
Abstract
More than two hundred individuals developed Creutzfeldt–Jakob disease (CJD) worldwide as a result of treatment, typically in childhood, with human cadaveric pituitary-derived growth hormone contaminated with prions1,2. Although such treatment ceased in 1985, iatrogenic CJD (iCJD) continues to emerge because of the prolonged incubation periods seen in human prion infections. Unexpectedly, in an autopsy study of eight individuals with iCJD, aged 36–51 years, in four we found moderate to severe grey matter and vascular amyloid-β (Aβ) pathology. The Aβ deposition in the grey matter was typical of that seen in Alzheimer’s disease and Aβ in the blood vessel walls was characteristic of cerebral amyloid angiopathy3 and did not co-localize with prion protein deposition. None of these patients had pathogenic mutations, APOE ε4 or other high-risk alleles4 associated with early-onset Alzheimer’s disease. Examination of a series of 116 patients with other prion diseases from a prospective observational cohort study5 showed minimal or no Aβ pathology in cases of similar age range, or a decade older, without APOE ε4 risk alleles. We also analysed pituitary glands from individuals with Aβ pathology and found marked Aβ deposition in multiple cases. Experimental seeding of Aβ pathology has been previously demonstrated in primates and transgenic mice by central nervous system or peripheral inoculation with Alzheimer’s disease brain homogenate6,7,8,9,10,11. The marked deposition of parenchymal and vascular Aβ in these relatively young patients with iCJD, in contrast with other prion disease patients and population controls, is consistent with iatrogenic transmission of Aβ pathology in addition to CJD and suggests that healthy exposed individuals may also be at risk of iatrogenic Alzheimer’s disease and cerebral amyloid angiopathy. These findings should also prompt investigation of whether other known iatrogenic routes of prion transmission may also be relevant to Aβ and other proteopathic seeds associated with neurodegenerative and other human diseases.
Similar content being viewed by others

Main
Human transmission of prion disease has occurred as a result of a range of medical and surgical procedures worldwide as well as by endocannibalism in Papua New Guinea, with incubation periods that can exceed five decades12,13. A well-recognized iatrogenic route of transmission was by treatment of persons of short stature with preparations of human growth hormone, extracted from large pools of cadaver-sourced pituitary glands, some of which were inadvertently prion-contaminated. Such treatments commenced in 1958 and ceased in 1985 following the reports of the occurrence of CJD amongst recipients. A review of all 1,848 patients who were treated with cadaveric-derived human growth hormone (c-hGH) in the United Kingdom from 1959 through 1985 found that 38 had developed CJD by the year 2000 with a peak incubation period of 20 years1. Multiple preparations using different extraction methods were used over this period and patients received batches from several preparations. One preparation (Wilhelmi) was common to all patients who developed iCJD and size-exclusion chromatography, used in non-Wilhelmi preparation methods, may have reduced prion contamination1. As of 2012, a total of 450 cases of iatrogenic CJD have been recognized worldwide after treatment with c-hGH or gonadotropin (226 cases), transplantation of dura mater (228) or cornea (2), and neurosurgery (4) or electroencephalography recording using invasive medical devices (2)2. In France, 119/1,880 (6.3%) recipients developed iCJD, in the UK 65/1,800 (3.6%) and in the USA 29/7,700 (0.4%)2,14.
