutionary timescales. In addition to the core genome that is conserved between species, differences in gene content can be linked to their specific biology. The genome suggests that P. malariae expresses a family of heterodimeric proteins on its surface that have structural similarities to a protein crucial for invasion of red blood cells. The data presented here provide insight into the evolution of the Plasmodium genus as a whole.
Similar content being viewed by others

Main
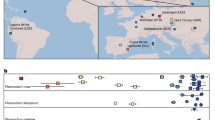
A reference genome of P. malariae was produced from clinically isolated parasites and sequenced using long-read sequencing technology (Table 1; Supplementary Information). The assembly surpasses available draft genome data for P. malariae7, especially in terms of contiguity (63 versus 7,270 scaffolds, N50 = 2.3 Mb versus 6.4 kb) (Supplementary Information; Extended Data Table 1) allowing large-scale structural changes to be accurately determined. Against a background of near-complete collinearity with P. vivax, we found a previously undescribed large reciprocal translocation between chromosomes 6 and 10 and a pericentric inversion of chromosome 5 (Extended Data Fig. 1a, b). Additional draft genomes for both species of P. ovale were assembled from P. falciparum co-infections and the genome of a parasite that we call ‘P. malariae-like’ was assembled from a chimpanzee co-infected with P. reichenowi (Fig. 1; Table 1; Extended Data Table 2; Supplementary Information).

Presence and absence of P. malariae (red), P. ovale (blue) or both (purple) by country on the basis of a literature review (Supplementary Information). Bar plots show proportion of P. falciparum infections with co-infections of P. malariae (red), P. ovale (blue), P. vivax (green), or two species (purple) on the basis of the Pf3K data set (Supplementary Information; Methods). Stars indicate origin of sample used for reference genome assembly and points show additional samples used. Map sourced from Wikipedia Commons (https://commons.wikimedia.org/wiki/File:BlankMap-World6.svg).
To investigate host-specific adaptation of parasites to human and chimpanzee hosts, we compared P. malariae to P. malariae-like. We found lower levels of nucleotide diversity in the human-infective species than in the chimpanzee-infective species (Table 1; Extended Data Fig. 2a). This mirrors the lower levels of nucleotide diversity in the human parasite P. falciparum than in its chimpanzee-infective relative P. reichenowi8. In both cases, the lack of diversity in human-infective species suggests recent population expansions. However, we found that a species that infects New World primates termed P. brasilianum was indistinguishable from P. malariae (Extended Data Fig. 2b), as previously suggested9. Thus host adaptation in the P. malariae lineage appears to be less restricted than in P. falciparum.
Using additional samples to calculate standard measures of molecular evolution (Methods; Supplementary Information), we identified a subset of genes under selection in both P. malariae and P. malariae-like and in an earlier study of P. falciparum and P. reichenowi10 (Extended Data Fig. 3a; Extended Data Table 3), showing some conservation of selection pressures in Plasmodium lineages and suggesting host-specific adaptation of parasites to human and chimpanzee hosts. There is evidence that five genes are under diversifying selection in both lineages (Extended Data Fig. 3b), including one encoding Merozoite surface protein 1 (msp1). Two genes expressed in blood stages are under significant balancing selection in both comparisons (Extended Data Fig. 3c): the genes encoding Apical membrane antigen 1 (ama1) and an uncharacterized conserved protein. Genes under significant selection in both comparisons are enriched for ‘pathogenesis’ and ‘entry into/exit from host cell’ Gene Ontology terms. The increased signatures of selection in the P. malariae lineage reflect a genome-wide increase in fixed nonsynonymous mutations in this species (Extended Data Fig. 3d). One of the genes with the highest ratio of nonsynonymous to synonymous mutations in the P. malariae and P. malariae-like comparison is reticulocyte-binding protein 1a (RBP1a), prompting us to hypothesize that human genes encoding transmembrane proteins that act as potential RBP1a receptors would have to be conserved in red blood cells between humans and New World primates but not with chimpanzees (Supplementary Information; Methods; Extended Data Table 4). As expected from the recent divergence of P. malariae from P. malariae-like, more genes were found with signatures of selection in P. malariae than between the two P. ovale species (Table 1; Extended Data Fig. 3a; Supplementary Information).
Using the added accuracy of manually curated gene sets for both P. malariae and P. o. curtisi (Table 1; Methods), maximum likelihood trees were constructed using 1,000 conserved single-copy core genes that are present in 12 selected Plasmodium species (Fig. 2; Supplementary Information). There is strong evidence that P. malariae forms an out-group both to rodent-infective species and to a primate-infective clade that includes P. vivax. This phylogenetic arrangement is identical to that found using apicoplast data6 but contrasts with other previous studies1,7,11 (Supplementary Information). However, our phylogenetic analysis is based on the most comprehensive amino acid alignment to date, which is enriched for neutrally evolving sites through stringent filtering and has been subjected to a number of different sensitivity tests (Supplementary Information). Assuming consistent mutation rates and generation times across the branches (Supplementary Information), we find that the relative split between the two P. ovale species is about five times earlier than the split between P. falciparum and P. reichenowi, whereas P. malariae and P. malariae-like seem to have split at a similar time to the latter two (Fig. 2).

Maximum likelihood phylogenetic tree of the Plasmodium genus, showing the P. malariae clade (red) and the P. ovale clade (blue) together with the divergence levels of the species as calibrated to the P. falciparum and P. reichenowi split (×). Using a previously published date of 3.0–5.5 million year ago for the P. falciparum and P. reichenowi split17, we thereby date the P. ovale split to 20.3 million years ago and the P. malariae split to 3.5 million years ago. Silhouettes show host specificity of the different species. Values at branching points are bootstrap values (Methods).
Manual curation of about 5,000 gene models of both P. malariae and P. o. curtisi enabled a detailed exploration of lineage-specific differences in gene content (Table 1), with some paralogous expansions being particularly notable (Extended Data Fig. 1c, d). Genes potentially involved in formation of hypnozoites in P. ovale—the lifecycle stage responsible for relapse infections—were also identified (Supplementary Information). The manual curation enabled pseudogenes that are differentially distributed between the two genomes and other human-infective Plasmodium species (Extended Data Table 5) to be analysed in ways not possible using computer-annotated draft genome data7. For instance, pseudogenes are found among a paralogously expanded family of invasion-associated RBPs (Extended Data Fig. 4a–e), and a homologue of a P. falciparum cyclin (PF3D7_1227500) is only pseudogenized in P. o. wallikeri and may be linked to the difference in relapse times between the two P. ovale species12.
