
Abstract
To explore the origins and consequences of tetraploidy in the African clawed frog, we sequenced the Xenopus laevis genome and compared it to the related diploid X. tropicalis genome. We characterize the allotetraploid origin of X. laevis by partitioning its genome into two homoeologous subgenomes, marked by distinct families of ‘fossil’ transposable elements. On the basis of the activity of these elements and the age of hundreds of unitary pseudogenes, we estimate that the two diploid progenitor species diverged around 34 million years ago (Ma) and combined to form an allotetraploid around 17–18 Ma. More than 56% of all genes were retained in two homoeologous copies. Protein function, gene expression, and the amount of conserved flanking sequence all correlate with retention rates. The subgenomes have evolved asymmetrically, with one chromosome set more often preserving the ancestral state and the other experiencing more gene loss, deletion, rearrangement, and reduced gene expression.
Similar content being viewed by others


Main
Ancient polyploidization events have shaped diverse eukaryotic genomes1, including two rounds of whole-genome duplication at the base of the vertebrate radiation2. While polyploidy is rare in amniotes, presumably owing to constraints on sex chromosome dosage3,4, it is common in fish5, amphibians6,7, and plants8. Polyploidy provides raw material for evolutionary diversification because gene duplicates can support new functions and networks9. However, the component subgenomes of a polyploid must cooperate to mediate potential incompatibilities of dosage, regulatory controls, protein–protein interactions and transposable element activity.
The African clawed frog X. laevis is one of a polyploid series that ranges from diploid to dodecaploid, and is therefore ideal for studying the impact of genome duplication10, especially given its status as a model for cell and developmental biology11. X. laevis has a chromosome number (2n = 36) nearly double that of the Western clawed frog Xenopus (formerly Silurana) tropicalis (2n = 20) and most other diploid frogs12, and is proposed to be an allotetraploid that arose via the interspecific hybridization of diploid progenitors with 2n = 18, followed by subsequent genome doubling to restore meiotic pairing and disomic inheritance10,13 (see Supplementary Note 1 and Extended Data Fig. 1 for discussion of the Xenopus allotetraploidy hypothesis).
Here we provide evidence for the allotetraploid hypothesis by tracing the origins of the X. laevis genome from its extinct progenitor diploids. The two subgenomes are distinct and maintain separate recombinational identities. Despite sharing the same nucleus, we find that the subgenomes have evolved asymmetrically: one of the two subgenomes has experienced more intrachromosomal rearrangement, gene loss by deletion and pseudogenization, and changes in levels of gene expression and in histone and DNA methylation. Superimposed on these global trends are local gene family expansions and the alteration of gene expression patterns.
Assembly, annotation and karyotype
We sequenced the genome of the X. laevis inbred ‘J’ strain by whole-genome shotgun methods in combination with long-insert clone-based end sequencing, (Supplementary Note 2) and organized the assembled sequences into chromosomes using fluorescence in situ hybridization (FISH) of 798 bacterial artificial chromosome clones (BACs) and in vivo and in vitro chromatin conformation capture analysis (Supplementary Note 3 and Methods). These complementary methods produced a high-quality chromosome-scale draft that includes all previously known X. laevis genes and assigns >91% of the assembled sequence (and 90% of the predicted protein-coding genes) to a chromosomal location.
We annotated 45,099 protein-coding genes and 342 microRNAs using RNA sequencing (RNA-seq) from 14 developmental stages (including the oocyte stage) and 14 adult tissues and organs (Supplementary Note 4), analysis of histone marks associated with transcription, and homology with X. tropicalis and other tetrapods (Supplementary Note 5 and Methods). Of the X. laevis protein-coding genes, 24,419 can be placed in 2:1 or 1:1 correspondence with 15,613 X. tropicalis genes, defining 8,806 homoeologous pairs of X. laevis genes with X. tropicalis orthologues and 6,807 single copy orthologues. The remaining genes are members of larger gene families (such as olfactory receptor genes) whose X. tropicalis orthology is more complex.
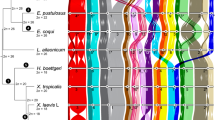
The X. laevis karyotype (Fig. 1a) reveals nine pairs of homoeologous chromosomes1,14,15. Each of the first eight pairs is co-orthologous to and named for a corresponding X. tropicalis chromosome, appending an L and S for the longer and shorter homoeologues, respectively16. XLA2L is the Z/W sex chromosome17, for which we determined a W-specific sequence in the q-subtelomeric region that includes the sex-determining gene dmw17 and a corresponding Z-specific haplotype. The homoeologous XLA2Sq, by contrast, has no such locus, and neither does XTR2 (Extended Data Fig. 2a and Supplementary Note 6). The ninth pair of homoeologues is a q–q fusion of proto-chromosomes homologous to XTR9 and XTR10, which probably occurred before allotetraploidization (Extended Data Fig. 2b–d and Supplementary Note 6). The S chromosomes are, on average, 13.2% shorter karyotypically16 and 17.3% shorter in assembled sequence than their L counterparts. The single nucleotide polymorphism rate in X. laevis is approximately 0.4%, far less than the approximately 6% divergence between homoeologous genes (Extended Data Fig. 1c and Supplementary Note 8.8).

a, Comparative cytogenetic map of XLA (Xenopus laevis) and XTR (Xenopus tropicalis) chromosomes. Magenta lines show relationships of chromosomal locations of 198 homoeologous gene pairs between XLA.L and XLA.S chromosomes, identified by FISH mapping using BAC clones (Supplementary Table 1 and Supplementary Note 3.1). Blue lines show relationships of chromosomal locations of orthologous genes between XTR chromosomes and (i) both XLA.L and XLA.S chromosomes (solid line) (lines between XLA.L and XLA.S are omitted), (ii) only XLA.L (dashed), or (iii) only XLA.S (dotted), which were taken from our previous studies14,15. Light blue lines indicate positional relationships of actr3 and lypd1 on XTR9q and rpl13a and rps11 on XTR10q with those on XLA9_10LS chromosomes (Supplementary Note 6.2). Double-headed arrows on the right of XLA.S chromosomes indicate the chromosomal regions in which inversions occurred. Ideograms of XTR and XLA chromosomes were taken from our previous reports15,16. b, Distribution of homoeologous genes (purple), singletons (grey) and subgenome-specific repeats across XLA1L (top) and XLA1S (bottom). Xl-TpL_harb is red, Xl-TpS_harb is blue, and Xl-TpS_mar is green. Purple lines mark homoeologous genes present in both L and S chromosomes, the black line marks the approximate centromere location on each chromosome. The homoeologous gene pairs, from left to right: rnf4, spcs3, intsl2, foxa1, sds, ap3s1, lifr, aqp7. Each bin is 3 Mb in size, with 0.5 Mb overlap with the previous bin. c, Chromosomal localization of the Xl-TpS_mar sequence with fluorescence in situ hybridization. Hybridization signals were only observed on the S chromosomes. Scale bar, 10 μm.
Subgenomes and timing of allotetraploidization
We reasoned that dispersed relicts of transposable elements specific to each progenitor would mark the descendent subgenomes in an allotetraploid (Fig. 2c and Extended Data Fig. 1). Three classes of DNA transposon relicts appeared almost exclusively on either the L or S chromosomes (Supplementary Note 7). Xl-TpL_harb and Xl-TpS_harb are subfamilies of miniature inverted-repeat transposable elements (MITE) of the PIF/harbinger superfamily18,19 whose relicts were almost completely restricted to L or S chromosomes, respectively (Fig. 1b and Extended Data Fig. 3a). Similarly, sequence relicts of the Tc1/mariner superfamily member Xl-TpS_mar (closely related to the fish MMTS subfamily20) were found almost exclusively on the S chromosomes (Fig. 1b), as confirmed by FISH analysis using Xl-TpS_mar as a probe (Fig. 1c and Supplementary Note 7.4; see Supplementary Note 7.3 for details on the rare elements that map to the opposite subgenome).

a, The distribution of pseudogene ages, as described in Supplementary Note 9 (top). Phylogenetic tree illustrating the different epochs in Xenopus (bottom), with times based on protein-coding gene phylogeny of pipids, including Xenopus, Pipa carvalhoi, Hymenochirus boettgeri and Rana pipiens (only Xenopus depicted). We date the speciation of X. tropicalis and the X. laevis ancestor at 48 Ma, the L and S polyploid progenitors at 34 Ma and the divergence of the polyploid Xenopus radiation at 17 Ma. Using these times as calibration points, we estimate bursts of transposon activity at 18 Ma (mariner, blue star) and 33–34 Ma (harbinger, red star). The purple star is the time of hybridization, around 17–18 Ma. b, Phylogenetic tree based on protein-coding genes of tetrapods, rooted by elephant shark (not shown). Alignments were done by MACSE (multiple alignment of coding sequences accounting for frameshifts and stop codons) and the maximum-likelihood tree was built by PhyML. Branch length scale shown at the bottom for 0.08 substitutions per site.The difference in branch length between Xenopus laevis-L and Xenopus laevis-S is similar to that seen between mouse and rat. Both subgenomes of X. laevis have longer branch lengths than X. tropicalis.