Since 2008, most UK patients with prion disease have been recruited into the National Prion Monitoring Cohort study5, including 22 of 24 recent patients with iatrogenic CJD (iCJD) related to treatment with c-hGH over this period, all of whom necessarily have very long incubation periods. Of this group of patients with iCJD, eight patients (referenced no.s 1, 2, 3, 4, 5, 6, 7, 8, Supplementary Information) aged 36–51 years, with an incubation period from first treatment to onset of 27.9–38.9 years (mean 33 years) and from last treatment to onset of 18.8–30.8 years (mean 25.5 years), underwent autopsy with extensive brain tissue sampling at our hospital. In all eight brain samples we confirmed prion disease with abnormal prion protein labelling of the neuropil, perineuronal network and in most cases microplaques as described previously15,16,17. However, four (no.s 4, 5, 6, 8) of the eight patients with iCJD also showed substantial amyloid-β (Aβ) deposition in the central nervous system parenchyma by histology (Fig. 1) and immunoblotting (Fig. 2). A further two brain samples (no.s 1, 3) had focal Aβ pathology in one of the brain regions; one showed Aβ entrapment in PrP plaques and only one was entirely negative for Aβ. Furthermore, there was widespread cortical and leptomeningeal cerebral Aβ angiopathy (CAA)3 in three patients (no.s 4, 6, 8) and focal CAA in one patient (no. 5) (Fig. 1). Such pathology is extremely rare in this age range, 10/290 in the equivalent 36–50 year age strata without CJD18, P = 0.0002, Fisher’s test. None of our patients with iCJD had pathogenic mutations in the prion protein gene (PRNP). We used a custom next generation sequencing panel4 to exclude mutation in any of 16 other genes associated with early-onset Alzheimer’s disease, CAA, or other neurodegenerative disorders, and none carried APOE ε4 or TREM2 R47H alleles (Supplementary Table 2). Although such observations are unprecedented in our wide experience of human prion diseases, we nevertheless considered whether prion disease itself might predispose to, or accelerate, Aβ pathology, for example by cross-seeding of protein aggregation or overload of clearance mechanisms for misfolded proteins. We therefore compared the Aβ pathology in the iCJD cohort with that of a cohort of 116 patients with other prion diseases who had undergone autopsy: sporadic CJD (sCJD) (n = 85, age 42–83), variant CJD (n = 2, age 25 and 36) and inherited prion diseases (IPD) (n = 29, age 29–86). None of the patients in the control cohorts had comparable Aβ pathology (Consortium to Establish a Registry for Alzheimer’s disease (CERAD) score, P = 0.001, CAA, P = 0.005, topographical Aβ score P = 0.02, and cumulative Aβ score P = 0.02 (rank sum test) and digital Aβ quantification P = 0.04 (t-test); all restricted to the strata aged 36–51 years (n = 19)) (Fig. 3, and Extended Data Figs 1 and 2 show similar results in adjusted analyses in the full cohort). Indeed none of 35 prion cases aged 52–60 had significant Aβ pathology, with the exception of two cases at ages 57 and 58 positive for APOE ε4 alleles. Instead, the sCJD cohort shows Aβ pathology in parenchyma and blood vessels to a similar extent/severity as seen in iCJD, only in a much older age group (Extended Data Figs 1 and 2), in keeping with the chance coincidence of late-onset Aβ pathology and sCJD as previously documented in a large study of 110 sCJD patients and 110 age-matched controls aged 27–84 (ref. 19) and a study of 2,661 individuals aged 26–95 (ref. 18). Further, we investigated whether prion and Aβ pathology co-localize in the iCJD cases. In our series there was a distinct absence of overlap of Aβ plaques and PrP (Fig. 1d, e) or Aβ CAA and vascular PrP (Fig. 1b, c), consistent with these pathologies developing independently.

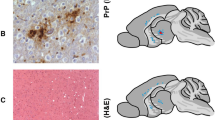
a, Frontal cortex with widespread diffuse Aβ deposition, formation of plaques, and widespread parenchymal and leptomeningeal CAA (patient no. 4). b, c, Non-colocalized deposition of Aβ and prion protein. Vessels with CAA do not entrap or co-seed prion protein. d, e, Adjacent histological sections stained for Aβ or prion protein show clearly separated plaques of both proteins (no. 5). f, An overlay with colour inversion of prion protein plaques highlights the separation. g, h, Dual labelling, confocal laser microscopy shows no co-localization of parenchymal Aβ plaques (no.s 5, 6) or CAA (no. 6). i, Aβ is detected in pituitary glands in patients with a high Aβ load in the brain. Scale bar corresponds to 200 μm in a, 100 μm in b–h, and 50 μm in i.

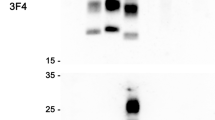
a–c, 10% (w/v) brain homogenates from patients with iCJD were analysed by enhanced chemiluminescence using anti-human Aβ monoclonal antibodies 6E10 that recognizes full-length APP and fragments that contain the epitope including Aβ (a) or 82E1 that specifically recognizes Aβ (b) or secondary antibody only (c). The identity of the patient brain sample is designated above each lane and the position of molecular mass markers is shown to the left. The equivalent of 5 µl 10% (w/v) brain homogenate was loaded per lane. The migration position of Aβ is indicated by the arrow. For gel source data, see Supplementary Fig. 1.

a, Widespread, moderate-to-severe early-onset CAA in three, and focal, mild CAA in one iCJD patient but only one focal, mild CAA in 19 controls. b, Significant differences of parenchymal Aβ accumulation (all central nervous system regions, see supplementary material). c, d, Cortical Aβ load was assessed semiquantitatively and quantitatively and again was significantly different between the iCJD and age-matched control cohort. For methods of quantification and calculations of significance levels see Supplementary Information.