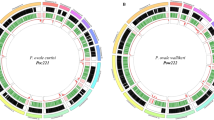
Large multigene families are a defining feature that distinguish the genomes of malaria species but are refractory to detailed analysis in non-curated draft genome data. In P. malariae and P. ovale, approximately 40% of the total genome is subtelomeric. However, the gene content of the subtelomeres differs substantially between the two species (Fig. 3a; Table 1). The breadth and sequence types of the pir gene repertoires of the P. ovale species are similar to P. vivax, whereas P. malariae contains only a restricted subset (Extended Data Fig. 5a). The ancient divergence of the two P. ovale species is supported by their pir repertoires being readily distinguishable (Extended Data Fig. 5b). Despite being sister taxa to P. ovale, the pir repertoire of rodent malaria parasites, however, appears to be completely different (Extended Data Fig. 5a). Moreover, almost 50% of pir genes in P. malariae are pseudogenes (compared to 25% in P. o. curtisi and 9% in P. vivax), suggesting an even smaller functional repertoire.

a, Gene network based on sequence similarity of all genes in P. malariae (red), P. ovale (blue), and P. vivax (green). Cluster 1 contains fam-l genes, cluster 2 contains fam-m genes, and cluster 3 contains surfins and STP1 genes. b, Chromosome 5 subtelomeric localization of fam-l and fam-m genes in doublets (blue brackets) on the telomere-facing strand. Also showing pseudogenes (grey) and hypothetical gene (blue). c, Predicted 3D structure of fam-l (above) and fam-m (below) overlaid with the RH5 crystal structure (Purple). Right-hand images show the protein rotated to the right.
The most notable difference in the subtelomeres of P. malariae is the presence of two large gene families that were not apparent in earlier partial genome data7, and which we have termed fam-l and fam-m (Fig. 3a). Proteins encoded by fam-l and fam-m show characteristics of proteins that are probably exported from the parasite to the infected red blood cell surface (PEXEL export signal, signal peptide, transmembrane domains, and a variable region). Most fam-l and fam-m genes face the telomeres and occur as doublets (Fig. 3b), and we found some evidence that they are co-evolving (Extended Data Fig. 5c, d). Proteins encoded by fam-l and fam-m genes may therefore form heterodimers, a feature not previously seen among subtelomeric gene families in other Plasmodium species. Finally, 3D structures of fam-l and fam-m proteins, predicted with high confidence (template modelling (TM)-score > 0.5)13, overlap the crystal structure of the P. falciparum RH5 protein (TM-score > 0.8)14, with 100% of the RH5 structure covered despite having only 10% sequence similarity (Fig. 3c). RH5 is the only known P. falciparum protein that is essential for erythrocyte invasion, through binding to basigin on the erythrocyte surface15. The RH5 kite-shaped fold is known to be present in RBP2a in P. vivax16, and may be a conserved structure necessary for the binding capabilities of all Rh and RBP genes. This suggests that fam-l and fam-m genes also have an adhesion role, possibly binding host receptors.
The present study highlights features of host-specific adaptations at several levels: ape to human, primate to rodent and within human hosts. As noted in previous comparative genomics studies involving host switches in the Plasmodium genus, invasion-related genes are consistently found to be rapidly evolving. The RBP family is highly expanded (Extended Data Fig. 4a) but its differential distribution across species suggests that RBP3 may be essential for invasion of normocytes (Extended Data Fig. 4c). In contrast to other studies1,7,11, we place the rodent malaria parasites as an outgroup to P. ovale but rooted by P. malariae. The rodent malaria parasites could therefore more closely model the biology of P. ovale than other human-infective species; this indicates that there must have been an ancestral host switch from primates to rodents. The relative dating of speciation events suggests that the move between non-human primates and humans occurred at approximately the same time in two well-separated lineages, suggesting that a common historical event may have promoted host switching and speciation in Plasmodium at the time. The much older speciation of the two P. ovale parasites and the fact that they have been considered identical until recently shows the limitation of morphology alone in species determination.
Owing to the importance of rapidly evolving multigene families and genome structure, high quality genomes for all human infective species of Plasmodium are desperately needed. Although P. malariae and P. ovale are known to be widespread and common in co-infections with P. falciparum (Fig. 1), their low parasitaemia levels have complicated their study and consequently little is known about them. The present study provides a high-quality reference genome for P. malariae, thereby providing a step forward in better understanding this elusive species.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Co-infection mining
We aligned the P. malariae (AB354570) and P. ovale (AB354571) mitochondrial genome sequences against those of P. falciparum18, P. vivax19, and P. knowlesi20 using MUSCLE21. For each species, we identified three 15-bp stretches within the Cox1 gene that contained two or more species-specific single-nucleotide polymorphisms (SNPs). We searched for these 15-bp species-specific barcodes within the sequencing reads of all 2,512 samples from the Pf3K global collection that mapped to the P. falciparum mitochondrial genome (http://www.malariagen.net). Samples that contained at least two sequencing reads matching one or more of the 15 bp barcodes for a specific species were considered to be positive for that species (Extended Data Table 2). We found good correspondence between the three different barcodes for each species, with over 80% of positive samples being positive for all three barcodes. We generated pseudo-barcodes by changing two randomly selected nucleotide bases at a time for 10 randomly selected 15 bp region in the P. vivax19 mitochondrial genome. We did not detect any positive hits using these pseudo-barcodes. As an additional negative control, we searched for P. knowlesi co-infections, but did not find any samples positive for this species as expected from the limited geographical range of this species. Two samples (PocGH01, PocGH02) had high numbers of sequencing reads for all three P. ovale barcodes and were used for reference genome assembly and SNP calling respectively.
Parasite material
All P. ovale samples were obtained from symptomatic patients diagnosed with a P. falciparum infection. The two P. o. curtisi samples (PocGH01, PocGH02) identified through co-infection mining (see above), were from two patients that tested positive on a CareStart (HRP2-based) rapid malaria diagnostic test kit at the Navrongo War Memorial hospital, Ghana. One P. o. wallikeri sample (PowCR01) and one P. o. curtisi sample (PocCR01) were from patients with uncomplicated malaria that tested positive by light microscopy at the Mile 16 Bolifamba Health Centre, Buea, Cameroon. The other P. o. wallikeri sample (PowCR02) was obtained from an individual with asymptomatic parasitaemia enrolled through a community survey in Mutengene, Cameroon. For all samples, following consent obtainment, about 2–5 ml of venous blood was obtained and then diluted with one volume of PBS. This was passed through CF11 cellulose powder columns to remove leukocytes before parasite DNA extraction. Ethical approval was obtained for all P. ovale samples used in this study.