The L and S chromosome sets therefore represent the descendants of two distinct diploid progenitors, confirming the allotetraploid hypothesis despite the absence of extant progenitor species. Analysis of synonymous divergence of protein-coding genes suggests that the L and S subgenomes diverged from each other around 34 Ma (T2) and from X. tropicalis around 48 Ma (T1) (Fig. 2a), consistent with prior gene-by-gene estimates from transcriptomes21,22,23,24 (Supplementary Note 8, Extended Data Fig. 4 and Methods). L- and S-specific transposable elements were active around 18–34 Ma, indicating that the two progenitors were independently evolving diploids during that period (Fig. 2a, Supplementary Note 7.5 and Extended Data Fig. 3). More recent transposon activity is more uniformly distributed across the L and S chromosomes (not shown). Finally, consistent with a common origin for tetraploid Xenopus species, we can clearly identify orthologues of L and S genes in whole-genome sequences of a related allotetraploid frog, X. borealis, and estimate the X. laevis–X. borealis divergence to be around 17 Ma (T3). These considerations constrain the allotetraploid event to around 17–18 Ma (T*). This timing is consistent with other estimates of the radiation of tetraploid Xenopus species, which are presumed to emerge from the bottleneck of a shared allotetraploid founder population23,24.
Karyotype stability
With the exception of the chromosome 9–10 fusion, X. laevis and X. tropicalis chromosomes have maintained conserved synteny since their divergence around 48 Ma (Fig. 1a, b). The absence of inter-chromosomal rearrangements is consistent with the relative stability of amphibian and avian karyotypes compared to those of mammals25, which typically show dozens of inter-chromosome rearrangements26. It also contrasts with many plant polyploids, which can show considerable inter-subgenome rearrangement27. The distribution of L- and S-specific repeats along entire chromosomes implies the absence of crossover recombination between homoeologues since allotetraploidization, presumably because the two progenitors were sufficiently diverged to avoid meiotic pairing between homoeologous chromosomes, though we cannot rule out very limited localized inter-homoeologue exchanges (Supplementary Note 7).
The extensive collinearity between homologous X. laevis L and X. tropicalis chromosomes (Fig. 1a) implies that they represent the ancestral chromosome organization. In contrast, the S subgenome shows extensive intra-chromosomal rearrangements, evident in the large inversions of XLA2S, XLA3S, XLA4S, XLA5S and XLA8S, as well as shorter rearrangements (Fig. 1a). The S subgenome has also experienced more deletions. For example, the 45S pre-ribosomal RNA gene cluster is found on X. laevis XLA3Lp, but its homoeologous locus on XLA3Sp is absent (Extended Data Fig. 5a). Extensive small-scale deletions (Extended Data Fig. 5b) reduce the length of S chromosomes relative to their L and X. tropicalis counterparts (see below).
Response of subgenomes to allotetraploidy
Redundant functional elements in a polyploid are expected to rapidly revert to single copies through the fixation of disabling mutations and/or loss28 unless prevented by neofunctionalization8, subfunctionalization26, or selection for gene dosage29. Differential gene loss between homoeologous chromosomes is sometimes referred to as ‘genome fractionation’30,31,32 (Supplementary Note 1). At least 56.4% of the protein-coding genes duplicated by allotetraploidization have been retained in the X. laevis genome (Supplementary Note 10; 60.2% if genes on unassigned short scaffolds are included). Previous studies that relied on cDNA21 and expressed sequence tag (EST) surveys22,33,34 observed far lower rates of retention, probably owing to sampling biases from gene expression (Supplementary Note 8.2).
Even higher retention rates were found for homoeologous microRNAs (156 out of 180, 86.7%), similar to the salmonid-specific duplication5, and both primary copies are expressed for intergenic homoeologous microRNAs (Supplementary Note 8.6 and Extended Data Fig. 5e). Pan-vertebrate putatively cis-regulatory conserved non-coding elements (CNEs)35 were also highly retained (541 out of 550, 98.4%; Supplementary Note 8.7 and Table 1). CNEs conserved between X. laevis and X. tropicalis, however, were retained at a significantly lower rate (49%, P ≤ 1 × 1050; Table 1 and Supplemental Table 3). Longer genes (by genomic span, exon number or coding length) were more likely to be retained (Wilcoxon signed-rank test, P ≤ 10−5; Supplementary Note 10.5 and Extended Data Fig. 5 h–j), broadly consistent with the idea that longer genes have more independently mutable functions and are therefore more susceptible to subfunctionalization and subsequent retention36.
Genes have been lost asymmetrically between the two subgenomes of X. laevis. Similar results have been reported for some plant polyploids30 but not in rainbow trout5. For X. laevis protein-coding genes with clear 1:1 or 2:1 orthologues in X. tropicalis, we found that significantly more genes were lost from the S subgenome (31.5%) than from the L subgenome (8.3%; χ2 test P = 2.23 × 10−50; Supplementary Table 2), with the same trend for other types of functional elements, such as histone H3 lysine 4 trimethylation (H3K4me3)-enriched promoters and p300-bound enhancers (Table 1). Across most of the genome, genes appeared to be lost independently of their neighbours, as runs of gene losses were nearly geometrically distributed (Fig. 3a, right). We did observe some large block deletions (for example, several olfactory clusters (Extended Data Fig. 5b) and a few unusually long blocks of functionally unrelated genes that were retained in two copies without loss (Fig. 3a, left)).

a, Distributions of consecutive retentions (left) and deletions (right) in the L (red) and S (blue) subgenomes. The distributions were fit using the equation y = a × (ebx) + c × (edx). The y axis is shown on a log scale. Significant differences were seen between L and S subgenomes in both distributions (Student’s t-test, retention, P = 3.6 × 10−22; deletion, P = 4.5 × 10−84). b, Evolutionary conservation of the Xenopus major histocompatibility complex (MHC) and differential MHC silencing on the two X. laevis subgenomes. Selected gene names shown above. The ‘Adaptive MHC’ encodes tightly-linked essential genes involved in antigen presentation to T cells; this group of genes is the primordial linkage group and has been preserved in most non-mammalian vertebrates, including Xenopus. Differential gene silencing is particularly pronounced, as four genes around the class I gene are functional on the S chromosome, but absent (dma and dmb (MHC-class II domain alpha and beta) or pseudogenes (ring3, really interesting new gene 3; lmp2, large multifunctional proteasome 2) on the L chromosome. The gene map is not to scale; pseudogenes are noted as indicated. HSA, Homo sapiens MHC; XLA Xenopus laevis MHC; GGA Gallus gallus (chicken) MHC. Refer to the Supplementary Table 8 for a more detailed MHC map. TAPBP, TAP binding protein, or tapasin; TAP2, antigen peptide transporter 2; CFB, complement factor B and TNFa, tumor necrosis factor α. c, Hox gene clusters. X. laevis retains eight hox clusters, consisting of pairs of hoxa, b, c and d clusters, on L and S chromosomes. even-skipped genes (evx1 or evx2) are positioned flanking hoxa and hoxd clusters. hox genes are classified into four groups namely, labial, proboscipedia, central and posterior groups. Note that hoxb2.L (2p, black) is a pseudogene. d, Syntenies around the mix gene family. Abbreviations for species and chromosome numbers: HSA1, H. sapiens;; GGA3 G. gallus (chicken); XTR5, X. tropicalis; XLA5L and XLA5S, X. laevisL and S subgenomes); DRE20, D. rerio (zebrafish);. Each Xenopus (sub)genome experienced its own independent expansion of the family (see Extended Data Fig. 5 for details).
Many lost genes were simply deleted, as demonstrated by significantly shorter distances between conserved flanking genes in the other subgenome and in X. tropicalis. Both the size and number of deletions were greater on the S subgenome (Extended Data Fig. 5c). We identified 985 ‘unitary’ (that is, non-retrotransposed) pseudogenes out of 1,531 loci examined in detail. This 64% detection rate was similar between subgenomes in X. laevis and comparable to that reported in trout5. Based on the accumulation of non-synonymous mutations37 we estimated that most of these pseudogenes escaped evolutionary constraint around 15 Ma (Fig. 2a and Extended Data Fig. 6), consistent with the onset of extensive redundancy in the allotetraploid, although the precision of our pseudogene age estimates is low (Supplementary Note 9). Most pseudogenes showed no evidence of expression, but of 769 pseudogenes longer than 100 bp, 133 (17.2%) showed residual expression (Extended Data Fig. 6). Conversely, among homoeologous gene pairs, we found 760 for which one member had little to no expression across our 28 sampled conditions. Although these retained some gene structure (start and stop codon, no frame shifts, good splice signals), they showed increased rates of amino acid change and appeared to be under relaxed selection (Extended Data Fig. 5f). We called these nominally dying genes ‘thanagenes’ (Supplementary Note 12.5). Reduced expression may be due to mutated cis-regulatory elements, as exemplified by the six6 gene pair (Fig. 4e, Extended Data Fig. 8 g–i and Supplementary Note 13.1).

