
Abstract
Large-scale reference data sets of human genetic variation are critical for the medical and functional interpretation of DNA sequence changes. Here we describe the aggregation and analysis of high-quality exome (protein-coding region) DNA sequence data for 60,706 individuals of diverse ancestries generated as part of the Exome Aggregation Consortium (ExAC). This catalogue of human genetic diversity contains an average of one variant every eight bases of the exome, and provides direct evidence for the presence of widespread mutational recurrence. We have used this catalogue to calculate objective metrics of pathogenicity for sequence variants, and to identify genes subject to strong selection against various classes of mutation; identifying 3,230 genes with near-complete depletion of predicted protein-truncating variants, with 72% of these genes having no currently established human disease phenotype. Finally, we demonstrate that these data can be used for the efficient filtering of candidate disease-causing variants, and for the discovery of human ‘knockout’ variants in protein-coding genes.
Similar content being viewed by others


Main
Over the last five years, the widespread availability of high-throughput DNA sequencing technologies has permitted the sequencing of the whole genomes or exomes of hundreds of thousands of humans. In theory, these data represent a powerful source of information about the global patterns of human genetic variation, but in practice, are difficult to access for practical, logistical, and ethical reasons; in addition, their utility is complicated by the heterogeneity in the experimental methodologies and variant calling pipelines used to generate them. Current publicly available data sets of human DNA sequence variation contain only a small fraction of all sequenced samples: the Exome Variant Server, created as part of the NHLBI Exome Sequencing Project (ESP)1, contains frequency information spanning 6,503 exomes; and the 1000 Genomes Project (1000G), which includes individual-level genotype data from whole-genome and exome sequence data for 2,504 individuals2.
Databases of genetic variation are important for our understanding of human population history and biology1,2,3,4,5, but also provide critical resources for the clinical interpretation of variants observed in patients who have rare Mendelian diseases6,7. The filtering of candidate variants by frequency in unselected individuals is a key step in any pipeline for the discovery of causal variants in Mendelian disease patients, and the efficacy of such filtering depends on both the size and the ancestral diversity of the available reference data.
Here we describe the joint variant calling and analysis of high-quality variant calls across 60,706 human exomes, assembled by the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org). This call set exceeds previously available exome-wide variant databases, by nearly an order of magnitude, providing substantially increased resolution for the analysis of very low-frequency genetic variants. We demonstrate the application of this data set to the analysis of patterns of genetic variation including the discovery of widespread mutational recurrence, the inference of gene-level constraint against truncating variation, the clinical interpretation of variation in Mendelian disease genes, and the discovery of human knockout variants in protein-coding genes.
The ExAC data set
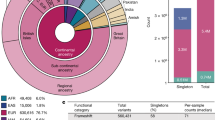
Sequencing data processing, variant calling, quality control and filtering was performed on over 91,000 exomes (see Methods), and sample filtering was performed to produce a final data set spanning 60,706 individuals (Fig. 1a). To identify the ancestry of each ExAC individual, we performed principal component analysis (PCA) to distinguish the major axes of geographic ancestry and to identify population clusters corresponding to individuals of European, African, South Asian, East Asian, and admixed American (hereafter referred to as Latino) ancestry (Fig. 1b; Supplementary Table 3); we note that the apparent separation between East Asian and other samples reflects a deficiency of Middle Eastern and Central Asian samples in the data set. We further separated Europeans into individuals of Finnish and non-Finnish ancestry given the enrichment of this bottlenecked population; the term European hereafter refers to non-Finnish European individuals.

a, The size and diversity of public reference exome data sets. ExAC exceeds previous data sets in size for all studied populations. b, Principal component analysis (PCA) dividing ExAC individuals into five continental populations. PC2 and PC3 are shown; additional PCs are in Extended Data Fig. 5a. c, The allele frequency spectrum of ExAC highlights that the majority of genetic variants are rare and novel (absent from prior databases of genetic variation, such as dbSNP). d, The proportion of possible variation observed by mutational context and functional class. Over half of all possible CpG transitions are observed. Error bars represent standard error of the mean. e, f, The number (e), and frequency distribution (proportion singleton; f) of indels, by size. Compared to in-frame indels, frameshift variants are less common (have a higher proportion of singletons, a proxy for predicted deleteriousness on gene product). Error bars indicate 95% confidence intervals.
We identified 10,195,872 candidate sequence variants in ExAC. We further applied stringent depth and site/genotype quality filters to define a subset of 7,404,909 high-quality variants, including 317,381 insertions or deletions (indels) (Supplementary Table 7), corresponding to one variant for every 8 base pairs (bp) within the exome intervals. The majority of these are very low-frequency variants absent from previous smaller call sets (Fig. 1c), of the high-quality variants, 99% have a frequency of <1%, 54% are singletons (variants seen only once in the data set), and 72% are absent from both 1000G and ESP data sets.