We then went on to examine pituitary glands for the presence of Aβ deposits. Pathological species of tau, Aβ and α-synuclein have been reported in the pituitary gland of patients with neurodegenerative disease and controls20. We examined 55 pituitary glands, 6 from patients without, and 49 from patients with cerebral Aβ pathology, and found in the latter group seven samples containing Aβ, confirming frequent Aβ in pituitaries of patients with Alzheimer’s disease-like pathology20 (Fig. 1i and Extended Data Fig. 3), consistent with the hypothesis that Aβ seeds have been iatrogenically transmitted to these patients with iCJD.
There has been longstanding interest as to whether other neurodegenerative diseases associated with the accumulation of aggregates of misfolded host proteins or amyloids might be transmissible in a ‘prion-like’ fashion21,22. Experimental seeding of Aβ pathology has previously been demonstrated in primates and transgenic mice by central nervous system inoculation with Alzheimer’s disease brain homogenate6,7,8,9,10. Of particular interest with respect to our findings is that peripheral (intraperitoneal) inoculation with Alzheimer’s disease brain extract into APP23 (ref. 11) transgenic mice has been demonstrated. While ageing APP23 mice show mostly parenchymal deposits, the intraperitoneally-seeded mice showed predominantly CAA, a feature seen in patients with iCJD who had significant Aβ pathology. This experimental study and our findings suggest that there are mechanisms to allow the transport of Aβ seeds as well as prions (and possibly other proteopathic seeds such as tau23) from the periphery to the brain24,25. While less than 4% of UK c-hGH treated individuals have developed iCJD, one out of eight patients with iCJD had focal, and three had widespread, moderate or severe CAA. Four patients had widespread parenchymal Aβ pathology and two further patients had focal cortical Aβ deposits. This might suggest that healthy individuals exposed to c-hGH are at high risk of developing early-onset Aβ pathology as this cohort ages.
Although none of the iCJD cases with Aβ pathology had hyperphosphorylated tau neurofibrillary tangle pathology characteristic of Alzheimer’s disease, it is possible that the full neuropathology of Alzheimer’s disease would have developed had these individuals not succumbed to prion disease at these relatively young ages. An earlier study concluded that c-hGH recipients did not seem to be at increased risk of Alzheimer’s disease, but this was based on death certificates only without autopsy data20. However, the severe CAA seen in the patients with iCJD in our study is unquestionably concerning and individuals with such pathology would be at increasing risk of cerebral haemorrhages had they lived longer. At-risk individuals, including patients who had received dura mater grafts26 could be screened by magnetic resonance imaging (MRI) for CAA-related pathologies (such as microbleeds) and by positron emission tomography (PET) for Aβ deposition27.
It is possible, however, that prions and Aβ seeds co-purify in the extraction methods used to prepare c-hGH, which might mean that there would be a relatively higher occurrence of Aβ pathology in those with iatrogenic prion infection. Analysis of any residual archival batches of c-hGH for both prions and Aβ seeds might be informative in this regard2. While our data argue against cross seeding, we cannot formally exclude the possibility that prions somehow seed Aβ deposition but do not co-localize with Aβ deposits. While there is no suggestion that Alzheimer’s disease is a contagious disease and no supportive evidence from epidemiological studies that Alzheimer’s disease is transmissible, notably by blood transfusion28,29, our findings should prompt consideration of whether other known iatrogenic routes of prion transmission, including surgical instruments and blood products, may also be relevant to Aβ and other proteopathic seeds seen in neurodegenerative diseases. Aβ seeds are known, like prions, to adhere to metal surfaces and to resist formaldehyde inactivation and conventional hospital sterilisation30.
Methods
No statistical methods were used to predetermine sample size, the experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Patient recruitment and genotyping
A national referral system for prion diseases was established by the Chief Medical Officer in the UK in 2004. UK neurologists were asked to refer all patients with suspected prion disease jointly to the National CJD Research and Surveillance Unit in Edinburgh and the NHS National Prion Clinic (NPC) in London. All patients with possible CJD who had received cadaver-derived growth hormone were referred to the NHS National Prion Clinic (London, UK) and since 2008 were recruited into the National Prion Monitoring Cohort study.
Next-generation sequencing to exclude mutations known to be causal of Aβ pathology
Deep next-generation sequencing using a custom panel was performed as described previously31. Analysis was done using NextGENe and Geneticist Assistant software (Softgenetics, USA). Variants were assessed for pathogenicity by reference to the published literature, control population allele frequencies (our primary database for allele frequency was the Broad Institute’s ExAC browser (http://exac.broadinstitute.org/)) and in silico predictive tools. The analysis methodology has been validated for the detection of APP duplication31, which was important to exclude. No causal mutations for dementia or Aβ pathology were detected, see Supplementary Table 2. As expected, several rare variants were detected which may modify the risk of various neurodegenerative diseases, see Supplementary Table 2.