a, Comparison of L and S gene loss by KEGG categories (left) and tissue-weighted gene co-expression network analysis (WGCNA) categories (right) (Supplementary Note 10.1). Blue line denotes expected L or S loss based on genome-wide average (56.4%). Red points denote functional categories showing a high degree of loss. Magenta points denote functional categories showing a high degree of retention (χ2 test, P < 0.01). b, Box plot of log10(LTPM/STPM) for homoeologous gene pairs, zoomed in to show medians. Ovary and maternally controlled developmental time points (left, light blue and dark blue bars, respectively), zygotically controlled developmental time points and adult tissues (right, red and green bars, respectively). Red line, equal ratio log10(1). On average, maternal datasets express the L gene of a homoeologous pair 12% more strongly than the S gene (median = 0%), whereas zygotic tissues and time points express the L gene of a homoeologous pair 25% more strongly than the S gene (median = 1.8%). The difference between the mean and medians is explained by many genes with large differences between homoeologues (Extended Data Fig. 8c). c, d, Developmental expression plot (left) and epigenetic landscape (right) surrounding hoxb4 (c) and numbl (d). Right, genomic profiles of H3K4me3 (green), p300 (yellow), RNA polymerase II (RNAP II; d, purple) and H3K36me3 (d, blue) ChIP–seq tracks, as well as DNA methylation levels determined by whole-genome bisulfite sequencing (grey). Gene annotation track shows hoxb4 (c) and numbl (d) genes on L (top) and S. Grey denotes conservation between L and S genomic sequences. d, The small amount of expression seen in maternal numbl and numbl.L is consistent between replicates. Gene expression is measured in transcripts per million mapped reads (TPM). e, Representative embryos with GFP expression, as detected by in situ hybridization at stages 32–33, driven by six6. L-CNE or six6. S-CNE linked to a basal promoter-GFP cassette (six6.L-CNE:GFP and six6. S-CNE:GFP, respectively). Embryos were 4,250–4,450 μm. Semi-quantitative image analysis revealed a substantial difference in average expression level; the expression driven by six6. S-CNE (n = 27) was 0.6-fold weaker than that by six6.L-CNE in the eye region (n = 32). Given eye-specific patterns of their endogenous expression, the six6 genes probably have additional silencers for restricting enhancer activity of the CNEs in the eye.
Although tetraploidy created two ‘copies’ of nearly every gene, additional gene copies were continually produced by tandem duplication (Fig. 3d and Extended Data Fig. 7). The number of tandem clusters was greater in X. tropicalis than in the X. laevis L subgenome, which in turn was greater than in the S subgenome (Supplementary Note 11.1). Although tandem duplication was faster in X. tropicalis than in X. laevis, there was also a higher rate of loss. Since tandem duplications and deletions occur by unequal crossing over during meiosis, these differing rates were consistent with the shorter generation time of X. tropicalis (Extended Data Fig. 7 f, g). The mean time to loss of an old tandem duplicate is around 40 Ma in X. laevis (on either subgenome) compared to around 16 Ma in X. tropicalis. Homoeologous gene loss and tandem duplication can combine to yield complex histories for some gene families. We discuss how these families contribute to the literature on whole-genome duplication evolution in Supplementary Notes 10 and 13.
Functional patterns of gene retention and loss
We found preferential retention or loss of many functional categories (Fig. 4a, Extended Data Figs 4e, 9, 10 and Supplementary Note 13). DNA binding proteins, components of developmentally regulated signalling (TGFβ, Wnt, Hedgehog and Hippo) and cell cycle regulation pathways were retained at a substantially higher rate (>90%) than average (Extended Data Fig. 10). Genes retained in multiple copies after the ancient vertebrate genome duplication were also more likely to be retained as homoeologues in X. laevis (Supplementary Note 10.4), similar to teleost and trout genome duplications5. We found nearly complete retention of 37 out of 38 duplicated genes in the four pairs of homoeologous Hox clusters, with a single pseudogene (Fig. 3c). High rates of homoeologue retention in most genes in these categories suggest that stoichiometrically controlled expression levels may be needed, or subfunctionalization of homoeologues may have occurred, either in their expression domain or in their target specificity.
Conversely, homoeologous genes in other functional categories have been lost at a higher rate, presumably because of a corresponding lack of selection for dosage. For example, genes involved in DNA repair were lost at a high rate (79%) (Supplementary Note 10.1). This is consistent with relaxed selection for repair in the immediate aftermath of allotetraploidy, when all genes were present in four copies per somatic cell5. Other metabolic categories were also prone to loss, presumably because single loci encoding enzymes were sufficient38. Genomic regions with notable loss include the major histocompatibility complex genes on the S subgenome (Fig. 3b) and several olfactory receptor clusters (Extended Data Fig. 5b). We hypothesize that homoeologous genes may be functionally incompatible in these cases, leading to en bloc deletion in response to selection pressure. Specific case studies of duplicate gene retention and loss are detailed in Extended Data Figs 9, 10 and Supplementary Note 13.
Evolution of gene expression
Gene expression is also a predictor of retention, whereby more highly expressed genes are more likely to be retained (Extended Data Fig. 8b), similar to results seen in Paramecium39,40. Developmentally regulated genes whose expression levels peak at the maternal zygotic transition (MZT) or during neural differentiation were retained at higher levels (P < 0.01), based on gene expression networks constructed from developmental and adult tissue expression (Methods, Fig. 4a (right), Extended Data Fig. 10e and Supplementary Note 12.3). We speculate that the exceptional retention of developmentally regulated genes is due to selection for stoichiometric dosage of these factors, and in some cases higher expression in the physically larger allotetraploid cells and embryos relative to those of diploid frogs, although a propensity36 of these genes for sub- or neofunctionalization could also have contributed. In the adult, genes whose expression peaks in the brain and eye were also retained at higher levels (Fig. 4b).
In X. laevis, the expression of homoeologues was highly correlated (Extended Data Fig. 8a), showing that the overall expression of homoeologues diverged similarly to that of orthologues between Xenopus species41. Many homoeologous pairs, however, were differentially expressed throughout development or across adult tissues, either in a spatiotemporal pattern (a form of subfunctionalization36; Supplementary Note 12.4 and Extended Data Fig. 8d–f) or in the same pattern but with differing expression levels. When homoeologous gene pairs were both expressed, the average L copy expression level was approximately 25% higher than that of the S copy consistently across adult tissues and after the MZT42 (Fig. 4b and Supplementary Note 12.2). Excess L expression, however, averaged only around 12% in oocyte and early pre-MZT stages, suggesting that the two subgenomes were more evenly expressed as maternal transcripts but developed an increased asymmetry after the MZT. Strikingly, we found 391 cases in which one homoeologue had no detectable maternal mRNA (oocytes, egg and stage 8; Fig. 4c, d and Extended Data Fig. 8c). Compared to similar transcript data from X. tropicalis, we found cases of an apparent loss of expression (‘maternal subfunctionalization’, that is, X. tropicalis and one X. laevis gene was expressed, whereas the other X. laevis gene was silenced pre-MZT) in 238 genes (for example numbl.S). We also found gains of expression (‘maternal neofunctionalization’, that is, the X. tropicalis gene was not expressed maternally, but one X. laevis gene was expressed) in 153 genes (for example hoxb4.L). We did not see such a large divergence in other expression domains (Supplementary Note 12.2 and Extended Data Fig. 8c), suggesting a higher level of plasticity of maternal mRNA regulation between X. laevis homoeologues, similar to the trend seen between Xenopus species41.
Overall, thousands of homoeologue pairs have either divergent spatiotemporal patterns or similar patterns with differing expression levels. Such homoeologue pairs differed in substitution rate and coding sequence length difference more than those that were similar in expression (Supplementary Note 12.4 and Extended Data Fig. 8d–f), a pattern that was also found in trout homoeologous pairs5. These expression differences can largely be attributed to changes in epigenetic regulation (Random Forest classification; ROC area under the curve 0.78), with changes in H3K4me3 and DNA methylation contributing the most explanatory power among our epigenetic variables (Supplementary Note 14). Detailed comparison of the two subgenomes will facilitate identification of specific sequences that control cis-regulatory differences between homoeologues.