The density of variation in ExAC is not uniform across the genome, and the observation of variants depends on factors such as mutational properties and selective pressures. In the ~45 million well-covered (80% of individuals with a minimum of 10× coverage) positions in ExAC, there are ~18 million possible synonymous variants, of which we observe 1.4 million (7.5%). However, we observe 63.1% of possible CpG transitions (C to T variants, in which the adjacent base is G), while only observing 3% of possible transversions and 9.2% of other possible transitions (Supplementary Table 9). A similar pattern is observed for missense and nonsense variants, with lower proportions due to selective pressures (Fig. 1d). Of 123,629 high-quality indels called in coding exons, 117,242 (95%) have a length <6 bases, with shorter deletions being the most common (Fig. 1e). Frameshifts are found in smaller numbers and are more likely to be singletons than in-frame indels (Fig. 1f), reflecting the influence of purifying selection.
Patterns of protein-coding variation
The density of protein-coding sequence variation in ExAC reveals a number of properties of human genetic variation that are undetectable in smaller data sets. For example, 7.9% of high-quality sites in ExAC are multiallelic (multiple different sequence variants observed at the same site), close to the Poisson expectation of 8.3%, given the observed density of variation, and far higher than that observed in previous data sets of 0.48% in the 1000G (exome intervals) and 0.43% in the ESP data sets.
The size of ExAC makes it possible to directly observe mutational recurrence: instances in which the same mutation has occurred multiple times independently throughout the history of the sequenced populations. For instance, among synonymous (non-protein-altering) variants, a class of variation expected to have undergone minimal selection, 43% of validated de novo events identified in external data sets of 1,756 parent-offspring trios8,9 are also observed independently in our data set (Fig. 2a), indicating a separate origin for the same variant within the demographic history of the two samples. This proportion is much higher for transition variants at CpG sites, well established to be the most highly mutable sites in the human genome10: 87% of previously reported de novo CpG transitions at synonymous sites are observed in ExAC, indicating that our sample sizes are beginning to approach saturation of this class of variation. This saturation is detectable by a change in the discovery rate at subsets of the ExAC data set, beginning at around 20,000 individuals (Fig. 2b), indicating that ExAC is the first human exome-wide data set, to our knowledge, large enough for this effect to be directly observed.

a, Proportion of validated de novo variants from two external data sets that are independently found in ExAC, separated by functional class and mutational context. Error bars represent standard error of the mean. Colours are consistent in a–d. b, Number of unique variants observed, by mutational context, as a function of number of individuals (downsampled from ExAC). CpG transitions, the most likely mutational event, begin reaching saturation at ~20,000 individuals. c, The site frequency spectrum is shown for each mutational context. d, For doubletons (variants with an allele count (AC) of 2), mutation rate is positively correlated with the likelihood of being found in two individuals of different continental populations. e, The mutability-adjusted proportion of singletons (MAPS) is shown across functional classes. Error bars represent standard error of the mean of the proportion of singletons.
Mutational recurrence has a marked effect on the frequency spectrum in the ExAC data, resulting in a depletion of singletons at sites with high mutation rates (Fig. 2c). We observe a correlation between singleton rates (the proportion of variants seen only once in ExAC) and site mutability inferred from sequence context11 (r = −0.98; P < 10−50; Extended Data Fig. 1d): sites with low predicted mutability have a singleton rate of 60%, compared to 20% for sites with the highest predicted rate (CpG transitions; Fig. 2c). Conversely, for synonymous variants, CpG variants are approximately twice as likely to rise to intermediate frequencies: 16% of CpG variants are found in at least 20 copies in ExAC, compared to 8% of transversions and non-CpG transitions, suggesting that synonymous CpG transitions have on average two independent mutational origins in the ExAC sample. Recurrence at highly mutable sites can further be observed by examining the population sharing of doubleton synonymous variants (variants occurring in only two individuals in ExAC). Low-mutability mutations (especially transversions), are more likely to be observed in a single population (representing a single mutational origin), whereas CpG transitions are more likely to be found in two separate populations (independent mutational events); as such, site mutability and probability of observation in two populations is significantly correlated (r = 0.884; Fig. 2d).
We also explored the prevalence and functional impact of multinucleotide polymorphisms (MNPs), in cases where multiple substitutions were observed within the same codon in at least one individual. We found 5,945 MNPs (mean = 23 per sample) in ExAC (Extended Data Fig. 2a), in which analysis of the underlying SNPs without correct haplotype phasing would result in altered interpretation. These include 647 instances in which the effect of a protein-truncating variant (PTV) is eliminated by an adjacent single nucleotide polymorphism (SNP) (referred to as a rescued PTV), and 131 instances in which underlying synonymous or missense variants result in PTV MNPs (referred to as a gained PTV). Our analysis also revealed 8 MNPs in disease-associated genes, resulting in either a rescued or gained PTV, and 10 MNPs that have previously been reported as disease-causing mutations (Supplementary Tables 10 and 11). These variants would be missed by virtually all currently available variant calling and annotation pipelines.