Autopsies and tissue preparation
Autopsies were carried out in a post mortem room designated for high risk autopsies. Informed consent to use the tissue for research was obtained in all cases. Ethical approval for these studies was obtained from the Local Research Ethics Committee of the UCL Institute of Neurology/National Hospital for Neurology and Neurosurgery. The anterior frontal, temporal, parietal and occipital cortex and the cerebellum (at the level of dentate nucleus) were dissected during the post mortem procedure and frozen. Samples of the following areas were taken and analysed: frontal, temporal, parietal, occipital, posterior frontal cortex including motor strip, basal ganglia, thalamus, hippocampus, brain stem including midbrain, and cerebellar hemisphere and vermis. Pituitary glands were taken in all cases.
Tissue samples were immersed in 10% buffered formalin and prion infectivity was inactivated by immersion into 98% formic acid for one hour. Tissue samples were processed to paraffin wax and tissue sections were routinely stained with haematoxylin and eosin.
Antibodies and immunohistochemistry
The following antibodies were used: Anti-PrP ICSM35 (D-Gen Ltd, London, UK32,33 1:1,000), Anti-phospho-Tau (AT-8, Innogenetics, 1:100) and anti-βA4 (DAKO 6F3D, 1:50). ICSM35 was stained on a Ventana Benchmark or Discovery automated immunohistochemical staining machine (ROCHE Burgess Hill, UK); βA4 and Tau were stained on a LEICA BondMax (LEICA Microsystems) or a Ventana automated staining instrument following the manufacturer’s guidelines, using biotinylated secondary antibodies and a horseradish-peroxidase-conjugated streptavidin complex and diaminobenzidine as a chromogen.
Immunoblot detection of Aβ in iCJD brain
Biochemical studies were carried out in a microbiological containment level 3 facility with strict adherence to safety protocols. Frozen brain tissue was available from seven of eight patients with growth hormone iCJD (cases 1 and 3–8). 10% (w/v) brain homogenates (grey matter; frontal cortex) were prepared in Dulbecco’s PBS lacking Ca2+ or Mg2+ ions using tissue grinders as described previously34. 20-μl aliquots were treated with 1 µl benzonase nuclease (purity >99%; 25 U ml−1; Novagen) for 15 min at 20 °C. Samples were then mixed with an equal volume of 2× SDS sample buffer (125 mM Tris-HCl, 20% (v/v) glycerol pH 6.8 containing 4% (w/v) SDS, 4% (v/v) 2-mercaptoethanol and 0.02% (w/v) bromophenol blue) and immediately transferred to a 100 °C heating block for 10 min. Electrophoresis was performed on 16% Tris-glycine gels (Invitrogen), run for 70 min at 200 V, before electroblotting to Immobilon P membrane (Millipore) for 16 h at 15 V as described previously34. Membranes were blocked in phosphate buffered saline (PBS) containing 0.05% (v/v) Tween 20 (PBST) and 5% (w/v) non-fat dried skimmed milk powder. Blots were then probed with anti-human Aβ monoclonal antibodies 6E10 (Covance) and 82E1 (IBL international, Hamburg, Germany) at final concentrations of 0.2 µg ml−1 in PBST for at least 1 h. After washing for 1 h with PBST the membranes were probed with a 1:10,000 dilution of alkaline-phosphatase-conjugated goat anti-mouse IgG secondary antibody (Sigma-Aldrich no. A2179) in PBST. After washing (90 min with PBST and 5 min with 20 mM Tris pH 9.8 containing 1 mM MgCl2) blots were incubated for 5 min in chemiluminescent substrate (CDP-Star; Tropix Inc.) and visualized on Biomax MR film (Carestream Health Inc.). Anti-human Aβ monoclonal antibody 82E1 recognizes an epitope specific to the amino terminus of Aβ while 6Ε10 recognizes an epitope spanning residues 3–8 of Aβ and cross-reacts with full-length APP or APP fragments that contain the epitope.