The two P. malariae-like samples, PmlGA01 and PmlGA02, were extracted from Chimpanzee blood (Pan troglodytes troglodytes, 9-year-old female and 11-year-old female, respectively) obtained during routine sanitary controls of animals living in a Gabonese sanctuary (Park of La Lékédi, Gabon). Blood collection was performed following international rules for animal health. Within six hours after collection, host white blood cell depletion was performed on fresh blood samples using the CF11 method22. After DNA extraction using the Qiagen blood and Tissue Kit and detection of P. malariae infections by Cytb PCR and sequencing23, the samples went through a whole-genome amplification step24.
One P. malariae sample, PmMA01, was collected from a patient with uncomplicated malaria in Faladje, Mali. Venous blood (2–5 ml) was depleted of leukocytes within 6 h of collection through CF11 cellulose powder columns as previously described25. The study protocol was approved by the Ethics Committee of Faculty of Medicine and Odontomatology and Faculty of Pharmacy, Bamako, Mali.
Four samples of P. malariae were obtained from travellers returning to Australia with malaria. PmUG01 and PmID01 were sourced from patients returning from Uganda and Papua Indonesia, respectively, who presented at the Royal Darwin Hospital, Darwin, with microscopy-positive P. malariae infection. PmMY01 was sourced from a patient presenting at the Queen Elizabeth Hospital, Sabah, Malaysia, with microscopy-positive P. malariae infection. Patient sample PmGN01 was collected from a patient who presented to Royal Brisbane and Women’s Hospital in 2013 on return from Guinea. Venous blood samples were subject to leukodepletion within 6 h of collection. PmUG01 was leukodepleted using a commercial Plasmodipur filter (EuroProxima, The Netherlands); custom-made cellulose-based filters were used for PmID01 and PmMY01, whereas PmGN01 was leukodepleted using an inline leukodepletion filter present in the venesection pack (Pall Leukotrap; WBT436CEA). DNA extraction was performed on filtered blood using commercial kits (QIAamp DNA Blood Midi kit, Qiagen). For samples PmUG01, PmID01 and PmMY01, ethical approval for the sample collection was obtained from the Human Research Ethics Committee of NT Department of Health and Families and Menzies School of Health Research (HREC-2010-1396 and HREC-2010-1431) and the Medical Research Ethics Committee, Ministry of Health Malaysia (NMRR-10-754-6684). For sample PmGN02, ethical approval was obtained from the Royal Brisbane and Women’s Hospital Human Research Ethics Committee (HREC/10/QRBW/379) and the Human Research Ethics Committee of the Queensland Institute of Medical Research (p1478).
Sample preparation and sequencing
One P. malariae sample, PmUG01, was selected for long-read sequencing, using Pacific Biosciences (PacBio), owing to its low host contamination and abundant DNA. Passing through a 25-mm blunt-ended needle, 6 μg of DNA was sheared to 20–25 kb. SMRT bell template libraries were generated using the PacBio issued protocol (20 kb Template Preparation using the BluePippin Size-Selection System). After a greater than 7 kb size-selection using the BluePippin Size-Selection System (Sage Science), the library was sequenced using P6 polymerase and chemistry version 4 (P6/C4) in 20 SMRT cells (Supplementary Information).
The remaining isolates were sequenced with Illumina Standard libraries of 200–300 bp fragments and amplification-free libraries of 400–600 bp fragments were prepared26 and sequenced on the Illumina HiSeq 2000 v3 or v4 and the MiSeq v2 according to the manufacturer’s standard protocol (Supplementary Information). Raw sequence data was deposited in the European Nucleotide Archive (Supplementary Information).
Genome assembly
The PacBio-sequenced P. malariae sample, PmUG01, was assembled using HGAP27 with an estimated genome size of 100 Mb to account for the host contamination (~85% human). The resulting assembly was corrected initially using Quiver27, followed by iCORN28. PmUG01 consisted of multiple haplotypes, with the majority haplotype being used for the iCORN28, and a coverage analysis was performed to remove duplicate contigs. Additional duplicated contigs were identified using a BLASTN29 search, with the shorter contigs being removed if they were fully contained within the longer contigs or merged with the longer contig if their contig ends overlapped. Host contamination was removed by manually filtering on GC, coverage, and BLASTN hits to the non-redundant nucleotide database29.
The Illumina-based genome assemblies for P. o. curtisi, P. o. wallikeri, and P. malariae-like were performed using MaSURCA30 for samples PocGH01, PowCR01, and PmlGA01 respectively. To confirm that the assemblies were indeed P. ovale, we mapped existing P. ovale capillary reads to the assemblies (http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?view=search, using as search terms: CENTER_PROJECT = “PO” and CENTER_NAME = “SC”). Before applying MaSURCA30, the samples were mapped to the P. falciparum 3D7 reference genome18 to remove contaminating reads. The draft assemblies were further improved by iterative uses of SSPACE31, GapFiller32 and IMAGE33. The resulting scaffolds were ordered using ABACAS34 against the P. vivax PVP01 (ref. 35) (http://www.genedb.org/Homepage/PvivaxP01) assembly (both P. ovale) or against the P. malariae PacBio assembly (P. malariae-like). The assemblies were manually filtered on GC, coverage, and BLASTN hits to the non-redundant nucleotide database29. iCORN28 was used to correct frameshifts. Finally, contigs shorter than 1 kb were removed.
Using two more samples, PocGH02 and PowCR02, additional draft assemblies of both P. ovale species were produced using MaSURCA30 followed by RATT36 to transfer the gene models from the high-quality assemblies.
The genome sequences and annotations are currently available on GeneDB for P. malariae (http://www.genedb.org/Homepage/Pmalariae) and for P. ovale curtisi (http://www.genedb.org/Homepage/Povale). The genome sequences have been deposited into the European Nucleotide Archive (http://www.ebi.ac.uk/ena). Accession numbers for all reads generated for this study can be found in Supplementary Information. Accession identifiers for the assembled genome sequences can be found in Supplementary Information.