Inferring variant deleteriousness and gene constraint
Deleterious variants are expected to have lower allele frequencies than neutral ones, due to negative selection. This theoretical property has been demonstrated previously in human population sequencing data12,13 and here (Fig. 1d, e). This allows inference of the degree of selection against specific functional classes of variation. However, mutational recurrence as described earlier indicates that allele frequencies observed in ExAC-scale samples are also skewed by mutation rate, with more mutable sites less likely to be singletons (Fig. 2c and Extended Data Fig. 1d). Mutation rate is in turn non-uniformly distributed across functional classes. For example, variants that result in the loss of a stop codon can never occur at CpG dinucleotides (Extended Data Fig. 1e). We corrected for mutation rates (Supplementary Information section 3.2) by creating a mutability-adjusted proportion singleton (MAPS) metric. This metric reflects (as expected), strong selection against predicted PTVs, as well as missense variants predicted by conservation-based methods to be deleterious (Fig. 2e).
The deep ascertainment of rare variation in ExAC also allows us to infer the extent of selection against variant categories on a per-gene basis by examining the proportion of variation that is missing compared to expectations under random mutation. Conceptually similar approaches have been applied to smaller exome data sets11,14, but have been underpowered, particularly when analysing the depletion of PTVs. We compared the observed number of rare (minor allele frequency (MAF) <0.1%) variants per gene to an expected number derived from a selection neutral, sequence-context based mutational model11. The model performs well in predicting the number of synonymous variants, which should be under minimal selection, per gene (r = 0.98; Extended Data Fig. 3b).
We quantified deviation from expectation with a Z score11, which for synonymous variants is centred at zero, but is significantly shifted towards higher values (greater constraint) for both missense and PTV (Wilcoxon P < 10−50 for both; Fig. 3a). The genes on the X chromosome are significantly more constrained than those on the autosomes for missense (P < 10−7) and loss-of-function mutations (P < 10−50), in line with previous work15. The high correlation between the observed and expected number of synonymous variants on the X chromosome (r = 0.97 versus 0.98 for autosomes) indicates that this difference in constraint is not due to a calibration issue. To reduce confounding by coding sequence length for PTVs, we developed an expectation-maximization algorithm (Supplementary Information section 4.4) using the observed and expected PTV counts within each gene to separate genes into three categories: null (observed ≈ expected), recessive (observed ≤ 50% of expected), and haploinsufficient (observed <10% of expected). This metric—the probability of being loss-of-function (LoF) intolerant (pLI)—separates genes of sufficient length into LoF intolerant (pLI ≥ 0.9, n = 3,230) or LoF tolerant (pLI ≤ 0.1, n = 10,374) categories. pLI is less correlated with coding sequence length (r = 0.17 as compared to 0.57 for the PTV Z score), outperforms the PTV Z score as an intolerance metric (Supplementary Table 15), and reveals the expected contrast between gene lists (Fig. 3b). pLI is positively correlated with the number of physical interaction partners of a gene product (P < 10−41). The most constrained pathways (highest median pLI for the genes in the pathway) are core biological processes (spliceosome, ribosome, and proteasome components; Kolmogorov–Smirnov test P < 10−6 for all), whereas olfactory receptors are among the least constrained pathways (Kolmogorov–Smirnov test P < 10−16), as demonstrated in Fig. 3b, and this is consistent with previous work5,16,17,18,19.

a, Histograms of constraint Z scores for 18,225 genes. This measure of departure of number of variants from expectation is normally distributed for synonymous variants, but right-shifted (higher constraint) for missense and protein-truncating variants (PTVs), indicating that more genes are intolerant to these classes of variation. b, The proportion of genes that are very probably intolerant of loss-of-function variation (pLI ≥ 0.9) is highest for ClinGen haploinsufficient (HI) genes, and stratifies by the severity and age of onset of the haploinsufficient phenotype. Genes essential in cell culture and dominant disease genes are likewise enriched for intolerant genes, whereas recessive disease genes and olfactory receptors have fewer intolerant genes. Black error bars indicate 95% confidence intervals. c, Synonymous Z scores show no correlation with the number of tissues in which a gene is expressed, but the most missense- and PTV-constrained genes tend to be expressed in more tissues. Thick black bars indicate the first to third quartiles, with the white circle marking the median. d, Highly missense- and PTV-constrained genes are less likely to have eQTLs discovered in GTEx as the average gene. Shaded regions around the lines indicate 95% confidence intervals. e, Highly missense- and PTV-constrained genes are more likely to be adjacent to genome-wide association study (GWAS) signals than the average gene. Shaded regions around the lines indicate 95% confidence intervals. f, MAPS (Fig. 2d) is shown for each functional category, broken down by constraint score bins as shown. Missense and PTV constraint score bins provide information about natural selection at least partially orthogonal to MAPS, PolyPhen, and CADD scores, indicating that this metric should be useful in identifying variants associated with deleterious phenotypes. Shaded regions around the lines indicate 95% confidence intervals. For panels a, c–f, variants are coloured with synonymous in grey, missense in orange, and protein-truncating in maroon.