Conclusion
The two subgenomes of Xenopus laevis have evolved asymmetrically, with the L subgenome more consistently resembling the ancestral condition and the S subgenome more disrupted by deletion and rearrangement. Asymmetric gene loss has been observed in allopolyploid plants30 and yeast43 at the segmental level, but it has not been shown directly that similarly fractionated segments derive from the same progenitor (Fig. 1c). Our results are consistent with the idea that optimized gene expression levels are an important force affecting gene retention following polyploidy39,40. The asymmetry between the L and S subgenomes could have been the result of an intrinsic difference between their diploid progenitors. Alternately, the remodelling of the S genome could have been a response to the L–S merger itself, a ‘genomic shock’44 resulting from the activation of transposable elements (Fig. 2a and Supplementary Note 8.5). The popularity of Xenopus as a model for the study of vertebrate development, cell biology and immunology is now extended to a model for vertebrate polyploidy.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Notation and terminology
‘Homoeologous’ chromosomes are anciently orthologous chromosomes that diverged by speciation but were reunited in the same nucleus by a polyploidization event. They are a special case of paralogues. Homoeologous genes are sometimes called ‘alloalleles’ to emphasize their role as alternate forms of a gene, but since homoeologues are unlinked and assort independently, we do not use this terminology. Similarly, loss of homoeologous genes is sometimes referred to as ‘diploidization’. We prefer the simpler and more descriptive term ‘gene loss’. Note that an allotetraploid such as Xenopus laevis has two related subgenomes, but these subgenomes are each transmitted to progeny via conventional disomic inheritance. So immediately after allotetraploidization, the new species is already genetically diploid. This is clearly the case for X. laevis, since we find no evidence for recombination between homoeologous chromosomes, which would create new sequences with mixed ‘L’ and ‘S’ type transposable elements.
Sequencing and assembly
DNA was extracted from the blood of a single female from the inbred J-strain for whole-genome shotgun sequencing. We generated 4.6 billion paired-end Illumina reads from a range of inserts and used Sanger dideoxy sequencing to obtain fosmid- and bacterial artificial chromosome (BAC)-end pairs and full BAC sequences. We used meraculous45 as the primary genome assembler. See supplementary notes for more detailed information.
Chromosome scale organization
We identified 798 BACs containing genes of interest distributed across the Xenopus genome and performed fluorescence FISH to assign these BACs to specific chromosomes based on Hoechst 33258-stained late-replication banding patterns (Supplementary Table 1). Tethered chromatin conformation capture (TCC)46 and in vitro chromatin conformation capture47 were performed as previously described, and assembled with HiRise47.
Characterization of sex locus
Sex determination in X. laevis follows a female heterogametic ZZ/ZW system48. We fully sequenced BAC clones representing both W and Z haplotypes, and identified both W- and Z-specific sequences (Extended Data Fig. 2a). The existence of the Z-specific sequence was unexpected and therefore verified by PCR analysis using specific primer sets and DNA from gynogenetic frogs having either W or Z loci.
Gene annotation
We made use of extensive previously generated transcriptome data for X. laevis and X. tropicalis, including 697,015 X. laevis EST sequences49. In addition, more than 1 billion RNA-seq reads were generated for this project from 14 oocyte/developmental stages and 14 adult tissues from J-strain X. laevis (Supplementary Note 4). These data were combined with homology and ab initio predictions using the Joint Genome Institute’s integrated gene call pipeline (see Supplementary Notes 4 and 8 for more details).
Characterization of subgenome-specific transposable elements
We found subgenome-specific repeats using a RepeatMasker50 result. The repeats were used to reconstruct full-length subgenome specific transposon sequences. The specific transposons, Xl-TpL_harb, Xl-TpS_harb and Xl-TpS_mar, were classified on the basis of their target site sequence and terminal inverted repeat (TIR) sequences. The coverage lengths of the transposons on each chromosome were calculated from the results of BLASTN search (E < 10−5) using the consensus sequences of the transposons as queries. The chromosomal distribution of the Xl-TpS_mar was revealed by a FISH analysis (Supplementary Note 7.4).
Phylogeny, divergence time, and evolutionary rates
We used Hymenochirus boettgeri, Pipa carvalhoi and Rana pipiens sequences as outgroups to estimate the evolutionary rate of duplicated genes in X. laevis and their relationship to X. tropicalis. See Supplementary Notes 7 and 8 for more detail.
Deletions and pseudogenes
Pseudogene sequences contain various defects including premature stop codons, frameshifts, disrupted splicing, and/or partial coding deletions. 985 pseudogenes were identified among 1,531 ‘2-1-2 regions’, with the others deleted or rendered unidentifiable by mutation. 368 out of 985 could be timed, based on the accumulation of non-synonymous and synonymous substitution between a pseudogene, its homoeologue and its orthologue in X. tropicalis, providing a time since the loss of constraint for each pseudogene37. See Supplementary Note 9 for additional details.
Functional annotation of genes
We used several bioinformatic methods and high-throughput datasets to assign functional annotations to Xenopus genes. Protein domains were assigned using InterPro (including PFAM and Panther)51 and KEGG52. Gene Ontology was assigned using InterPro2Go51. We identified genes that encode mitochondrial proteins by mapping the MitoCarta53 database from mouse to the most recent X. tropicalis proteome. Xenopus genes associated with germ plasm were manually curated using the extensive Xenopus literature (Supplementary Note 13).
Gene expression
We analysed transcriptome data generated for 14 oocyte/developmental stages and 14 adult tissues in duplicate except for oocyte stages (see Supplementary Note 4). Expression levels were measured by mapping paired-end RNA-seq reads to predicted full length cDNA and reporting transcripts per one million mapped reads (TPM). We consider the limit of detectable expression to be TPM >0.5. Co-expression modules were defined by weighted gene correlation network analysis (WGCNA) clustering54 (Supplementary Note 12).
Epigenetic analysis
We determined DNA methylation levels (DNAme) by whole genome bisulfite sequencing and used ChIP–seq to generate profiles of the promoter mark histone H3 lysine 4 trimethylation (H3K4me3), the transcription elongation mark H3K36me3, as well as RNA polymerase II (RNAPII) and the enhancer-associated co-activator p300. To test which regulatory features would contribute most to the L versus S expression differences, we applied a Random Forest machine learning algorithm to analyse differential expression between the L and S homoeologues (See Supplementary Note 14).
Data availability
The XENLAv9.1 genome assembly and annotation are deposited at NCBI (accession LYTH00000000. The DNA read libraries of X. laevis and X. borealis were deposited at the Sequence Read Archive under accessions SRP071264 and SRP070985, respectively. Datasets of the X. laevis RNA-seq short reads were deposited in NCBI Gene Expression Omnibus (accession number GSE73430 for stages, GSE73419 for tissues). Datasets of the Hymenochirus RNA-seq short reads were deposited in NCBI GEO (accession number GSE76089). The epigenetic data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE76059 for ChIP–seq. MethylC-seq data are accessible through GEO Series accession number GSE76247. The sequence data from BAC and fosmid clones have been deposited to DDBJ/GenBank/EMBL under the accession numbers: (i) GA131508–GA227532, GA228275–GA244139, GA244852–GA274229, GA274976–GA275712, GA277157–GA344957, GA345673–GA350926 and GA351685–GA393223 for the XLB1 end-sequences; (ii) GA720358–GA756840 for the XLB2 end-sequences; (iii) GA756841–GA867435 for the XLFIC end-sequences and (iv) AP012997–AP013026,AP014660–AP014679, AP017316 and AP017317 for the finished BAC/fosmid sequences.