Gene annotation
RATT36 was used to transfer gene models on the basis of synteny conserved with other sequenced Plasmodium species (P. falciparum18, P. vivax19, P. berghei37, and P. gallinaceum (http://www.genedb.org/Homepage/Pgallinaceum). In addition, genes were predicted ab initio using AUGUSTUS38, trained on a set consisting of manually curated P. malariae and P. ovale genes respectively. Non-coding RNAs and tRNAs were identified using Rfam 12.0 (ref. 39). Gene models were then manually curated for both the P. malariae and P. o. curtisi reference genomes, using Artemis40 and the Artemis Comparison Tool41. These tools were also used to manually identify deleted and disrupted genes (Extended Data Table 5). The P. malariae-like and P. o. wallikeri genomes were both annotated using the Companion tool42.
Phylogenetics
Following orthologue assignment using BLASTP29 and OrthoMCL43, amino acid sequences of 1,000 core genes from 12 Plasmodium species (P. gallinaceum44, P. falciparum18, P. reichenowi10, P. knowlesi20, P. vivax P01 (ref. 35), P. cynomolgi45, P. chabaudi37, P. berghei37, and the four assemblies produced in this study) were aligned using MUSCLE21. The alignments were cleaned using GBlocks46 with default parameters to remove non-informative and gapped sites. The cleaned non-zero length alignments were then concatenated. This resulted in an alignment of 421,988 amino acid sites per species. The optimal substitution model for each gene partition was determined by running RAxML47 for each gene separately using all implemented substitution models. The substitution models with the minimum Akaike Information Criterion on a guide maximum parsimony tree were used for each gene partition. A maximum likelihood phylogenetic tree was constructed using RAxML47 version 8.2.4. with 100 bootstraps48 (Fig. 2). To confirm this tree, we used different phylogenetic tools including PhyloBayes49 and PhyML50, a number of different substitution models within RAxML, starting the tree search from the commonly accepted phylogenetic tree, and removing sites in the alignment which supported significantly different trees (1). Figtree was used to display and colour the tree (http://tree.bio.ed.ac.uk/software/figtree/).
A phylogenetic tree of four P. malariae (PmID01, PmGN01, PmGN02, PmMY01) and all P. malariae-like samples (PmlGA01, PmlGA02) was generated using PhyML50 on the basis of all P. malariae genes. For each sample, the raw SNPs, as called using the SNP calling pipeline (see below), were mapped onto all genes to morph them into sample-specific gene copies using BCFtools51. Amino acid alignments for all genes were concatenated and cleaned using GBlocks46 with default parameters.
Divergence dating
Species divergence times were estimated using the Bayesian inference tool G-PhoCS52, a software which uses unlinked neutrally evolving loci and a given phylogeny to estimate demographic parameters. One additional sample per assembly (PmGN01 for P. malariae, PocGH02 for P. o. curtisi, PowCR02 for P. o. wallikeri, and PmlGA02 for P. malariae-like) was used to morph the respective assembly using iCORN28. Regions in the genomes without mapping were masked, as iCORN28 would not have morphed them. Unassigned contigs and subtelomeric regions were removed for this analysis owing to the difficulty of alignment. Repetitive regions in the chromosomes of the four assemblies and the four morphed samples were masked using Dustmasker53 and then the chromosomes were aligned using FSA54. The P. o. wallikeri and the P. o. curtisi chromosomes were aligned against each other, as were the P. malariae and P. malariae-like chromosomes. The alignments were split into 1 kb loci, removing those that contained gaps, masked regions, and coding regions to conform with the neutral loci prediction of G-PhoCS52. G-PhoCS52 was run for one million Markov Chain Monte Carlo (MCMC)-iterations with a sample-skip of 1,000 and a burn-in of 10,000 for each of the two-species pairs. Follow-up analyses using Tracer (http://beast.bio.ed.ac.uk/Tracer) confirmed that this was sufficient for convergence of the MCMC chain in all cases. In the model, we assumed a variable mutation rate across loci and allowed for on-going gene flow between the populations. The tau values obtained from this were 0.0049 for P. malariae and 0.0434 for P. ovale.
The tau values were used to calculate the date of the split, using the formula (tau × G) / m, where G is the generation time in years and m is the mutation rate. Testing a number of different generation time and mutation rate estimates in order to optimize the P. falciparum and P. reichenowi split to 4 million years ago as estimated previously17, we found a mutation rate of 3.8 × 10−10 SNPs per site per lifecycle55 and a generation time of 65 days56 to generate this previously published date17. For P. malariae, a generation time of 100 days was used owing to the longer intra-erythrocytic cycle.
3D structure prediction
The I-TASSER57 version 4.4 online web server58 (http://www.zhanglab.ccmb.med.umich.edu/I-TASSER) was used for 3D protein structure prediction. Predicted structures with a TM-score of over 0.5 were considered reliable as suggested in the I-TASSER user guidelines13. TM-align14, as implemented in I-TASSER58, was used to overlay the predicted protein structure with existing published protein structures.
Hypnozoite gene search
Using the OrthoMCL43 clustering between all sequenced Plasmodium species used for the phylogenetic analysis (see above), we examined clusters containing only genes of hypnozoite-forming species: P. vivax P01 genes, P. cynomolgi45 genes and genes of both of the P. ovale species.
Additionally, we examined P. o. curtisi orthologues of previously published hypnozoite gene candidates45, looking in the 1 kb 5′ upstream region for any of the four ApiAP2 motifs59 involved in sporozoite regulation and expression: GCATGC (PF3D7_1466400), GCCCCG (PF3D7_1342900), TAAGCC (PF3D7_1342900), and TGTTAC (PF3D7_0420300) (Supplementary Information).
Gene family analysis
All P. malariae, P. ovale, and P. vivax P01 genes were compared pairwise using BLASTP29, with genes having a minimum local BLAST hit of 50% identity over 150 amino acids or more being considered connected. These gene connections were visualized in Gephi60 using a Fruchterman-Reingold61 layout and with unconnected genes removed.
P. malariae, P. o. curtisi and P. o. wallikeri protein sequences for Plasmodium interspersed repeat (pir) genes, excluding pseudogenes, were combined with those from P. vivax P01 (ref. 35), P. knowlesi20, P. chabaudi AS v3 (http://www.genedb.org/Homepage/Pchabaudi), P. yoelii yoelii 17X v2 (ref. 37), and P. berghei v3 (ref. 62). Sequences were clustered using tribeMCL63 with blast e-value 0.01 and inflation 2. This resulted in 152 subfamilies. We then excluded clusters with one member. The number of genes per species in each subfamily were plotted in a heatmap using the heatmap.2 function in ggplots in R-3.1.2.
The pir genes from two P. o. curtisi and two P. o. wallikeri assemblies (two high-quality and two draft genome assemblies) were compared pairwise using BLASTP29 with a 99% identity over a minimum of 150 amino acids cutoff. The gene–gene connections were visualized in Gephi60 using a Fruchterman–Reingold61 layout after removing unconnected genes.