Crucially, we note that LoF-intolerant genes include virtually all known severe haploinsufficient human disease genes (Fig. 3b), but that 72% of LoF-intolerant genes have not yet been assigned a human disease phenotype despite clear evidence for extreme selective constraint (Supplementary Table 13). We note that this extreme constraint does not necessarily reflect a lethal disease or status as a disease gene (for example, BRCA1 has a pLI of 0), but probably points to genes in which heterozygous loss of function confers some non-trivial survival or reproductive disadvantage.
The most highly constrained missense (top 25% missense Z scores) and PTV (pLI ≥ 0.9) genes show higher expression levels and broader tissue expression than the least constrained genes20 (Fig. 3c). These most highly constrained genes are also depleted for expression quantitative trait loci (eQTLs) (P < 10−9 for missense and PTV; Fig. 3d), yet are enriched within genome-wide significant trait-associated loci (χ2 test, P < 10−14, Fig. 3e). Genes intolerant of PTV variation would be expected to be dosage-sensitive, as in such genes natural selection does not tolerate a 50% deficit in expression due to the loss of single allele. It is thus unsurprising that these genes are also depleted of common genetic variants that have a large enough effect on expression to be detected as eQTLs with current limited sample sizes. However, smaller changes in the expression of these genes, through weaker eQTLs or functional variants, are more likely to contribute to medically relevant phenotypes.
Finally, we investigated how these constraint metrics would stratify mutational classes according to their frequency spectrum, corrected for mutability as in the previous section (Fig. 3f). The effect was most dramatic when considering nonsense variants in the LoF-intolerant set of genes. For missense variants, the missense Z score offers information orthogonal to Polyphen2 and CADD classifications, which are measures predicting the likely deleteriousness of variants, indicating that gene-level measures of constraint offer additional information to variant-level metrics in assessing potential pathogenicity.
ExAC improves variant interpretation in rare disease
We assessed the value of ExAC as a reference data set for clinical sequencing approaches, which typically prioritize or filter potentially deleterious variants on the basis of functional consequence and allele frequency6. Filtering on ExAC reduced the number of candidate protein-altering variants by sevenfold compared to the ESP data set, and was most powerful when the highest allele frequency in any one population (‘popmax’) was used rather than the average (‘global’) allele frequency (Fig. 4a). ESP is not well-powered to filter at 0.1% allele frequency without removing many genuinely rare variants, as allele frequency estimates based on low allele counts are both upward-biased and imprecise (Fig. 4b). We thus expect that ExAC will provide a very substantial boost in the power and accuracy of variant filtering in Mendelian disease projects.

a, Predicted missense and protein-truncating variants in 500 randomly chosen ExAC individuals were filtered based on allele frequency (AF) information from ESP, or from the remaining ExAC individuals. At a 0.1% allele frequency filter, ExAC provides greater power to remove candidate variants, leaving an average of 154 variants for analysis, compared to 1,090 after filtering against ESP. Popmax allele frequency also provides greater power than global allele frequency, particularly when populations are unequally sampled. b, Estimates of allele frequency in Europeans based on ESP are more precise at higher allele frequencies. Sampling variance and ascertainment bias make allele frequency estimates unreliable, posing problems for Mendelian variant filtration. 69% of ESP European singletons are not seen a second time in ExAC (tall bar at left), illustrating the dangers of filtering on very low allele counts. c, Allele frequency spectrum of disease-causing variants in the Human Gene Mutation Database (HGMD) and/or pathogenic or probable pathogenic variants in ClinVar for well-characterized autosomal dominant and autosomal recessive disease genes28. Most are not found in ExAC; however, many of the reportedly pathogenic variants found in ExAC are at too high a frequency to be consistent with disease prevalence and penetrance. d, Literature review of variants with >1% global allele frequency or >1% Latin American or South Asian population allele frequency confirmed there is insufficient evidence for pathogenicity for the majority of these variants. Variants were reclassified by American College of Medical Genetics and Genomics (ACMG) guidelines24.
Previous large-scale sequencing studies have repeatedly shown that some purported Mendelian disease-causing genetic variants are implausibly common in the population21,22,23 (Fig. 4c). The average ExAC participant harbours ~54 variants reported as disease-causing in two widely used databases of disease-causing variants (Supplementary Information section 5.2). Most (~41) of these are high-quality genotypes but with implausibly high (>1%) popmax allele frequencies. We therefore hypothesized that most of the supposed burden of Mendelian disease alleles per person is due not to genotyping error, but rather to misclassification in the literature and/or in databases.
We manually curated the evidence of pathogenicity for 192 previously reported pathogenic variants with allele frequency >1% either globally or in South Asian or Latino individuals, populations that are underrepresented in previous reference databases. Nine variants had sufficient data to support disease association, typically with either mild or incompletely penetrant disease effects; the remainder either had insufficient evidence for pathogenicity, no claim of pathogenicity, or were benign traits (Supplementary Information section 5.3). It is difficult to prove the absence of any disease association, and incomplete penetrance or genetic modifiers may contribute in some cases. Nonetheless, the high cumulative allele frequency of these variants combined with their limited original evidence for pathogenicity suggest little contribution to disease, and 163 variants met American College of Medical Genetics criteria24 for reclassification as benign or probably benign (Fig. 4d). A total of 126 of these 163 have been reclassified in source databases as of December 2015 (Supplementary Table 20). Supporting functional data were reported for 18 of these variants, highlighting the need to review cautiously even variants with experimental support.