Examination of prion pathology
In all iCJD cases there was variably prominent microvacuolar change in the neocortices, deep grey nuclei and cerebellar cortex. Immunostaining for the abnormal prion protein revealed synaptic labelling in all grey matter areas examined. In all but one case, there were also microplaques in all grey matter structures. Variability in the intensity of the immunoreactivity for the abnormal prion protein was evident but detailed comparison between the cases and separately within each case was not feasible as prolonged formalin fixation in some cases significantly attenuated the immunoreactivity. It was apparent that more prominent microvacuolar change and synaptic labelling for abnormal prion protein was more intense in the pre-central gyrus and parietal lobe when compared to the anterior frontal and occipital cortices. Deep cortical layers showed more severe changes. In all cases the microvacuolar degeneration and prion protein deposits in the deep grey nuclei and hippocampal formation was prominent. It was most severe in the caudate nucleus and putamen, and appeared less severe in thalamus and it was least prominent in the globus pallidus. In the cerebellar vermis there was marked granule cell atrophy and often widespread loss of Purkinje cells accompanied by severe Bergmann gliosis, while cerebellar hemispherical cortex showed only patchy loss of Purkinje cells and no significant granule cell loss. Microvacuolar degeneration in the molecular layer was more prominent in the vermis than in the cerebellar hemisphere. No apparent difference in prion protein deposition was seen in vermis and hemisphere. In the dentate nucleus variably intense synaptic prion protein immunoreactivity was present, while the cyto-architecture of the nucleus was well preserved.
Examination, classification and quantification of Aβ pathology
All brains were examined according to the ABC classification35, which assesses the topographic progression of Aβ pathology in the brain (Thal phases36), topographic progression of Tau neurofibrillary tangle pathology (Braak and Braak37) and the density of mature (senile), neuritic plaques in the neocortex (Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria38,39). To allow a more detailed assessment of neocortical Aβ the original Thal phases were modified as follows. Phase 0, no cortical Aβ; phase 0.5, 1–2 neocortical regions affected; phase 1, 3–4 neocortical regions involved; phases 2–5 were scored as published36. In addition we have carried out a semiquantitative assessment of neocortical Aβ load on a standardised region within frontal, temporal, parietal and occipital lobes, and scored as follows. 0, entirely negative; 1, a single small deposit; 2, multiple small deposits, disseminated; 3, multiple small deposits, plus an area with a larger patch; 4, diffuse moderate numbers of deposits; 5, diffuse, frequent numbers of deposits. For each case a cumulative score (0–20) of total semiquantitatively assessed Aβ load in the neocortex was calculated. Cerebral amyloid angiopathy (CAA) was graded (0–3) according to the Vonsattel criteria3. CAA was assessed in leptomeninges and parenchyma of all hemispheric lobes and cerebellum with summary score (0–30) calculated for each case.
Image acquisition and processing
Histological slides were digitised on a LEICA SCN400F scanner (LEICA Milton Keynes, UK) at ×40 magnification and 65% image compression setting during export. Slides were archived and managed on LEICA Slidepath (LEICA Milton Keynes, UK). For the preparation of light microscopy images, 1,024 × 1,024 pixel sized image captures were taken, after matching paired images (Aβ and prion staining) in Slidepath, and overlays in Fig. 1f were prepared using the colour conversion function in conjunction with the image overlay in Slidepath. Laser scanning microscopy of double immunofluorescent tissue preparations was on a ZEISS LSM710 confocal microscope (ZEISS Cambridge, UK). Publication figures were assembled in Adobe Photoshop. Data plots were generated using Prism 5 (GraphPad Software, Inc., La Jolla, USA).
Digital image analysis for cortical Aβ quantification
From all cases Aβ immunostained slides from frontal, temporal, parietal and occipital lobes were digitised as described above. Digital image analysis on 496 whole slides was performed using Definiens Developer 2.3 (Definiens, Munich, Germany). Initial tissue identification was performed at a resolution corresponding to 5× image magnification and stain detection was performed at ×10 resolution. Tissue detection and initial segmentation was done to identify all tissue within the image, separating the sample from background and non-tissue regions for further analysis. This separation was based on identification of the highly homologous relatively bright/white region of background present at the perimeter of each image. A composite raster image produced by selecting the lowest pixel value from the three comprising colour layers (RGB colour model) provided a greyscale representation of brightness. The mean brightness of this background region was used to exclude all background regions from further analysis.