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
References
Van de Peer, Y., Maere, S. & Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732 (2009)
Holland, P. W., Garcia-Fernàndez, J., Williams, N. A. & Sidow, A. Gene duplications and the origins of vertebrate development. Development Suppl., 125–133 (1994)
Muller, H. J. Why polyploidy is rarer in animals than in plants. Am. Nat. 59, 346–353 (1925)
Orr, H. A. ‘Why polyploidy is rarer in animals than in plants’ revisited. Am. Nat. 136, 759–770 (1990)
Berthelot, C. et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 5, 3657 (2014)
Woods, I. G. et al. The zebrafish gene map defines ancestral vertebrate chromosomes. Genome Res. 15, 1307–1314 (2005)
Glasauer, S. M. K. & Neuhauss, S. C. F. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics 289, 1045–1060 (2014)
Otto, S. P. The evolutionary consequences of polyploidy. Cell 131, 452–462 (2007)
Ohno, S. Evolution by Gene Duplication (Springer, 1970)
Kobel, H. R. & Du Pasquier, L. Genetics of polyploid Xenopus. Trends Genet. 2, 310–315 (1986)
Harland, R. M. & Grainger, R. M. Xenopus research: metamorphosed by genetics and genomics. Trends Genet. 27, 507–515 (2011)
Kuramoto, M. A list of chromosome numbers of anuran amphibians. Bull. Fukuoka Univ. Educ. 39, 83–127 (1990)
Bisbee, C. A., Baker, M. A., Wilson, A. C., Haji-Azimi, I. & Fischberg, M. Albumin phylogeny for clawed frogs (Xenopus). Science 195, 785–787 (1977)
Uno, Y., Nishida, C., Takagi, C., Ueno, N. & Matsuda, Y. Homoeologous chromosomes of Xenopus laevis are highly conserved after whole-genome duplication. Heredity 111, 430–436 (2013)
Uno, Y. et al. Inference of the protokaryotypes of amniotes and tetrapods and the evolutionary processes of microchromosomes from comparative gene mapping. PLoS One 7, e53027 (2012)
Matsuda, Y. et al. A new nomenclature of Xenopus laevis chromosomes based on the phylogenetic relationship to Silurana/Xenopus tropicalis. Cytogenet. Genome Res. 145, 187–191 (2015)
Yoshimoto, S. et al. A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc. Natl Acad. Sci. USA 105, 2469–2474 (2008)
Zhang, X. et al. P instability factor: an active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. Proc. Natl Acad. Sci. USA 98, 12572–12577 (2001)
Jurka, J. & Kapitonov, V. V. PIFs meet Tourists and Harbingers: a superfamily reunion. Proc. Natl Acad. Sci. USA 98, 12315–12316 (2001)
Ahn, S. J., Kim, M.-S., Jang, J. H., Lim, S. U. & Lee, H. H. MMTS, a new subfamily of Tc1-like transposons. Mol. Cells 26, 387–395 (2008)
Morin, R. D. et al. Sequencing and analysis of 10,967 full-length cDNA clones from Xenopus laevis and Xenopus tropicalis reveals post-tetraploidization transcriptome remodeling. Genome Res. 16, 796–803 (2006)
Hellsten, U. et al. Accelerated gene evolution and subfunctionalization in the pseudotetraploid frog Xenopus laevis. BMC Biol. 5, 31 (2007)
Bewick, A. J., Chain, F. J. J., Heled, J. & Evans, B. J. The pipid root. Syst. Biol. 61, 913–926 (2012)
Cannatella, D. Xenopus in space and time: fossils, node calibrations, tip-dating, and paleobiogeography. Cytogenet. Genome Res. 145, 283–301 (2015)
Voss, S. R. et al. Origin of amphibian and avian chromosomes by fission, fusion, and retention of ancestral chromosomes. Genome Res. 21, 1306–1312 (2011)
Ferguson-Smith, M. A. & Trifonov, V. Mammalian karyotype evolution. Nat. Rev. Genet. 8, 950–962 (2007)
Langham, R. J. et al. Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 166, 935–945 (2004)
Haldane, J. B. S. The part played by recurrent mutation in evolution. Am. Nat. 67, 5–19 (1933)
Birchler, J. A. & Veitia, R. A. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl Acad. Sci. USA 109, 14746–14753 (2012)
Schnable, J. C., Springer, N. M. & Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl Acad. Sci. USA 108, 4069–4074 (2011)
Sankoff, D., Zheng, C. & Wang, B. A model for biased fractionation after whole genome duplication. BMC Genomics 13 (Suppl. 1), S8 (2012)
Garsmeur, O. et al. Two evolutionarily distinct classes of paleopolyploidy. Mol. Biol. Evol. 31, 448–454 (2014)
Sémon, M. & Wolfe, K. H. Preferential subfunctionalization of slow-evolving genes after allopolyploidization in Xenopus laevis. Proc. Natl Acad. Sci. USA 105, 8333–8338 (2008)
Chain, F. J. J., Dushoff, J. & Evans, B. J. The odds of duplicate gene persistence after polyploidization. BMC Genomics 12, 599 (2011)
Lee, A. P., Kerk, S. Y., Tan, Y. Y., Brenner, S. & Venkatesh, B. Ancient vertebrate conserved noncoding elements have been evolving rapidly in teleost fishes. Mol. Biol. Evol. 28, 1205–1215 (2011)
Force, A. et al. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545 (1999)
Meredith, R. W., Gatesy, J., Murphy, W. J., Ryder, O. A. & Springer, M. S. Molecular decay of the tooth gene Enamelin (ENAM) mirrors the loss of enamel in the fossil record of placental mammals. PLoS Genet. 5, e1000634 (2009)
Kondrashov, F. A. & Koonin, E. V. A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends Genet. 20, 287–290 (2004)
Aury, J.-M. et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444, 171–178 (2006)
Gout, J.-F., Kahn, D., Duret, L. & Paramecium Post-Genomics Consortium. The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution. PLoS Genet. 6, e1000944 (2010)
Yanai, I., Peshkin, L., Jorgensen, P. & Kirschner, M. W. Mapping gene expression in two Xenopus species: evolutionary constraints and developmental flexibility. Dev. Cell 20, 483–496 (2011)
Langley, A. R., Smith, J. C., Stemple, D. L. & Harvey, S. A. New insights into the maternal to zygotic transition. Development 141, 3834–3841 (2014)
Marcet-Houben, M. & Gabaldón, T. Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker’s yeast lineage. PLoS Biol. 13, e1002220 (2015)
McClintock, B. The significance of responses of the genome to challenge. Science 226, 792–801 (1984)
Chapman, J. A. et al. Meraculous: de novo genome assembly with short paired-end reads. PLoS One 6, e23501 (2011)
Chen, L. et al. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–98 (2011)
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016)
Chang, C. Y. & Witschi, E. Genic control and hormonal reversal of sex differentiation in Xenopus. Proc. Soc. Exp. Biol. Med. 93, 140–144 (1956)
Gilchrist, M. J. From expression cloning to gene modeling: the development of Xenopus gene sequence resources. Genesis 50, 143–154 (2012)
Smit, A. F. A., Hubley, R & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org.
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015)
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014)
Calvo, S. E., Clauser, K. R. & Mootha, V. K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257 (2016)
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008)
Edwards, N. S. & Murray, A. W. Identification of Xenopus CENP-A and an associated centromeric DNA repeat. Mol. Biol. Cell 16, 1800–1810 (2005)
McLysaght, A. et al. Ohnologs are overrepresented in pathogenic copy number mutations. Proc. Natl. Acad. Sci. USA 111, 361–366 (2014)
Tan, M. H. et al. RNA sequencing reveals a diverse and dynamic repertoire of the Xenopus tropicalis transcriptome over development. Genome Res. 23, 201–216 (2013)
Acknowledgements
Please see Supplementary Note 15 for funding information.
Author information
Authors and Affiliations
Contributions
R.M.H., M.T., D.S.R., G.J.C.V., A.Fuj., A.S., A.M.S., T.Kw., Y.U., A.Fuk., M.K. and H.Og. provided project leadership, with additional project management from Y.M., M.A., Y.Iz., N.U., J.Sh., J.B.W., E.M.M., J.Sc., A.M.Z., P.D.V. and M.I. Y.Iz. and J.R. inbred J strain frogs. A.T., C.H., A.Fuj., J.G., J.C., J.K., J.Sh., T.Mit. and J.B.L. generated genome sequence data. J.A.C., A.M.S., T.Kw., J.J., A.Fuk., M.T. and J.Sc. performed genome assembly and validation. S.T., T.Kw., A.M.S., Y.S., T.T., A.T., A.S. and M.T. generated and analysed the transcriptome data. A.M.S., T.Kw., S.J.v.H. and S.S. generated the annotations. Manual validation of annotation was done by H.Og., S.T., A.Fuk., A.S., M.K., H.Oc., T.T., T.Mic., M.W., T.Ki., Y.O., S.Ma., Y.H., T.N., Y.Y., J.F., K.B., V.L., D.W.D., M.T. and K.K. K.M., A.M.S. and R.H. generated the Hymenochirus transcriptome data. A.M.S. performed the phylogenetic analysis, with contributions from S.V.M. and U.H. M.W., A.Fuk., S.Ma., Y.U., Y.M. and M.T. performed the chromosome structure analysis. A.M.S., A.H., O.S., J.C. and Y.U. studied the transposable elements. BAC-FISH was performed by Y.U., A.Fuk., M.K., A.T., S.T., H.Og., H.Oc., Y.K., T.T., T.M., M.W., T.Ki., Y.O., Y.H., T.S.Y., C.T., T.N., A.S., Y.M., N.U., M.A., Y.Iz., A.Fuj. and M.T. I.Q., S.H., N.P. and J.St. generated and analysed the chromatin conformation capture data and their use in long-range scaffolding. H.Og. and H.Oc. performed the transgenic enhancer analysis. S.J.v.H, G.G., S.S.P., I.v.K., O.B., R.L., and G.J.C.V. generated and analysed the epigenetic data. A.S., A.M.S., T.Ki., M.K., M.T., Y.O., T.T., A.Fuk., M.W., T.Mic., D.W.H., T.N. and L.D. conducted the gene and pathway analysis. D.S.R., A.M.S., T.Kw., R.M.H., M.T., A.S., Y.U., G.J.C.V., M.K., U.H., S.J.v.H., A.Fuk., A.H., O.S., H.Og., T.T., I.Q., J.K., Y.O., S.T., M.W., T.Mic., A.T., H.Oc., T.Ki., S.Maw., Y.S., T.N., Y.Iz. and M.F.F. wrote the paper and supplementary notes, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
Dovetail Genomics LLC is a commercial entity developing genome assembly methods. N.P. and J.St. are employees of Dovetail Genomics, and D.S.R. is a scientific advisor to and minor investor in Dovetail.
Additional information
NCBI (LYTH00000000). Sequence Read Archive (SRP071264, SRP070985). NCBI Gene Expression Omnibus ( GSE73430, GSE73419, GSE76089, GSE76059, GSE76247). DDBJ/GenBank/EMBL ( AP017316 and AP017317).