We also sought phenotypic data for a subset of ExAC participants homozygous for reported severe recessive disease variants, again enabling reclassification of some variants as benign. North American Indian childhood cirrhosis is a recessive disease of cirrhotic liver failure during childhood requiring liver transplant for survival to adulthood, previously reported to be caused by CIRH1A p.R565W25 (CIRH1A is also known as UTP4). ExAC ontains 222 heterozygous and 4 homozygous Latino individuals, with a population allele frequency of 1.92%. The 4 homozygotes had no history of liver disease and recontact in two individuals revealed normal liver function (Supplementary Table 22). Thus, despite the rigorous linkage and Sanger sequencing efforts that led to the original report of pathogenicity, the ExAC data demonstrate that this variant is either benign or insufficient to cause disease, highlighting the importance of matched reference populations.
The above curation efforts confirm the importance of allele frequency filtering in analysis of candidate disease variants6,26,27. However, literature and database errors are prevalent even at lower allele frequencies: the average ExAC individual contains 0.89 (<1% popmax allele frequency) reportedly Mendelian variants in well-characterized dominant disease genes28, and 0.21 at <0.1% popmax allele frequency. This inflation probably results from a combination of false reports of pathogenicity and incomplete penetrance, as we have recently shown for PRNP29. The abundance of rare functional variation in many disease genes in ExAC is a reminder that such variants should not be assumed to be causal or highly penetrant without careful segregation or case-control analysis7,24.
Effect of rare protein-truncating variants
We investigated the distribution of PTVs, variants predicted to disrupt protein-coding genes through the introduction of a stop codon, frameshift, or the disruption of an essential splice site; such variants are expected to be enriched for complete loss of function of the affected genes. Naturally occurring PTVs in humans provide a model for the functional impact of gene inactivation, and have been used to identify many genes in which LoF causes severe disease30, as well as rare cases where LoF is protective against disease31.
Among the 7,404,909 high-quality variants in ExAC, we found 179,774 high-confidence PTVs (as defined in Supplementary Information section 6), 121,309 of which are singletons. This corresponds to an average of 85 heterozygous and 35 homozygous PTVs per individual (Fig. 5a). The diverse nature of the cohort enables the discovery of substantial numbers of new PTVs: out of 58,435 PTVs with an allele count greater than one, 33,625 occur in only one population. However, although PTVs as a category are extremely rare, the majority of the PTVs found in any one person are common, and each individual has only ~2 singleton PTVs, of which 0.14 are found in PTV-constrained genes (pLI > 0.9). ExAC recapitulates known aspects of population demographic models, including an increase in intermediate-frequency (1–5%) PTVs in Finland32 and relatively common (>1%) PTVs in Africans (Fig. 5b). However, these differences are diminished when considering only LoF-constrained (pLI > 0.9) genes (Extended Data Fig. 4).

a, The average ExAC individual has 85 heterozygous and 35 homozygous protein-truncating variants (PTVs), of which 18 and 0.19 are rare (<1% allele frequency), respectively. Error bars represent standard deviation. b, Breakdown of PTVs per individual (a) by popmax allele frequency bin. Across all populations, most PTVs found in a given individual are common (>5% allele frequency). c, d, Number of genes with at least one PTV (c), or homozygous PTV (d), as a function of number of individuals, downsampled from ExAC. The South Asian population is broken down by consanguinity (inbreeding coefficient, F). At 60,000 individuals for ExAC, the plots in c, d, extend to 15,750 with at least one PTV and 1,550 genes with at least one homozygous PTV. Dotted line represents all ExAC samples.
Using a sub-sampling approach, we show that the discovery of both heterozygous (Fig. 5c) and homozygous (Fig. 5d) PTVs scales very differently across human populations, with implications for the design of large-scale sequencing studies to ascertain human knockouts, as described later.
Discussion
Here we describe the generation and analysis of the most comprehensive catalogue (to our knowledge) of human protein-coding genetic variation to date, incorporating high-quality exome sequencing data from 60,706 individuals of diverse geographic ancestry. The resulting call set provides unprecedented resolution for the analysis of low-frequency protein-coding variants in human populations, as well as a public resource (http://exac.broadinstitute.org) for the clinical interpretation of genetic variants observed in disease patients.
The very large sample size of ExAC also provides opportunities for a high-resolution analysis of the sensitivity of human genes to functional variation. Although previous sample sizes have been adequately powered for the assessment of gene-level intolerance to missense variation11,14, ExAC provides sufficient power for the first time to investigate genic intolerance to PTVs, highlighting 3,230 highly LoF-intolerant genes, 72% of which have no established human disease phenotype in the OMIM or ClinVar databases of observed human genetic mutations. Although this extreme depletion of PTVs will probably highlight genes in which loss of a single copy has been reproductively disadvantageous over recent human history, not all high pLI genes will lead to lethal disease. Additionally, disease genes—particularly those that act after post-reproductive age—do not necessarily have high pLI values (for example, the pLI of BRCA1 is 0). In separate work33 we show that ExAC similarly provides power to identify genes intolerant of copy number variation. Quantification of genic intolerance to both classes of variation will provide added power to disease studies.