Mirror tree analysis
Using Artemis40, 79 fam-m and fam-l doublets that were confidently predicted as being paired-up were manually selected on the basis of their dispersal throughout the subtelomeres of different chromosomes. The Mirrortree64 web server (http://csbg.cnb.csic.es/mtserver/) was used to construct mirror trees for these 79 doublets. 35 doublets with recent branching from another doublet were manually selected to enrich for genes under recent selection (Extended Data Fig. 5c). To control for chance signals of co-evolution on the basis of their subtelomeric location, the same methodology was repeated by choosing 79 pir genes in close proximity of fam-m genes as ‘pseudo-doublets’ for analysis and paired up in the Mirrortree64 web server (Extended Data Fig. 5d).
Reticulocyte-binding protein (RBP) phylogenetic plot
Full-length RBP genes were manually inspected using ACT41 and verified to either be functional or pseudogenized by identification of sequencing reads in other samples that confirm mutations inducing premature stop codons or frameshifts. All functional RBPs were aligned using MUSCLE21 and cleaned using GBlocks46 with default parameters. PhyML50 was used to construct a phylogenetic tree of the different RBPs (Extended Data Fig. 4a). Figtree was used to colour the tree (http://tree.bio.ed.ac.uk/software/figtree/).
SNP calling
Additional P. malariae (PmMY01, PmID01, PmMA01, PmGN01) and P. o. curtisi (PocGH01, PocGH02, PocCR01) samples were mapped back against the reference genomes using SMALT (-y 0.8, -i 300) (Supplementary Information). As outgroups, P. malariae-like (PmlGA01, PmlGA02) and P. o. wallikeri (PowCR01, PowCR02) were also mapped against the P. malariae and P. o. curtisi genomes respectively. The resulting ‘bam’ formatted files were merged for either of the two genomes, and GATK65 Unified Genotyper was used to call SNPs from the merged bam files (Supplementary Information). As per GATK65 best practices, SNPs were filtered by quality of depth (QD > 2), depth of coverage (DP > 10), mapping quality (MQ > 20), and strand bias (FS < 60). Additionally, all sites for which we had missing data for any of the samples or for which we had heterozygous calls were filtered. Finally, we filtered sites that were masked using Dustmasker53 to remove repetitive and difficult to map regions. The same methodology was also applied to two P. vivax samples (SRR3400910 and SRR332566) and two P. falciparum Pf3K field samples (PF0066-C and PF0038-C, see https://www.malariagen.net/projects/pf3k) for comparative purposes.
Molecular evolution analysis
To calculate the nucleotide diversity for the different species, we extracted all filtered SNPs in the genomes excluding the subtelomeres. We then counted the number of pairwise differences between the different samples divided by the resulting genome size, comprising three comparisons for species with three samples (P. malariae, P. o. curtisi, P. vivax, P. falciparum) and one comparison for species with two samples (P. o. wallikeri, P. malariae-like). These estimates were then averaged by species (Table 1).
The filtered SNPs were used to morph the reference genomes using BCFtools51 for each sample, from which sample-specific gene models were obtained. Nucleotide alignments of each gene were then generated. Codons with alignment positions that were masked using Dustmasker53 were excluded. For each alignment (that is, gene), we calculated Hudson–Kreitman–Aguadé (HKA)66, McDonald–Kreitman (MK)67, and Ka/Ks68 values (see below). Subtelomeric gene families and pseudogenes were excluded from the analysis. The results were analysed and plotted in RStudio (http://www.rstudio.com/).
For the HKA66, we counted the proportion of pairwise nucleotide differences intra-specifically (that is, within P. malariae and within P. o. curtisi) and inter-specifically (that is, between P. malariae and P. malariae-like, between P. o. curtisi and P. o. wallikeri). The intraspecific comparisons were averaged to obtain the nucleotide diversity π of the genes and these were divided by the average interspecific comparisons, the nucleotide divergence, to get the HKA ratio (HKAr) for each gene.
The MK test67 was performed for each gene by obtaining the number of fixed and polymorphic changes, as well as a P value, as previously described69 and then calculating the skew as log2(((Npoly + 1) / (Spoly + 1)) / ((Nfix + 1) / (Sfix + 1))), where Npoly and Nfix are polymorphic and fixed non-synonymous substitutions respectively, and Spoly and Sfix refer to the synonymous substitutions.
To calculate the average Ka/Ks ratio68, we took the cleaned alignments of the MK test, extracting the pairwise sequences of P. malariae and P. malariae-like (and of P. o. curtisi and P. o. wallikeri). The ‘Bio::Align::DNAStatistics’ module was used to calculate the Ka/Ks values for each pair70, averaging across samples within a species.
Using existing RNA sequencing data from seven different life-cycle stages in P. falciparum71, reads were mapped against spliced gene sequences (exons, but not UTRs) from the P. falciparum 3D7 reference genome18 using Bowtie2 (ref. 72) v2.1.0 (-a -X 800 –x). Read counts per transcript were estimated using eXpress v1.3.0 (ref. 73). Genes with an effective length cutoff below 10 in any sample were removed. Summing over transcripts generated read counts per gene. Numbers were averaged for all gametocyte stages and for all blood stages. Genes with no stage having 10 or more reads were classified as being expressed elsewhere. Genes in P. malariae and P. ovale were classified by the maximum expression stage of their P. falciparum orthologue if the difference between the maximum expression stage and the second highest stage was larger than the difference between the second and third highest stage, otherwise the gene was classified as having no peak expression.
The Gene Ontology term enrichment analysis was performed in R, using TopGO74. As a Gene Ontology database, the predicted Gene Ontology terms from the P. falciparum 3D7 genes orthologous to the P. malariae and P. o. curtisi genes included in the analysis were used. Collated tables for all molecular evolution measures for all genes can be found in Source Data File 1 for P. malariae and P. o. curtisi.
RBP1a receptor search
To find the putative RBP1a receptor, we performed an OrthoMCL43 clustering between human, chimpanzee75, and common marmoset76 genes. P. brasilianum (P. malariae) results in chronic infection in the common marmoset77. Genes without transmembrane domains as well as those annotated as ‘predicted’ were removed. To limit false positives, all remaining genes were searched against the chimpanzee genes using BLASTP29 with a threshold of 1 × 10−10.