Reviewer Information Nature thanks C. Amemiya, S. Burgess and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Allotetraploidy and assembly.
a–e, Scenarios for allotetraploid formation from distinct ancestral diploid species A and B. Horizontal single lines indicate normal gametes, horizontal double lines indicate unreduced gametes; black square represents fertilization; vertical double lines indicate spontaneous (somatic) genome doubling. a, (i) Fusion of unreduced gametes from species A and B. (ii) Interspecific hybridization followed by spontaneous doubling. (iii) Fusion of unreduced gametes produced by interspecific hybrids. (iv) Interspecific hybrids produce unreduced gametes, which fuse with normal gametes from species A. The resulting triploid again produces unreduced gametes, which fuse with normal gametes from species B. (v) Unreduced gamete from species A fuses with normal gamete from species B. The resulting AAB triploid produces unreduced gametes that are fertilized by normal gametes species B. See Supplementary Note 1.1 for a more detailed discussion. b, History of the J strain. See Supplementary Note 2.1 for details. The years of events and generation numbers (such as frog transfer to another institute, establishment of homozygosity, construction of materials) are indicated in the scheme. Generation numbers are estimates due to loss of old breeding records. c, The nucleotide distance of orthologues (green), homoeologues (red) and alleles (blue) is discussed in Supplementary Note 8.7. The distances are shown on a log scale to differentiate between the distributions. d, Frequency histogram showing the number of 51-mers with specified count in the shotgun dataset. The prominent peak implies that each genomic locus is sampled 29× in 51-mers. Note the absence of a feature at twice this depth, indicating that homoeologous features with high identity are rare. e, Cumulative proportion of 51-mers as a function of relative depth (that is, depth/29). Relative depth provides an estimate of genomic copy number. The rapid rise at relative depth 1 implies that 70–75% of the X. laevis genome is a single copy with respect to 51-mers. The remainder of the genome is primarily concentrated in repetitive sequences with copy number > 100. Note logarithmic scale. f, The contact map of 85,260 TCC read pairs for JGIv72.000090484.chr4S. Read pairs were binned at 10-kb intervals. For each read pair, the forward and reverse reads map with a map quality score of at least 20. g, The contact map of 85,260 Chicago read pairs for JGIv72.000090484.chr4S, a 3.1-Mb scaffold in the XENLA_JGI_v72 assembly. h, The insert distribution of TCC and Chicago read pairs that map to the same scaffold of XENLA_JGI_v72 with a map quality score of at least 20. The x axis is the read pair separation distance. The y axis is the counts for that bin divided by the total number of reads. The bins are 1 kb.
Extended Data Figure 2 Chromosome structure.
a, Structure of the sex chromosome of X. laevis (XLA2L) and comparison with XLA2S and XTR2. The W version of XLA2L harbours a W-specific sequence containing the female sex-determining gene dmw (red) while Z has a different Z-specific sequence (blue). Pentagon arrows and black triangles indicate genes and olfactory receptor genes, respectively. Their tips correspond to their 3′-ends. b, Alignment of the q-terminal regions of XTR9 and 10 with corresponding regions of XLA9_10L and XLA9_10S. Genes near the q-terminal regions of XTR 9 and XTR10 were missing in the X. tropicalis genome assembly v9, but rps11, rpl13a, lypd1 and actr3 were expected to be located there based on the synteny with human chromosomes, and then verified by cDNA FISH (upper panels). Small triangles on XLA9_10L and S indicate the distribution of gene models showing both identity and coverage greater than 30%, against the human and chicken peptide sequences from Ensembl, in the region ±2 Mb from the prospective 9/10 junction. HSA, human chromosome; GGA, chicken chromosome. The magnified view represents syntenic genes to scale with colours corresponding to human genes. c, The orders of orthologous genes across XTR9, XTR10, XLA9_10L and XLA9_10S. Green arrowheads: positions of centromeres in XTR9 and 10 predicted by examination of the cytogenetic chromosome length ratio of p versus q arms15. Blue arrowheads: positions of centromere repeats, frog centromeric repeat-1 (ref. 55), in XLA9_10L and S. Magenta and yellow ellipses, chromosomal locations of snrpn (magenta) and stau1 (yellow) from X. tropicalis v9 and X. laevis v9.1 assemblies. Red ellipses, chromosomal locations of four genes, rps11, rpl13a, lypd1 and actr3. XTR9 is inverted to facilitate comparison. Blue bidirectional arrows indicate the homologous regions where pericentric inversions may have occurred on proto-chromosomes (see Extended Data Fig. 2d). d, Schematic representation for the two hypothetical processes of chromosomal rearrangements (fusion and inversion) that occurred between the hypothetical proto-XTR9 and 10 to produce proto-XLA9_10, and eventually XLA9_10L and S. The process of chromosome rearrangements is explained parsimoniously in two different ways (left and right panels), starting from proto-XTR9 and 10. Actual and hypothetical ancestral chromosomal locations of snrpn and stau1 are shown by magenta and yellow circles, respectively. Note that the chromosomal locations of these genes on the proto-XTR10 differ between the two models. Chromosome segments homologous to XTR9 and XTR10 are shown in red and blue, respectively. XTR9 is inverted to facilitate comparison. Bidirectional arrows indicate the regions where pericentric inversions may have occurred. Black arrows indicate the direction of chromosomal evolution.
Extended Data Figure 3 Transposons.
a, Density of the subgenome-specific transposons on each chromosome (coverage length of transposable element (bp)/chromosome length (Mbp)). The coverage lengths of transposons were calculated from the results of BLASTN search (E-value cutoff 10−5) using the consensus sequences as queries. b, Jukes-Cantor distances across non-CpG sites, corrected as in Supplementary Note 7.5. Distances between X. tropicalis and X. laevis transposons consensus sequences are shown. The X. laevis-specific transposon differences are each individual transposon sequence against the consensus sequence for that subfamily. c, Phylogenetic tree of Xl-TpS_mar transposon expansions in the X. laevis genome, built using Jukes–Cantor corrected distances (Supplementary Note 7.5). Sub-clusters with enough members to determine accurate timings are highlighted. The scale bar represents the corrected Jukes–Cantor distance of 0.08 substitutions per site.
Extended Data Figure 4 Phylogeny.
a, Phylogenetic tree of pan-vertebrate conserved non-coding elements (pvCNEs), rooted by elephant shark. Alignments were done by MUSCLE, and the maximum-likelihood tree was built by PhyML. Branch length scale shown at the bottom. The difference in branch lengths of tetrapods follows the same topology as the protein-coding tree (Fig. 2b). b, Complete phylogenetic tree from Fig. 2a, with divergence times computed by r8s. c, Distribution of synonymous and non-synonymous rates Ks and Ka on specific subgenomes during the time between L and S speciation, before X. laevis and X. borealis speciation. We find accelerated mutations rates between T2 and T3 in Ks and Ka (P = 1.4 × 10−5 (left), 8.6 × 10−3 (right)). d, Distribution of Ks and Ka on specific subgenomes during the time after X. laevis and X. borealis speciation. We do not find significantly accelerated substitution rates (P = 0.10 (left) and P = 0.03 (right)). e, Table showing the number of homoeologues and singletons identified as homoeologues from the ancient vertebrate duplication (or ohnologues as they were historically called)56, 79.9% of ohnologues retain both copies in X. laevis today, significantly more than the 54.3% of the rest of the genome after excluding ohnologues (χ2 test P = 4.44 × 10−69). f, Table showing the branch lengths of bootstrapped maximum likelihood trees described in Supplementary Note 12.5. The columns refer to the X. tropicalis (XTR), L chromosome of X. laevis (XLA.L), S chromosome of X. laevis (XLA.S) and XLA.L/XLA.S branch lengths respectively. The first row shows triplets where all genes show expression, the second row shows triplets where L is a thanagene, and the third row shows triplets where S is a thanagene. The L branch length is significantly smaller when all genes are expressed, or when S is a thanagene (Wilcoxon signed-rank test, P = 1.7 × 10−216 and 6.4 × 10−212 respectively). The S branch length is smaller when L is a thanagene (P = 2.4 × 10−223). The ratio of branch lengths (L/S) is significantly different for either L or S thanagene datasets compared to when all genes are expressed (P = 3.55 × 10−214 and 7.48 × 10−220 respectively). The ratio is also different between the two thanagene datasets (P = 1.79 × 10−217).