Stain detection (brown) is based on the transformation of the RGB colour model to a HSD representation40. This provides a raster image of the intensity of each colour of interest (brown and blue). A series of dynamic thresholds (Tx) are then used to identify areas of interest (Ax). Initially, following exclusion of intensely stained areas with values greater than 1 arbitrary unit (au) (values range from 0au to 3au in HSD images), the 5th centile ( ) of brown stain intensity was calculated as a baseline. This represents the Tbrown stain separating the top 5% of Atissue. The standard deviation (C5δ) within the lower 95% of Atissue was used to update the Tbrown stain as
) of brown stain intensity was calculated as a baseline. This represents the Tbrown stain separating the top 5% of Atissue. The standard deviation (C5δ) within the lower 95% of Atissue was used to update the Tbrown stain as  with all pixels above this threshold classed as ‘stain’ (Astain) and those below as ‘unstained’ (Aunstained). Astain was excluded if the intensity of blue staining was not significantly lower than the level of brown stain (difference less than 0.1au) to remove generically dark areas. The remaining Astain were further categorised using thresholds based on the mean (
with all pixels above this threshold classed as ‘stain’ (Astain) and those below as ‘unstained’ (Aunstained). Astain was excluded if the intensity of blue staining was not significantly lower than the level of brown stain (difference less than 0.1au) to remove generically dark areas. The remaining Astain were further categorised using thresholds based on the mean ( ) and standard deviation (Bδ) of brown staining within the Aunstained: Tbrown =
) and standard deviation (Bδ) of brown staining within the Aunstained: Tbrown =  (lower threshold); Tdark brown =
(lower threshold); Tdark brown =  (upper threshold), to give Aunstained ≤ Tbrown > Alight brown ≤ Tdark brown > AAβ deposit. Artefacts were then identified as Astain with area greater than 1 mm2, or an area greater than 0.1 mm2 with a standard deviation of brown staining below 0.2au. These Aartefacts were then expanded to include surrounding pixels with brown staining greater than
(upper threshold), to give Aunstained ≤ Tbrown > Alight brown ≤ Tdark brown > AAβ deposit. Artefacts were then identified as Astain with area greater than 1 mm2, or an area greater than 0.1 mm2 with a standard deviation of brown staining below 0.2au. These Aartefacts were then expanded to include surrounding pixels with brown staining greater than  . This excludes large areas of homogenous staining and areas of more diffuse, non-specific chromogen deposit.
. This excludes large areas of homogenous staining and areas of more diffuse, non-specific chromogen deposit.
The white matter region within the tissue was then manually selected by an expert neuropathologist (Z.J., S.B.). This white matter was excluded from calculation of proportional coverage of AAβ deposit within Atissue.
Change history
11 September 2015
Due to an administrative error, an incorrect version of the Competing Financial Interests (CFI) statement was published in relation to this paper. Although the published CFI statement did reference the authors' affiliation with D-Gen, it did not contain all of the information provided to Nature by the authors about the interests of the company. As soon as Nature was made aware of the error, we acted quickly to correct the text and expand the CFI statement. The CFI statement for this paper as originally published was "J.C. is a Director and J.C. and J.D.F.W. are shareholders of D-Gen Limited, which supplies antibody ICSM35." The updated CFI statement is “J.C. is a Director and J.C. and J.D.F.W. are shareholders of D-Gen Limited, an academic spin-out company working in the field of prion disease diagnosis, decontamination and therapeutics. D-Gen supplied antibody ICSM35”.
References
Swerdlow, A. J., Higgins, C. D., Adlard, P., Jones, M. E. & Preece, M. A. Creutzfeldt-Jakob disease in United Kingdom patients treated with human pituitary growth hormone. Neurology 61, 783–791 (2003)
Brown, P. et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg. Infect. Dis. 18, 901–907 (2012)
Vonsattel, J. P. et al. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann. Neurol. 30, 637–649 (1991)
Beck, J. et al. Validation of next-generation sequencing technologies in genetic diagnosis of dementia. Neurobiol. Aging 35, 261–265 (2014)
Rudge, P. et al. Iatrogenic CJD due to pituitary-derived growth hormone with genetically determined incubation times of up to 40 years. Brain. http://dx.doi.org/10.1093/brain/awv235 (2015)
Baker, H. F., Ridley, R. M., Duchen, L. W., Crow, T. J. & Bruton, C. J. Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol. Neurobiol. 8, 25–39 (1994)
Eisele, Y. S. et al. Induction of cerebral β-amyloidosis: intracerebral versus systemic Aβ inoculation. Proc. Natl Acad. Sci. USA 106, 12926–12931 (2009)
Hamaguchi, T. et al. The presence of Aβ seeds, and not age per se, is critical to the initiation of Aβ deposition in the brain. Acta Neuropathol. 123, 31–37 (2012)
Heilbronner, G. et al. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 14, 1017–1022 (2013)
Meyer-Luehmann, M. et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 (2006)
Eisele, Y. S. et al. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 330, 980–982 (2010)