Data deposition statement
All raw data has been deposited as described in Supplementary Information. Assembled genome sequences can be found under the study PRJEB14392 (http://www.ebi.ac.uk/ena/data/view/PRJEB14392). The individual accession numbers are as follows for PmUG01 (contig accession: FLRL01000001–FLRL01000047; chromosome accession: LT594622–LT594637), PocGH01 (contig accession: FLRI01000001–FLRI01000638; chromosome accession: LT594582–LT594597), PowCR01 (contig accession: FLRJ01000001–FLRJ01000771; chromosome accession: LT594505-LT594520) and PmlGA01 (contig accession: FLRK01000001–FLRK01000035; chromosome accession: LT594489–LT594503).
References
Sutherland, C. J. et al. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J. Infect. Dis. 201, 1544–1550 (2010)
Collins, W. E. & Jeffery, G. M. Plasmodium malariae: parasite and disease. Clin. Microbiol. Rev. 20, 579–592 (2007)
Collins, W. E. & Jeffery, G. M. Plasmodium ovale: parasite and disease. Clin. Microbiol. Rev. 18, 570–581 (2005)
Doderer-Lang, C. et al. The ears of the African elephant: unexpected high seroprevalence of Plasmodium ovale and Plasmodium malariae in healthy populations in Western Africa. Malar. J. 13, 240 (2014)
Bousema, T., Okell, L., Felger, I. & Drakeley, C. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat. Rev. Microbiol. 12, 833–840 (2014)
Arisue, N. et al. The Plasmodium apicoplast genome: conserved structure and close relationship of P. ovale to rodent malaria parasites. Mol. Biol. Evol. 29, 2095–2099 (2012)
Ansari, H. R. et al. Genome-scale comparison of expanded gene families in Plasmodium ovale wallikeri and Plasmodium ovale curtisi with Plasmodium malariae and with other Plasmodium species. Int. J. Parasitol. 46, 685–696 (2016)
Sundararaman, S. A. et al. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat. Commun. 7, 11078 (2016)
Lalremruata, A. et al. Natural infection of Plasmodium brasilianum in humans: Man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine 2, 1186–1192 (2015)
Otto, T. D. et al. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat. Commun. 5, 4754 (2014)
Schaer, J. et al. High diversity of West African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc. Natl Acad. Sci. USA 110, 17415–17419 (2013)
Nolder, D. et al. An observational study of malaria in British travellers: Plasmodium ovale wallikeri and Plasmodium ovale curtisi differ significantly in the duration of latency. BMJ Open 3, e002711 (2013)
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015)
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005)
Crosnier, C. et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480, 534–537 (2011). 10.1038/nature10606
Gruszczyk, J. et al. Structurally conserved erythrocyte-binding domain in Plasmodium provides a versatile scaffold for alternate receptor engagement. Proc. Natl Acad. Sci. USA 113, E191–E200 (2016)
Silva, J. C., Egan, A., Arze, C., Spouge, J. L. & Harris, D. G. A new method for estimating species age supports the coexistence of malaria parasites and their Mammalian hosts. Mol. Biol. Evol. 32, 1354–1364 (2015)
Gardner, M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002)
Carlton, J. M. et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763 (2008)
Pain, A. et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455, 799–803 (2008)
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
Auburn, S. et al. An effective method to purify Plasmodium falciparum DNA directly from clinical blood samples for whole genome high-throughput sequencing. PLoS One 6, e22213 (2011)
Ollomo, B. et al. A new malaria agent in African hominids. PLoS Pathog. 5, e1000446 (2009)
Oyola, S. O. et al. Optimized whole-genome amplification strategy for extremely AT-biased template. DNA Res. 21, 661–671 (2014)
Kamau, E. et al. K13-propeller polymorphisms in Plasmodium falciparum parasites from sub-Saharan Africa. J. Infect. Dis. 211, 1352–1355 (2015)
Bronner, I. F., Quail, M. A., Turner, D. J. & Swerdlow, H. Improved protocols for illumina sequencing. Curr. Protoc. Hum. Genet. 80, 11–42 (2014)
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013)
Otto, T. D., Sanders, M., Berriman, M. & Newbold, C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26, 1704–1707 (2010)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677 (2013)
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011)
Nadalin, F., Vezzi, F. & Policriti, A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13 (Suppl 14), S8 (2012)
Tsai, I. J., Otto, T. D. & Berriman, M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 11, R41 (2010)
Assefa, S., Keane, T. M., Otto, T. D., Newbold, C. & Berriman, M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969 (2009)
Auburn, S. et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. http://dx.doi.org/10.12688/wellcomeopenres.9876.1 (2016)
Otto, T. D., Dillon, G. P., Degrave, W. S. & Berriman, M. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 39, e57 (2011)
Otto, T. D. et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. 12, 86 (2014)
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006)
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137 (2015)
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000)
Carver, T. J. et al. ACT: the Artemis Comparison Tool. Bioinformatics 21, 3422–3423 (2005)
Steinbiss, S. et al. Companion: a web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 44 (W1), W29–W34 (2016)
Li, L., Stoeckert, C. J., Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003)
Boehme, U. et al. Complete avian malaria parasite genomes reveal host-specific parasite evolution in birds and mammals. Preprint at: http://biorxiv.org/content/early/2016/11/09/086504 (2016)
Tachibana, S. et al. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat. Genet. 44, 1051–1055 (2012)
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007)
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463 (2005)
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771 (2008)
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009)
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010)
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
Gronau, I., Hubisz, M. J., Gulko, B., Danko, C. G. & Siepel, A. Bayesian inference of ancient human demography from individual genome sequences. Nat. Genet. 43, 1031–1034 (2011)
Morgulis, A., Gertz, E. M., Schäffer, A. A. & Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040 (2006)