Extended Data Figure 5 Structural evolution.
a, Chromosomal locations of the 45S pre-ribosomal RNA gene (rna45s), which encodes a precursor RNA for 18S, 5.8S and 28S rRNAs, was determined using pHr21Ab (5.8-kb for the 5′ portion) and pHr14E3 (7.3-kb for the 3′ portion) fragments as FISH probes. DNA fragments used for the probes were provided by National Institutes of Biomedical Innovation, Health and Nutrition, Osaka, and labelled with biotin-16-dUTP (Roche Diagnostics) by nick translation. After hybridization, the slides were incubated with FITC-avidin (Vector Laboratories). Hybridization signals (arrows) were detected to the short arm of XLA3L, but not XLA3S. Scale bar, 5 μm. b, A large deletion including an olfactory receptor gene (or) cluster. Schematic structures of or gene clusters and adjacent genes on the 8th chromosomes of X. tropicalis (XTR8) and X. laevis (XLA8L and XLA8S). Chromosomal locations: XTR8: 107,524,547–108,927,581; XLA8L: 105,062,063–106,610,199; XLA8S: 91,630,596–92,060,451. Horizontal bars, genomic DNA sequences; triangles, genes. Outside of or gene cluster, only representative genes are shown. The size of the triangle is to scale. The orientation of triangles indicates 5′ to 3′ direction of genes. Thin lines connect orthologous/homoeologous genes. Magenta triangles, or genes; green triangles, pseudogenes (point-mutated or truncated or genes). The number of or genes is shown underneath gene clusters. Dotted lines, a deleted region in XLA8S compared to XLA8L. The centromere is located on the left side and the telomere is on the right. c, The relative frequency (left panel) and size (right panel) of genomic regions deleted in the S (blue) and L (green) chromosomes respectively. Both subgenomes experienced sequence loss through deletions, but the deletions on the S subgenome are larger and have been more frequent. Deletions were called based on the progressive Cactus sequence alignment between the X. laevis L and S subgenomes and the X. tropicalis genome. Chromosome 9_10 of X. laevis was split into 9 and 10 on the basis of alignment with the X. tropicalis chromosomes. Sequences from L that were not present on S, but could at least partially be identified in X. tropicalis, and consisted of gaps for no more than 25% of their length, were called as deleted regions in S. The same procedure was followed for deleted regions in L. d, Identification of triplet loci is described in Supplementary Note 8.1. Loci were classified into groups based on the presence of gene 2 in both X. laevis subgenomes (homoeologue retained), versus those that had a pseudogene in the middle (pseudogene) or no remnant of the middle gene as assessed by Exonerate (deletion). To normalize the intergenic lengths, we divided the nucleotide distance between genes 1 and 3 in either X. laevis subgenome by the orthologous distance in X. tropicalis. The median of the normalized ratio distribution is plotted on the bar chart. On average, S deletions appear to be larger than L deletions (52.9% versus 80.2% of the size of the orthologous X. tropicalis region, respectively). e, The number of RNA-seq reads aligning ±1 kb of precursor miRNA loci (red) was compared to the read count for 10,000 random unannotated 2.1 kb regions of the genome (blue). All 83 homoeologous, intergenic miRNA pairs showed alignment within their regions, as opposed to 4,127 out of 10,000 (41.27%) of the randomly chosen intergenic sequences. The putative primary-miRNA loci also have a higher read count than the expressed randomly chosen regions (Wilcoxon signed-rank test, P = 1.4 × 10−38). f, The Cactus alignment was parsed to identify flanking CNE around each X. tropicalis gene. The number of CNEs >50 bp in length for singletons is shown in red, homoeologues in blue. Kolmogorov-Smirnov test P = 10−11. g, The average distance to the nearest gene was computed for each chromosomal locus in X. tropicalis. The average intergenic distance for those with a single X. laevis gene is shown in red, those with two shown in blue. Wilcoxon signed-rank test (P = 9.8 × 10−24). h, The distribution of gene retention by genomic footprint of the X. tropicalis orthologue. We define genomic footprint as the genomic distance from the start signal of the coding sequence (CDS) to the stop signal, including introns. The x axis shows log10(genomic footprint), the y axis the retention rate of each bin. The error bars are the standard deviation of the total divided by the number of genes in each bin. We tested for significant differences in length between homoeologues and singletons by a Wilcoxon signed-rank test (P = 2.4 × 10−96). i. The distribution of gene retention by CDS length of the X. tropicalis orthologue. The x axis shows log10 (CDS length), the y axis the retention rate of each bin. The error bars are the standard deviation of the total divided by the number of genes in each bin. We tested for significant differences in length between homoeologues and singletons by a Wilcoxon signed-rank test (P = 1.7 × 10−21). j, The distribution of gene retention by exon number of the X. tropicalis orthologue. The x axis shows number of exons; the y axis the retention rate of each bin. The error bars are the standard deviation of the total divided by the number of genes in each bin. We tested for significant differences in length between homoeologues and singletons by a Wilcoxon signed-rank test (P = 3.2 × 10−8).
Extended Data Figure 6 Pseudogenes.
a, Illustration of htt.S pseudogene alignment to X. tropicalis htt and the extant X. laevis htt.L, translated to amino acids. The amino acid position is shown at the beginning of each line. Missing codons are marked by dashes. Frameshifts and premature stops are marked by X and *, respectively (and pointed to with red arrows). The first exon of the pseudogene is completely missing from the S chromosome (top). The characteristic poly-Q region is maintained by both htt and htt.L. An exon with conservation in the pseudogene (bottom), illustrating that despite many frameshifts, premature stops, the lack of a proper start and insertions of new sequence, we identify many codons in the pseudogene that occur in large conserved blocks. b, Illustration of our model to compute pseudogene ages. The star represents the point of nonfunctionalization for a locus that is currently a pseudogene. We assume the expected rate of nonsynonymous changes can be estimated by the Ka of the extant gene and X. tropicalis. We then compare the Ks and Ka of the pseudogene sequence to estimate the time of nonfunctionalization. See Supplementary Note 9 for a more detailed discussion. c, Estimated epochs of pseudogenization for 430 genes are indistinguishable from a burst of pseudogenization >10 Ma (Ks > 0.03). See Supplementary Note 9 for a more detailed discussion. d. Correlation of pseudogene expression with its extant homoeologue. The little expression seen in pseudogenes tends to be uncorrelated with the extant homoeologue. e, Histogram of pseudogene expression values across all 28 tissues and developmental stages (red) compared to all extant genes (blue). The pseudogenes are rarely expressed and tend to be expressed at lower levels than extant protein-coding genes. f, Histograms of expression variance of pseudogenes (red) compared to extant genes (blue). The small amount of pseudogene expression observed does not tend to vary across tissues and developmental stages in the same way that extant genes do.
Extended Data Figure 7 Tandem duplications.
a, Phylogenetic trees of the mix/bix cluster. Nucleotide sequences were aligned using MUSCLE and a phylogenetic diagram was generated by the ML method with 1,000 bootstraps (MEGA6). Circles with different colours represent X. laevis L genes (magenta), X. laevis S genes (blue) and X. tropicalis genes (green). The table shows the correspondence of bix gene names proposed in this study and previously used (synonyms). b, FISH analysis showing XLA3S-specific deletion of the nodal5 gene cluster. One unit of the nodal5 gene region, including exons, introns and an intergenic region was used as a probe for FISH (counterstained with Hoechst). Arrows indicate the hybridization signals of nodal5s. Scale bar, 5 μm. c, Comparison of the nodal5 gene cluster. Genome sequencing revealed that nodal5.e1.L~.e5.L (pink) and nodal6.L are clustered. Amplification of nodal5 gene in XLA3L and loss of this cluster in XLA3S were confirmed. Pseudogenes (nodal5p1.L~p4.L and nodal5p1.S) are indicated in black. The nodal5 cluster of X. tropicalis does not contain any pseudogene. d, The X. laevis L chromosome has four complete copies of nodal3 (nodal3.e1.L~.e4.L), whereas the gene cluster is lost from the X. laevis S chromosome. A truncated nodal3 gene (nodal3p1.L) is likely to be a pseudogene and highly degenerate pseudogenes (nodal3p2.L and nodal3p3.L) also exist on the L chromosome. e, Like nodal3, vg1 is lost from the S chromosome although there is a pseudogene (vg1p.S). vg1 is specifically amplified on the X. laevis L chromosome (vg1.e1.L~.e3.L) in comparison with X. tropicalis. An amino acid change (Ser20 to Pro20) in Vg1 protein has been shown to result in functional differences (Supplementary Note 13.9). vg1 and derrière are orthologous to mammalian gdf1. f, Fraction of all genes duplicated and retained to present epoch per 1 expected 4DTV (fourfold degenerate transversion) at different epochs (semi-log scale). Shown also are linear fits, which would be consistent with constant birth- and death-rate models (first epoch is omitted from both fitted datasets, as is second epoch from X. laevis). See Supplementary Note 11 for a more detailed discussion. g, Same as f, but for ‘short genes’ (CDS <600 bp) and ‘long genes’ (CDS >1,200 bp) separately. The loss rate of new duplicates appears to be similar. If the extra copy of a newly duplicated gene was lost when the first 100% disabling mutation occurred, we would expect, on average, the longer genes to be lost.