The ExAC resource provides the largest database to date (to our knowledge) for the estimation of allele frequency for protein-coding genetic variants, providing a powerful filter for analysis of candidate pathogenic variants in severe Mendelian diseases. Frequency data from ESP1 have been widely used for this purpose, but those data are limited by population diversity and by resolution at allele frequencies ≤ 0.1%. ExAC therefore provides substantially improved power for Mendelian analyses, although it is still limited in power at lower allele frequencies, emphasizing the need for more sophisticated pathogenic variant filtering strategies alongside on-going data aggregation efforts.
We show that different populations confer different advantages in the discovery of gene-disrupting PTVs, providing guidance for the identification of human knockouts to understand gene function. Sampling multiple populations would probably be a fruitful strategy for a researcher investigating common PTV variation. However, discovery of homozygous PTVs is markedly enhanced in the South Asian samples, which come primarily from a Pakistani cohort with 38.3% of individuals self-reporting as having closely related parents, emphasizing the extreme value of consanguineous cohorts for human knockout discovery34,35,36 (Fig. 5d). Other approaches to enriching for homozygosity of rare PTVs, such as focusing on bottlenecked populations, have already proved fruitful32,34.
Even with this large collection of jointly processed exomes, many limitations remain. First, most ExAC individuals were ascertained for biomedically important disease; although we have attempted to exclude severe paediatric diseases, the inclusion of both cases and controls for several polygenic disorders means that ExAC certainly contains disease-associated variants37. Second, future reference databases would benefit from including a broader sampling of human diversity, especially from under-represented Middle Eastern and African populations. Third, the inclusion of whole genomes will also be critical to investigate additional classes of functional variation and identify non-coding constrained regions. Finally, and most critically, detailed phenotype data are unavailable for the vast majority of ExAC samples; future initiatives that assemble sequence and clinical data from very large-scale cohorts will be required to fully translate human genetic findings into biological and clinical understanding.
Although the ExAC data set exceeds the scale of previously available frequency reference data sets, much remains to be gained by further increases in sample size. Indeed, the fact that even the rarest transversions have mutational rates11 on the order of 1 × 10−9 implies that the vast majority of possible non-lethal SNVs probably exist in some living human. ExAC already includes >63% of all possible protein-coding CpG transitions at well-covered synonymous sites; orders-of-magnitude increases in sample size will eventually lead to saturation of other classes of variation.
ExAC was made possible by the willingness of multiple large disease-focused consortia to share their raw data, and by the availability of the software and computational resources required to create a harmonized variant call set on the scale of tens of thousands of samples. The creation of yet larger reference variant databases will require continued emphasis on the value of genomic data sharing.
Methods
Variant discovery
We assembled approximately 1 petabyte of raw sequencing data (FASTQ files) from 91,796 individual exomes drawn from a wide range of primarily disease-focused consortia (Supplementary Table 2). We processed these exomes through a single informatic pipeline and performed joint variant calling of single nucleotide variants (SNVs) and indels across all samples using a new version of the Genome Analysis Toolkit (GATK) HaplotypeCaller pipeline. Variant discovery was performed within a defined exome region that includes Gencode v19 coding regions and flanking 50 bases. At each site, sequence information from all individuals was used to assess the evidence for the presence of a variant in each individual. Full details of data processing, variant calling and resources are described in the Supplementary Information sections 1.1–1.4.
Quality assessment
We leveraged a variety of sources of internal and external validation data to calibrate filters and evaluate the quality of filtered variants (Supplementary Table 7). We adjusted the standard GATK variant site filtering38 to increase the number of singleton variants that pass this filter, while maintaining a singleton transmission rate of 50.1%, very near the expected 50%, within sequenced trios. We then used the remaining passing variants to assess depth and genotype quality filters compared to >10,000 samples that had been directly genotyped using SNP arrays (Illumina HumanExome) and achieved 97–99% heterozygous concordance, consistent with known error rates for rare variants in chip-based genotyping39. Relative to a ‘platinum standard’ genome sequenced using five different technologies40, we achieved sensitivity of 99.8% and false discovery rates (FDR) of 0.056% for single nucleotide variants (SNVs), and corresponding rates of 95.1% and 2.17% for insertions and deletions (indels), respectively. Lastly, we compared 13 representative non-Finnish European exomes included in the call set with their corresponding 30× PCR-free genome. The overall SNV and indel FDR was 0.14% and 4.71%, respectively, while for SNV singletons it was 0.389%. The overall FDR by annotation classes missense, synonymous and protein truncating variants (including indels) were 0.076%, 0.055% and 0.471% respectively (Supplementary Tables 5 and 6). Full details of quality assessments are described in Supplementary Information section 1.6.