Bradley, R. K. et al. Fast statistical alignment. PLOS Comput. Biol. 5, e1000392 (2009)
Claessens, A. et al. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 10, e1004812 (2014)
Daniels, R. F. et al. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc. Natl Acad. Sci. USA 112, 7067–7072 (2015)
Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69 (Suppl 8), 108–117 (2007)
Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008)
Campbell, T. L., De Silva, E. K., Olszewski, K. L., Elemento, O. & Llinás, M. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 6, e1001165 (2010)
Bastian, M. H. S. & Jacomy M. Gephi: an open source software for exporing and manipulating networks. International AAAI Conference on Weblogs and Social Media (2009)
Fruchterman, T. M. J. & Reingold, E. M. Graph drawing by force-directed placement. Softw. Pract. Exper. 21, 1129–1164 (1991)
Fougère, A. et al.. Variant exported blood-stage proteins encoded by Plasmodium multigene families are expressed in liver stages where they are exported into the parasitophorous vacuole. PLoS Pathog. 12, e1005917 (2016)
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002)
Ochoa, D. & Pazos, F. Studying the co-evolution of protein families with the Mirrortree web server. Bioinformatics 26, 1370–1371 (2010)
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010)
Innan, H. Modified Hudson–Kreitman–Aguade test and two-dimensional evaluation of neutrality tests. Genetics 173, 1725–1733 (2006)
Kreitman, M. & Hudson, R. R. Inferring the evolutionary histories of the Adh and Adh-dup loci in Drosophila melanogaster from patterns of polymorphism and divergence. Genetics 127, 565–582 (1991)
Nekrutenko, A., Makova, K. D. & Li, W. H. The KA/KS ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res. 12, 198–202 (2002)
Holloway, A. K., Lawniczak, M. K., Mezey, J. G., Begun, D. J. & Jones, C. D. Adaptive gene expression divergence inferred from population genomics. PLoS Genet. 3, 2007–2013 (2007)
Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986)
López-Barragán, M. J. et al. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics 12, 587 (2011)
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012)
Roberts, A. & Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 10, 71–73 (2013)
Alexa, A. R., J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.24.0 (2016)
Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005)
Marmoset Genome Sequencing and Analysis Consortium. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 46, 850–857 (2014)
Wedderburn, N., Mitchell, G. H. & Davies, D. R. Plasmodium brasilianum in the common marmoset Callithrix jacchus. Parasitology 90, 573–578 (1985)
Acknowledgements
This work was supported by the Medical Research Council (MR/J004111/1; MR/L008661/1) and the Wellcome Trust (098051). S.A. and R.N.P. are funded by the Wellcome Trust (091625). F.R., B.O. and F.P. are financed by the ANR JCJC 2012 ORIGIN, the LMI Zofac, as well as by CNRS, IRD, and CIRMF. C.I.N. is funded by a Wellcome Trust Investigator Award (104792/Z/14/Z). A.A.D. is funded as a Sanger International Fellow. J.S.M. and N.M.A. are supported by NHMRC Practitioner Fellowships (1041802; 1042072). The authors thank E. Willaume from the Park of La Lékédi and the people involved in the sanitary controls of the chimpanzees. The authors also thank P. Griffin and S. Sekuloski for assistance in collection of P. malariae isolate PmGN01.
Author information
Authors and Affiliations
Contributions
G.G.R. carried out the sequence assembly, genome annotation and all of the data analysis; U.C.B. performed manual gene curation; M.S. coordinated sequencing; A.J.R., J.A.C., M. M. and F.P. performed data analysis; G.G.R., T.O.A., L.A.E., J.W.B., D.P.K., C.I.N., M.B. and T.D.O. designed the P. ovale project; G.G.R., F.R., B.O., F.P., C.I.N., M.B. and T.D.O. designed the P. malariae-like project; G.G.R., A.A.D., O.M.A, N.M.A., S.A., R.N.P., J.S.M., C.I.N., M.B. and T.D.O. designed the P. malariae project; G.G.R., C.I.N., M.B., T.D.O. wrote the manuscript; all authors read and critically revised the manuscript; and C.I.N., M.B., T.D.O. directed the overall study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks D. Neafsey, D. Wirth and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Large genomic changes in the P. malariae and P. ovale genome sequences.
a, Artemis Comparison Tool (ACT)41 view showing reciprocal translocation of chromosomes 6 and 10 in P. malariae. The red lines indicate blast similarities, chromosome 6 in orange and chromosome 10 in brown. b, ACT41 view showing a pericentric inversion in chromosome 5 of P. malariae. Red lines indicate blast similarities and blue lines indicate inverted blast hits. c, Expansion of 22 copies (20 functional) of Pfg27 in P. malariae (top) compared to a single copy in P. falciparum (bottom) with red lines indicating blast similarities. Functional genes are in red and pseudogenes in grey. This compares to only 17 Pfg27 copies described previously7, which were found on six separate contigs, while all Pfg27 copies described here are found on the same contig. d, Expansion of PVP01_1270800 (PF3D7_1475900 in P. falciparum), a gene with no known function, in P. o. curtisi and P. o. wallikeri, with different copy numbers in each, compared to the one copy in P. vivax. Functional genes shown in orange and pseudogenes shown in grey. This gene family was recently named KELT7, and we confirm the 8 copies present in P. o. wallikeri, but show that P. o. curtisi has 9 copies, two of which are pseudogenes.
Extended Data Figure 2 P. malariae-like has significantly longer branch lengths than P. malariae, and P. brasilianum is identical to P. malariae.
a, A phylogenetic tree of all P. malariae and P. malariae-like samples generated using PhyML50 on the basis of all P. malariae genes. P. malariae samples are indicated by a green bar and P. malariae-like samples are indicated by a purple bar. Silhouettes represent host infectivity. b, A PhyML50 phylogenetic tree of all P. brasilianum 18S rRNA sequences9, indicated by a red bar and red tip branches, and the corresponding 18S rRNA sequences from the P. malariae and P. malariae-like assemblies, labelled as such. Silhouettes represent the host origin for each sample.
Extended Data Figure 3 Different population genetics in P. malariae and P. o. curtisi.