Extended Data Figure 8 Gene expression analysis.
a, Pairwise Pearson correlation distributions between homoeologous genes (red) and all genes (blue). Left histogram, stage data; right, adult data. The x axis shows the correlation; the y axis the percentage of data. The homoeologous genes have a correlation distribution closer to one owing to the fact that these were recently the same locus. X. laevis TPM values of 0.5 were lowered to 0. Any gene with no TPM >0 was removed from analysis. We then added 0.1 to all TPM values and log transformed (log10) them. b, Scatter plot comparing binned genes by their median X. tropicalis expression57 to the retention rate of their X. laevis (co)-orthologues. Error bars are the standard deviation for the whole dataset divided by the square root of the number of genes analysed in a bin. We assessed significance by a Wilcoxon signed-rank test of the homoeologous and singleton distributions, P = 6.31 × 10−113. c, Full version of the box plot shown in Fig. 4c. The difference between subgenomes is difficult to see at this magnification, illustrating that many loci deviate from the whole genome median of preferring the L homoeologue. There were some L outliers expressed 104 as much as their S homoeologues, whereas no S genes showed such a strong trend. These differences are discussed in more detail in Supplementary Note 12. d, Box plot of 4DTV by homoeologue class defined in Supplementary Note 12.4. Significant differences are marked by a red asterisk (Wilcoxon signed-rank test, P < 10−5). The high correlation, similar expression (HCSE) group showed lower sequence change than other groups (P = 3.7 × 10−12) and the no correlation, different expression (NCDE) group showed high rates of sequence change (P = 5.6 × 10−14). e, Box plot of CDS length difference between X. laevis homoeologues by homoeologue class defined in Supplementary Note 12.4. Significant differences are marked by a red asterisk (Wilcoxon signed-rank test, P < 10−5). The HCSE group showed smaller CDS length differences than other groups (P = 2.4 × 10−13) and the NCDE group showed large differences in homoeologue CDS length (P = 2.1 × 10−32). f, Box plot of Ka/Ks between X. laevis homoeologues by homoeologue class defined in Supplementary Note 12.4. Significant differences are marked by a red asterisk (t-test P < 10−5). The HCSE group showed lower non-synonymous sequence change than other groups (P = 8.2 × 10−19) and the NCDE and no correlation, similar expression (NCSE) groups showed higher rates of non-synonymous sequence changes (P = 2.0 × 10−12 and P = 7.0 × 10−9 respectively). g, RNA-seq analysis of six6.L (red) and six6.S (blue) during X. laevis development (left) and in adult tissues (right). Expression levels of six6.S were lower than those of six6.L at most developmental stages and in adult tissues. h, Diagram of Homo sapiens, X. tropicalis and X. laevis six6 loci (upper panel). Magenta and black boxes indicate CNEs and exons, respectively. The phylogenetic tree analyses of H. sapiens, X. tropicalis and X. laevis six6 CNEs (lower left panel) and Six6 proteins (lower right panel). Notably, six6.S is more diverged from X. tropicalis six6 than six6.L, both in the encoded protein sequences and in CNEs within 3 kb of the transcription start sites. Materials, methods and the CNE locations on genome assemblies are described in Supplementary Note 13.1. i, On the basis of chromatin state properties, a Random Forest machine-learning algorithm can accurately predict L versus S expression bias. The classification is based on all genes with greater than threefold expression difference at NF stage 10.5 (a set of 1,129 genes). The mean (dotted black line) of the ROC area under the curve is 0.778 (tenfold cross-validation). Features were selected using Linear Support Vector Classification and are shown in j. j, Relative importance (based on Gini impurity) of selected features used in the Random Forest classification. All features used in the classification are shown. Among various variables, the ratios of H3K4me3 and DNA methylation at the promoter contributed most to the decision tree model. A difference in p300 binding in the genomic region surrounding the gene also contributed to the Random Forest classification, as did the presence or absence of a number of specific transcription factor motifs in the promoter.
Extended Data Figure 9 Examples of pathway responses.
a, The Wnt pathway. Left panel, several key components of the canonical Wnt pathway in the X. laevis genome. The numbers in brackets show the number of paralogues. Components that have homoeologous pair of genes or singletons are shown in blue or red, respectively. Each gene (wnt: 21 genes, LRP: 2 genes, Fzd: 10 genes, Dvl: 3 genes, Frat(GBP): 1 gene, GSK3: 2 genes, Axin: 2 genes, bcatenin: 1 gene, APC: 2 genes, TCF/LEF:4 genes) was classified into 4 groups according to subcellular localization, and the number of singleton and homoeologue retained genes is shown by pie charts. Right panel, syntenies around four singleton genes. b, Cell cycle regulation. Upper right panel, diagram of the cell cycle and regulatory proteins critical to each phase. Cyclin H (ccnh) and Cdk7 constitute Cdk-activating kinase (CAK), a key factor required for activation of all Cdks. Genes encoding Cyclin H and Cdk7 (red), but not other regulators (blue), became singletons. Upper left panel, pie charts show the numbers of homoeologous pairs (blue) and singletons (red) in each functional category as indicated. Lower left panel, syntenies of ccnh and cdk7 loci in X. tropicalis and X. laevis. Lower right table, individual genes used for drawing the pie charts are shown in the table. c, The Hippo pathway. Upper panel, Hippo pathway components and retention of their homoeologous gene pairs. All genes for Hippo pathway components as indicated were identified in the whole genome of X. laevis. Blue icons indicate that both of the homoeologous genes are expressed in normal development and adult organs. The red icon, Taz, indicates a singleton. Yap is interchangeable with Taz in most cases, but TAZ, but not YAP, serves as a mediator of Wnt signalling (broken line). Pie charts show the numbers of homoeologue pairs (blue) and singleton (red) in each category of the Hippo pathway components classified according to subcellular localization. Lower panel, comparative analysis of syntenies around the taz gene. X. tropicalis scaffold247 is not incorporated into the chromosome-scale assembly (v9) and hence its chromosomal location is not known yet. The p arm termini of XLA8L and XLA8S are on the left. See Supplemental Note 13 for further details.
Extended Data Figure 10 Pathways continued.
a, The TGFβ pathway. Pie charts indicate the ratio of differentially expressed homoeologous pairs (orange) and singletons (red). Many of the extracellular regulatory factors are either differentially regulated or became singletons. Genes for a type I receptor, co-receptors and an inhibitory Smad are also differentially regulated. Multicopy genes such as nodal3, nodal5 and vg1 are not counted as singletons, even though those genes are deleted on S chromosomes. Instead, these and duplicated chordin genes are categorized into differentially regulated genes. b, The sonic hedgehog pathway. Upper panel, the simplified hedgehog pathway known in Shh signalling is schematically shown. Most signalling components are encoded by both homoeologous genes, whereas Hhat (shown in red) is encoded by a singleton gene. Where paralogues exist, the numbers of paralogues are shown in parentheses. In the left cell, the Shh precursor (Hh precursor) is matured through the process involving Hhat and Hhatl and secreted. In the right cell, the binding of Shh (Hh) to Ptch1 (Ptch) receptor inhibits Ptch1-mediated repression of Smo, leading to Smo activation and subsequent inhibition of PKA; otherwise PKA converts Gli activators to truncated repressors. As a consequence, Gli proteins activate target genes, such as Ptch1 and Hhip. The transmembrane protein Hhip binds Shh and suppresses Shh activity. Lower panel, schematic comparison of syntenies around hhat genes of X. tropicalis chromosome 5 (top) and X. laevis 5L chromosome (middle) and the corresponding region of X. laevis 5S chromosome (bottom). The diagram is not drawn to scale. c, Deletion rates on L (x axis) versus S (y axis) for different Pfam groups. For Pfam groups we computed the number of X. laevis single-copy genes (singletons) versus homoeologue pairs and computed the fraction retained. The line of expected L/S loss is based on the genome-wide average (56.4%). Red points show groups with high or low rates of loss (P < .01). See Supplementary Table 5 for more information. d, Deletion rates on L (x axis) versus S (y axis) for different stage weighted gene correlation network analysis (WGCNA)54 groups (visualized as a heatmap in Fig. 4a). For stage WGCNA groups we computed the number of X. laevis single-copy genes (singletons) versus homoeologue pairs and computed the fraction retained. The line of expected L/S loss is based on the genome-wide average (56.4%). Red points show groups with high or low rates of loss (P < .01). e, Deletion rates on L (x axis) versus S (y axis) for different GO groups. For GO groups we computed the number of X. laevis single-copy genes (singletons) versus homoeologue pairs and computed the fraction retained. The line of expected L/S loss is based on the genome-wide average (56.4%). Red points show groups with high or low rates of loss (P < 0.01). See Supplementary Table 5 for more information.
Supplementary information
Supplementary Information
This file contains Supplementary Notes 1-15, which detail analyses from the main text and additional references (see Contents for more details). (PDF 2532 kb)
Supplementary Tables
This zipped file contains Supplementary Table 1-11. (ZIP 32343 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Session, A., Uno, Y., Kwon, T. et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343 (2016). https://doi.org/10.1038/nature19840
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature19840
This article is cited by
-
Noncanonical function of folate through folate receptor 1 during neural tube formation
Nature Communications (2024)
-
Conserved chromatin and repetitive patterns reveal slow genome evolution in frogs
Nature Communications (2024)
-
Deep transcriptome profiling reveals limited conservation of A-to-I RNA editing in Xenopus
BMC Biology (2023)
-
Comparative genomics reveals insights into anuran genome size evolution
BMC Genomics (2023)
-
A proteasomal β5 subunit of Haemonchus contortus with a role in the growth, development and life span
Parasites & Vectors (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