Sample filtering
The 91,796 samples were filtered based on two criteria. First, samples that were outliers for key metrics were removed (Extended Data Fig. 5b). Second, in order to generate allele frequencies based on independent observations without enrichment of Mendelian disease alleles, we restricted the final release data set to unrelated adults with high-quality sequence data and without severe paediatric disease. After filtering, only 60,706 samples remained, consisting of ~77% of Agilent (33 Mb target) and ~12% of Illumina (37.7 Mb target) exome captures. Full details of the filtering process are described in Supplementary Information section 1.7.
ExAC data release
For each variant, summary data for genotype quality, allele depth and population specific allele counts were calculated before removing all genotype data. This variant summary file was then functionally annotated using variant effect predictor (VEP) with the LOFTEE plugin. This data set can be accessed via the ExAC Browser (http://exac.broadinstitute.org), or downloaded from: (ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3/ExAC.r0.3.sites.vep.vcf.gz). Full details regarding the annotation of the ExAC data set are described in the Supplementary Information sections 1.9–1.10.
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
References
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220 (2012)
The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature 526, 68–74 (2015)
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011)
Stoneking, M. & Krause, J. Learning about human population history from ancient and modern genomes. Nature Rev. Genet. 12, 603–614 (2011)
MacArthur, D. G. et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012)
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Rev. Genet. 12, 745–755 (2011)
MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014)
The Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015)
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014)
Cooper, D. N. & Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 78, 151–155 (1988)
Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nature Genet. 46, 944–950 (2014)
Tennessen, J. A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012)
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nature Genet. 47, 435–444 (2015)
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S. & Goldstein, D. B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9, e1003709 (2013)
Vicoso, B. & Charlesworth, B. Evolution on the X chromosome: unusual patterns and processes. Nature Rev. Genet. 7, 645–653 (2006)
Jeong, H., Mason, S. P., Barabási, A. L. & Oltvai, Z. N. Lethality and centrality in protein networks. Nature 411, 41–42 (2001)
Goh, K.-I. et al. The human disease network. Proc. Natl Acad. Sci. USA 104, 8685–8690 (2007)
Rolland, T. et al. A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014)
Itan, Y. et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl Acad. Sci. USA 112, 13615–13620 (2015)
The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015)
Bell, C. J. et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 3, 65ra4 (2011)
Xue, Y. et al. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 91, 1022–1032 (2012)
Piton, A., Redin, C. & Mandel, J.-L. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 93, 368–383 (2013)
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015)
Chagnon, P. et al. A missense mutation (R565W) in Cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am. J. Hum. Genet. 71, 1443–1449 (2002)
Stenson, P. D. et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9 (2014)
Dewey, F. E. et al. Sequence to medical phenotypes: a framework for interpretation of human whole genome DNA sequence data. PLoS Genet. 11, e1005496 (2015)
Blekhman, R. et al. Natural selection on genes that underlie human disease susceptibility. Curr. Biol. 18, 883–889 (2008)
Minikel, E. V. et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 8, 322ra9 (2016)
Chong, J. X. et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 97, 199–215 (2015)
Kathiresan, S. Developing medicines that mimic the natural successes of the human genome: lessons from NPC1L1, HMGCR, PCSK9, APOC3, and CETP . J. Am. Coll. Cardiol. 65, 1562–1566 (2015)
Lim, E. T. et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 10, e1004494 (2014)
Ruderfer, D. M. et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nature Genet. http://dx.doi.org/10.1038/ng.3638 (2016)
Sulem, P. et al. Identification of a large set of rare complete human knockouts. Nature Genet. 47, 448–452 (2015)
Narasimhan, V. M. et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science http://dx.doi.org/10.1126/science.aac8624 (2016)
Saleheen, D. et al. Human knockouts in a cohort with a high rate of consanguinity. Preprint at bioRxiv http://dx.doi.org/10.1101/031518 (2015)
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nature Neurosci. 18, 631–636 (2015)
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 43, 491–498 (2011)
Voight, B. F. et al. The Metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 8, e1002793 (2012)
Zook, J. M. et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nature Biotechnol. 32, 246–251 (2014)
Acknowledgements
We would like to thank the scientific community for their support and comments on bioRxiv, Twitter, and other public forums, and B. Bulik-Sullivan, J. Bloom and R. Walters for their help with mathematical notation. The full acknowledgements are detailed in Supplementary Information section 8.
Author information
Authors and Affiliations
Consortia
Contributions
M.Le., K.J.K., E.V.M., K.E.S., E.B., T.F., A.H.O., J.S.W., A.J.H., B.B.C., T.T., D.P.B., J.A.K., L.E.D., K.E., F.Z., J.Z., E.P., M.J.D. and D.G.M. contributed to the analysis and writing of the manuscript. M.Le., E.B., T.F., K.J.K., E.V.M., F.Z., D.P.B., J.B., D.N.C., N.D., M.D., R.D., J.F., M.F., L.G., J.G., N.G., D.H., A.K., M.I.K., A.L.M., P.N., L.O., G.M.P., R.P., M.A.R., V.R., S.A.R., D.M.R., K.S., P.D.S., C.S., B.P.T., G.T., M.T.T., B.W., H.W., D.Y., S.B.G., M.J.D. and D.G.M. contributed to the production of the ExAC data set. D.M.A., D.A., M.B., J.D., S.D., R.E., J.C.F., S.B.G., G.G., S.J.G., C.M.H., S.K., M.La., S.M., M.I.M., D.M., R.M., B.M.N., A.P., S.M.P., D.S., J.M.S., P.S., P.F.S., J.T., M.T.T., H.C.W., J.G.W., M.J.D. and D.G.M. contributed to the design and conduct of the various exome sequencing studies and review of the manuscript.