a, HKA ratio and MK Skew for both P. malariae versus P. malariae-like (left) and P. o. curtisi versus P. o. wallikeri (right). Genes with high HKAr values (>0.15, vertical line) are coloured by the peak expression of their orthologue in P. falciparum71 (red, blood stage; green, gametocyte; blue, ookinete; yellow, other; grey, no peak expression) (Methods). Genes with high HKAr and a significant MK skew (square symbols): (1) merozoite surface protein 9, PF3D7_1228600; (2) rRNA (adenosine-2'-O-)-methyltransferase, PF3D7_1429400; (3) merozoite surface protein 1, PF3D7_0930300; (4) formin 1, PF3D7_0530900. b, Gene-wide HKAr values for the P. falciparum to P. reichenowi comparison, described earlier10, versus HKAr for the P. o. curtisi to P. o. wallikeri (blue) and the P. malariae to P. malariae-like (red) comparisons. Five genes show significant HKAr (> 0.15) values for both comparisons: (1) ferrodoxin reductase-like protein (PF3D7_0720400); (2) EGF-like membrane protein (PF3D7_0623300); (3) ADP/ATP carrier protein (PF3D7_1004800); (4) merozoite surface protein 1 (PF3D7_0930300); (5) conserved Plasmodium protein (PF3D7_0311000). c, log2 of P values of gene-wide MK tests for the P. falciparum to P. reichenowi comparison10 by P. o. curtisi to P. o. wallikeri (blue) and P. malariae to P. malariae-like (red) comparisons. Three genes have significant MK skews (log2(P) < −3) for both comparisons: (1) glideosome-associated connectyor (PF3D7_1361800); (2) apical membrane antigen 1 (PF3D7_1133400); (3) NAD(P)H-dependent glutamate synthase (PF3D7_1435300). d, Nonsynonymous versus synonymous fixed mutations per gene for both the P. o. curtisi to P. o. wallikeri (blue) and the P. malariae to P. malariae-like (red) comparisons. Whereas the former has most genes centred around the x = y line, the latter has most genes below this line with more nonsynonymous than synonymous mutations, indicative of an ancestral bottleneck. e, Bar plot of proportion of P. malariae (above) and P. o. curtisi (below) genes expressed at different stages (no peak expression (grey), ookinete (blue), gametocyte (green), intraerythrocytic (red), and other stage (yellow)) binned by Ka/Ks, with the number of genes in each bin displayed (n). P. o. curtisi genes with very high Ka/Ks values (>2.5) are enriched for genes with peak expression in gametocyte.
Extended Data Figure 4 Reticulocyte-binding protein changes in P. malariae and P. ovale.
a, Phylogenetic tree of all full-length functional RBPs in P. malariae (red branches), P. o. curtisi (blue branches without stars), P. o. wallikeri (blue branches with stars), and P. vivax (green branches). Brackets indicate the different subclasses of RBPs: RBP1a, RBP1b, RBP2 and RBP3. b, ACT41 view of functional (orange) and pseudogenized (grey) RBP1a and RBP1b in five species (P. vivax, P. o. curtisi, P. o. wallikeri, P. malariae and P. malariae-like). Blue indicates assembly gaps. Red bars between species indicate level of sequence similarity, with darker colour indicating higher similarity. c, Number of RBP genes in each of the three RBP classes (RBP1, RBP2, RBP3) by species (P. vivax, P. o. curtisi, P. o. wallikeri, P. cynomolgi, P. malariae, P. knowlesi) grouped by erythrocyte invasion preference (reticulocyte versus normocyte). d, PhyML50 generated phylogenetic tree of all RBP genes over 1 kb long in P. o. curtisi and P. o. wallikeri. Pseudogenes are denoted with (P). Multiple functional RBP2 genes match up with pseudogenized copies in the other genome. e, ACT41 view of RBP1b in red for P. o. curtisi (bottom) and the corresponding disrupted open reading frame in P. o. wallikeri (top), with black ticks indicating stop codons. Reads (in blue) from an additional P. o. wallikeri sample (PowCR02) confirm the bases introducing the frameshift (green box) and premature stop codon (yellow box) in RBP1b.
Extended Data Figure 5 Subtelomeric gene family dynamics in P. ovale and P. malariae.
a, Heat map showing the sharing of pir subfamilies between different species based on tribeMCL63. Columns show pir subfamilies and rows show species. Colours indicate the number of genes classified into each subfamily for each species. Subfamilies were ordered by size, species were ordered for clarity. pir genes in rodent-infective species fall into a small number of well-defined families. Those in P. vivax, P. malariae and P. ovale, however, are much more diverse. There is little overlap between rodent subfamilies and human-infective subfamilies, despite P. ovale being a sister taxa to the rodent-infecting species. P. knowlesi has some sharing with other species, but its largest families are species specific, suggesting it has undergone specialization of its pir repertoire. b, Gene network of pir genes for both high-quality assemblies of P. o. curtisi (dark red) and P. o. wallikeri (dark blue) and draft assemblies of each (light red and light blue respectively). pir genes with BLASTP29 identity hits of 99% and over 150 amino acids become connected in the graph. Genes without connections were removed. There is one connection between the two species (circled in black and with a zoomed in version), 801 between the P. o. curtisi assemblies, 524 between the P. o. wallikeri assemblies, 527 on average within each P. o. curtisi assembly, and 423 on average within each P. o. wallikeri assembly. This indicates that there is considerably less sharing of pir genes between the two P. ovale species than within each species, as expected if the two do not recombine with each other. c, Mirror tree64 for 79 fam-m and fam-l doublets, where the two phylogenetic trees correspond to either of the families with lines connecting branch tips of the same doublet. 35 branches (red) were manually selected owing to exhibiting recent branching. Inset below shows the correlations as calculated by the Mirrortree webserver64 between the two trees for all branches (above, correlation = 0.19, P < 0.001) and red branches (below, correlation = 0.53, P < 0.001). This shows that the two families are co-evolving, especially when doublets that recently branched are selected, suggesting that the co-evolution may be disrupted over longer periods of time, potentially through recombination. d, Mirror tree64 for 79 pir and fam-m pseudo-doublets (Methods), where the two phylogenetic trees correspond to either of the families with lines connecting branch tips of the same doublet. We manually selected 35 branches (red) as they exhibited recent branching. Inset shows the correlations as calculated by the Mirrortree webserver64 between the two trees for all branches (above, correlation = −0.09, P > 0.05) and red branches (below, correlation = −0.10, P > 0.05). This shows that the two families are not co-evolving, and that subtelomeric location does not produce sporadic signals of co-evolution.
Supplementary information
Supplementary Information
This file contains Supplementary Text, Supplementary Tables 1-9 and Supplementary References – see contents for details. (PDF 672 kb)
Supplementary Data
This file contains a summary of the molecular evolutionary analysis of the P. malariae genome and a summary of the molecular evolutionary analysis of the P. ovale curtisi genome. (XLSX 4004 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rutledge, G., Böhme, U., Sanders, M. et al. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature 542, 101–104 (2017). https://doi.org/10.1038/nature21038
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature21038
This article is cited by
-
Detection of P. malariae using a new rapid isothermal amplification lateral flow assay
Malaria Journal (2024)
-
New reference genomes to distinguish the sympatric malaria parasites, Plasmodium ovale curtisi and Plasmodium ovale wallikeri
Scientific Reports (2024)
-
Genome-wide liver transcriptomic profiling of a malaria mouse model reveals disturbed immune and metabolic responses
Parasites & Vectors (2023)
-
Why Plasmodium vivax and Plasmodium falciparum are so different? A tale of two clades and their species diversities
Malaria Journal (2022)
-
Systems biology of malaria explored with nonhuman primates
Malaria Journal (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}