Collinge, J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev. Neurosci. 24, 519–550 (2001)
Collinge, J. et al. Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet 367, 2068–2074 (2006)
Abrams, J. Y. et al. Lower risk of Creutzfeldt-Jakob disease in pituitary growth hormone recipients initiating treatment after 1977. J. Clin. Endocrinol. Metab. 96, E1666–E1669 (2011)
Brandner, S. et al. Central and peripheral pathology of kuru: pathological analysis of a recent case and comparison with other forms of human prion disease. Phil. Trans. R. Soc. Lond. B 363, 3755–3763 (2008)
Ellison, D. et al. Neuropathology: A reference Text of CNS Pathology. 3rd edn (Elsevier, 2012)
Parchi, P. et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 124, 517–529 (2012)
Braak, H. & Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357 (1997)
Hainfellner, J. A. et al. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 96, 116–122 (1998)
Irwin, D. J. et al. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 70, 462–468 (2013)
Collinge, J. & Clarke, A. R. A general model of prion strains and their pathogenicity. Science 318, 930–936 (2007)
Gajdusek, D. C. Transmissible and non-transmissible amyloidoses: autocatalytic post-translational conversion of host precursor proteins to beta-pleated sheet configurations. J. Neuroimmunol. 20, 95–110 (1988)
Clavaguera, F. et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 127, 299–301 (2014)
Beekes, M., Thomzig, A., Schulz-Schaeffer, W. J. & Burger, R. Is there a risk of prion-like disease transmission by Alzheimer- or Parkinson-associated protein particles? Acta Neuropathol. 128, 463–476 (2014)
Bolmont, T. et al. Induction of tau pathology by intracerebral infusion of amyloid-β-containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. Am. J. Pathol. 171, 2012–2020 (2007)
Preusser, M. et al. Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jakob disease after dural grafting. J. Neurol. Neurosurg. Psychiatry 77, 413–416 (2006)
Rowe, C. C. et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 7, 129–135 (2008)
Daviglus, M. L. et al. Risk factors and preventive interventions for Alzheimer disease: state of the science. Arch. Neurol. 68, 1185–1190 (2011)
O’Meara, E. S. et al. Alzheimer’s disease and history of blood transfusion by apolipoprotein-E genotype. Neuroepidemiology 16, 86–93 (1997)
Fritschi, S. K. et al. Aβ seeds resist inactivation by formaldehyde. Acta Neuropathol. 128, 477–484 (2014)
Beck, J. et al. Validation of next-generation sequencing technologies in genetic diagnosis of dementia. Neurobiol. Aging 35, 261–265 (2014)
Isaacs, A. M. et al. Lack of TAR-DNA binding protein-43 (TDP-43) pathology in human prion diseases. Neuropathol. Appl. Neurobiol. 34, 446–456 (2008)
Khalili-Shirazi, A. et al. PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J. Gen. Virol. 86, 2635–2644 (2005)
Wadsworth, J. D. et al. Molecular diagnosis of human prion disease. Methods Mol. Biol. 459, 197–227 (2008)
Hyman, B. T. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13 (2012)
Thal, D. R., Rüb, U., Orantes, M. & Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800 (2002)
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H. & Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 (2006)
Mirra, S. S. et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486 (1991)
Heyman, A., Fillenbaum, G. G. & Mirra, S. S. Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): clinical, neuropsychological, and neuropathological components. Aging (Milano) 2, 415–424 (1990)
van Der Laak, J. A., Pahlplatz, M. M., Hanselaar, A. G. & de Wilde, P. C. Hue-saturation-density (HSD) model for stain recognition in digital images from transmitted light microscopy. Cytometry 39, 275–284 (2000)
Acknowledgements
This work was funded by the UK Medical Research Council and the National Institute of Health Research (NIHR) UCLH/UCL Biomedical Research Centre and Dementia Biomedical Research Unit. We are grateful to all patients and their relatives and carers for their participation in the National Prion Monitoring Cohort and for consent to autopsy and use of tissues for this research. We thank all physicians who contributed information and the National CJD Research and Surveillance Unit for coordination of patient referral and tissue sharing under the National referral scheme. We also thank J. Broni, L. Brock and T. Wilkins for histological assistance and B. Peters, R. Sinclair, C.-W. Lok and M. Karseras for support with post mortem examinations at UCLH. We thank G. Adamson, T. Campbell, J. Uphill and R. Druyeh for assistance with genetic analyses.
Author information
Authors and Affiliations
Contributions
Z.J. and S.B. performed autopsies and neuropathological diagnosis and analysis of cases. J.K., S.M., P.R. and J.C. diagnosed and recruited patients to the study and analysed clinical and genetic data. A.S.W. performed statistical analysis and modelling. J.D.F.W. and A.J.N. performed western blots, M.E., F.L., A.R.-L. and J.L. performed immunohistochemistry and analysed data, J.C. and S.B. oversaw the study and drafted the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
J.C. is a Director and J.C. and J.D.F.W. are shareholders of D-Gen Limited, an academic spin-out company working in the field of prion disease diagnosis, decontamination and therapeutics. D-Gen supplied antibody ICSM35.