Corresponding author
Ethics declarations
Competing interests
P.F.S. is a scientific advisor to Pfizer.
Additional information
ExAC data set is publicly available at (http://exac.broadinstitute.org).
Reviewer Information Nature thanks L. Biesecker, J. Shedure and the other anonymous reviewer(s) for their contribution to the peer review of this work.
A list of participants and their affiliations appears in the Supplementary Information
Extended data figures and tables
Extended Data Figure 1 The effect of recurrence across different mutation and functional classes.
a, TiTv (transition to transversion) ratio of synonymous variants at downsampled intervals of ExAC. The TiTv is relatively stable at previous sample sizes (<5,000), but changes drastically at larger sample sizes. b, For synonymous doubleton variants, mutability of each trinucleotide context is correlated with mean Euclidean distance of individuals that share the doubleton. Transversion (red), and non-CpG transition (green) doubletons are more likely to be found in closer PCA space (more similar ancestries) than CpG transitions (blue). c, The proportion singleton among various functional categories. The functional category stop lost has a higher singleton rate than nonsense. Error bars represent standard error of the mean. d, Among synonymous variants, mutability of each trinucleotide context is correlated with proportion singleton, suggesting CpG transitions (blue) are more likely to have multiple independent origins driving their allele frequency up. e, The proportion singleton metric from c, broken down by transversions, non-CpG transitions, and CpG variants. Notably, there is a wide variation in singleton rates among mutational contexts in functional classes, and there are no stop-lost (variants that result in the loss of a stop codon) CpG transitions. Error bars represent standard error of the mean.
Extended Data Figure 2 Multi-nucleotide variants discovered in the ExAC data set.
a, Number of MNPs per impact on the variant interpretation. b, Distribution of the number of MNPs per sample where phasing changes interpretation, separated by allele frequency. Common >1%, rare <1%. MNPs comprised of a rare and common allele are considered rare as this defines the frequency of the MNP.
Extended Data Figure 3 Relationships between depth and observed versus expected variants, as well as correlations between observed and expected variant counts for synonymous, missense, and protein-truncating.
a, The relationship between the median depth of exons (bins of 2) and the sum of all observed synonymous variants in those exons divided by the sum of all expected synonymous variants. The curve was used to determine the appropriate depth adjustment for expected variant counts. For the rest of the panels, the correlation between the depth-adjusted expected variants counts and observed are depicted for synonymous (b), missense (c), and protein-truncating (d). The black line indicates a perfect correlation (slope = 1). Axes have been trimmed to remove TTN.
Extended Data Figure 4 Number of protein-truncating variants in constrained genes per individual by allele frequency bin.
Equivalent to Fig. 5b limited to constrained (pLI ≥ 0.9) genes.
Extended Data Figure 5 Principal component analysis (PCA) and key metrics used to filter samples.
a, Principal component analysis using a set of 5,400 common exome SNPs. Individuals are coloured by their distance from each of the population cluster centres using the first 4 principal components. b, The metrics number of variants, TiTv, alternate heterozygous/homozygous (HetHom) ratio and indel (InsDel) ratio. Populations are Latino (red), African (purple), European (blue), South Asian (yellow) and East Asian (green).
Related audio
41586_2016_BFnature19057_MOESM242_ESM.mp3
The ExAC database is the largest collection of human protein-coding variation yet, providing scientific and clinical insight.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data, Supplementary References, Supplementary Tables 1-5, 7-8, 10-12, 14-15, 17-18, 21-25, (see separate zipped file for Tables 6, 9, 13, 16, 19 and 20) and Supplementary Figures 1-5 - see Contents on pages 8-9 for more details. (PDF 6547 kb)
Supplementary Tables
This zipped file contains Supplementary Tables 6, 9, 13, 16, 19 and 20. (ZIP 5918 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lek, M., Karczewski, K., Minikel, E. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). https://doi.org/10.1038/nature19057
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature19057
This article is cited by
-
MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach
Genome Medicine (2024)
-
Explicable prioritization of genetic variants by integration of rule-based and machine learning algorithms for diagnosis of rare Mendelian disorders
Human Genomics (2024)
-
Statistical methods for assessing the effects of de novo variants on birth defects
Human Genomics (2024)
-
Single-cell RNA sequencing in donor and end-stage heart failure patients identifies NLRP3 as a therapeutic target for arrhythmogenic right ventricular cardiomyopathy
BMC Medicine (2024)
-
Diverse functions of cytochrome c in cell death and disease
Cell Death & Differentiation (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