Extended data figures and tables
Extended Data Figure 1 Vertical scatter plot of Aβ pathology in iCJD brain samples, age-matched and older controls.
a, CAA summary score as described in Methods. iCJD (age range 36–51) with three highly scoring brain samples with CAA. In the age-matched control groups, no comparable pathology was found. Significant CAA was only seen in a cohort including older individuals carrying the APOE ε4 allele. The outlier in the sCJD group (pink triangle in the sCJD > 52 year group) had a surgical intervention 40 years before death, and in addition to CJD also had severe CAA. b, Topographical Aβ deposition, assessed according to a scheme modified from the Thal classification as described in the Methods section. In the group of individuals of 51 years and below, significant Aβ deposition is seen in the iCJD group, but not in age-matched prion diseases of different aetiology. c, Cortical Aβ deposition, assessed according to CERAD. In the group of individuals of 51 years and below, mature (neuritic) plaques are seen in the iCJD group, but not in age-matched prion diseases of different aetiology. Only in the cohort comprising much older individuals is there an increase of cortical mature plaques. d, e, Semiquantitative assessment and quantification of neocortical Aβ using Definiens Developer image analysis shows a separation that is similar to that shown in a and b. APOE ε4 genotype was unavailable in nine of the 85 patients with sCJD and these were not included in the graphs. Note the logarithmic scale in e.
Extended Data Figure 2 Scattergram of Aβ pathology in iCJD brain samples compared with other prion diseases.
Plot of severity scores of CAA or parenchymal Aβ against the age of individuals in the cohorts, demonstrating early-onset of CAA and grey matter Aβ pathology in the iCJD cohort. a, Early-onset of CAA. The outlier in the sCJD group (pink triangle) had a surgical intervention 40 years before death, and in addition to CJD also had severe CAA. b, c, Early detection of grey matter Aβ by a topographical assessment (Thal phase) and using CERAD Criteria. d, e, Semiquantitative assessment and quantification of neocortical Aβ. APOE ε4 genotype was unavailable in nine of the 85 patients with sCJD and these were not included in the graph. Note the logarithmic scale in e.
Extended Data Figure 3 Topographical Aβ of the brain samples corresponding to the pituitaries analysed for Aβ.
Plot of the Thal phases (topographical Aβ deposition) of the brain samples corresponding to the 55 pituitary glands examined for the presence of Aβ. Aβ was assessed in 55 pituitary glands from patients with iCJD (n = 5, age range 36–47), IPD (n = 4, age range 51–95), sCJD (n = 41, age range 54–89) and non-CJD controls (n = 5, age range 72–90) (groups shown on x axis). In six patients from iCJD, IPD and sCJD groups no Aβ deposits were found in the brain or pituitary gland. In 49 patients from all groups (iCJD, IPD, sCJD and non-CJD) there were variably frequent Aβ deposits in the brain parenchyma, corresponding to Thal phases 1–5 (distribution shown on y axis). Of these 49 cases, six cases (IPD n = 1, sCJD n = 4, and non-CJD n = 1) showed Aβ deposits also in the pituitary glands (positive cases highlighted in red) and in one patient from sCJD group Aβ deposits were seen in the brain tissue attached to the pituitary gland (highlighted in blue).
Supplementary information
Supplementary Information
This file contains Supplementary Data for patients 1-8, Supplementary Tables 1-2 and additional references. (PDF 284 kb)
Rights and permissions
About this article

Cite this article
Jaunmuktane, Z., Mead, S., Ellis, M. et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250 (2015). https://doi.org/10.1038/nature15369
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature15369
This article is cited by
-
Iatrogenic Alzheimer’s disease in recipients of cadaveric pituitary-derived growth hormone
Nature Medicine (2024)
-
Signs of ‘transmissible’ Alzheimer’s seen in people who received growth hormone
Nature (2024)
-
Cryo-EM structures of Aβ40 filaments from the leptomeninges of individuals with Alzheimer’s disease and cerebral amyloid angiopathy
Acta Neuropathologica Communications (2023)
-
Alleviating the unwanted effects of oxidative stress on Aβ clearance: a review of related concepts and strategies for the development of computational modelling
Translational Neurodegeneration (2023)
-
Border-associated macrophages promote cerebral amyloid angiopathy and cognitive impairment through vascular oxidative stress
Molecular Neurodegeneration (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
