
Abstract
An epidemic of Ebola virus disease of unprecedented scale has been ongoing for more than a year in West Africa. As of 29 April 2015, there have been 26,277 reported total cases (of which 14,895 have been laboratory confirmed) resulting in 10,899 deaths1. The source of the outbreak was traced to the prefecture of Guéckédou in the forested region of southeastern Guinea2,3. The virus later spread to the capital, Conakry, and to the neighbouring countries of Sierra Leone, Liberia, Nigeria, Senegal and Mali1. In March 2014, when the first cases were detected in Conakry, the Institut Pasteur of Dakar, Senegal, deployed a mobile laboratory in Donka hospital to provide diagnostic services to the greater Conakry urban area and other regions of Guinea. Through this process we sampled 85 Ebola viruses (EBOV) from patients infected from July to November 2014, and report their full genome sequences here. Phylogenetic analysis reveals the sustained transmission of three distinct viral lineages co-circulating in Guinea, including the urban setting of Conakry and its surroundings. One lineage is unique to Guinea and closely related to the earliest sampled viruses of the epidemic. A second lineage contains viruses probably reintroduced from neighbouring Sierra Leone on multiple occasions, while a third lineage later spread from Guinea to Mali. Each lineage is defined by multiple mutations, including non-synonymous changes in the virion protein 35 (VP35), glycoprotein (GP) and RNA-dependent RNA polymerase (L) proteins. The viral GP is characterized by a glycosylation site modification and mutations in the mucin-like domain that could modify the outer shape of the virion. These data illustrate the ongoing ability of EBOV to develop lineage-specific and potentially phenotypically important variation.
Similar content being viewed by others

Main
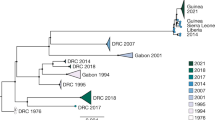
We combined our 85 Guinean EBOV sequences (Extended Data Table 1) with 110 publicly available 2014 EBOV genome sequences sampled from Guinea, Mali and Sierra Leone, producing a total data set of 195 sequences. Phylogenetic analysis reveals greater genetic diversity than previously described, with the presence of three distinct lineages, in contrast to the relatively limited variation documented early in the Sierra Leone outbreak4 (Fig. 1). The first lineage (denoted GUI-1) represents a cluster of sequences only found in Guinea, although from all urban and rural regions sampled in this country, and that is most closely related to the earliest viruses sampled in March 2014 (ref. 2). This lineage co-circulated in the greater Conakry region with viruses of the remaining two lineages described below. Notably, GUI-1 is characterized by multiple non-synonymous mutations in the nucleoprotein (NP), VP35 and GP such that it may also be phenotypically distinct, although this will require future experimental verification (Fig. 2a).

Tip times are scaled to the date of sampling (with a timescale shown on the x axis), and colour-coded according to the geographic location of sampling (at the district level for Guinea, and country level for Sierra Leone and Mali).

a, Mutations found in at least two separate sequences, showing one patient per row. Grey blocks indicate identity with the Kissidougou Guinean sequence (GenBank accession KJ660346). The top row shows the type of mutation (dark grey, intergenic; green, synonymous; red, non-synonymous), with the genomic location indicated above. Cluster assignment is shown at the left. b, The geographic distribution of EBOV variants, coloured by clusters. c, Number of Ebola virus disease patients sequenced per ten days, coloured by cluster.
These data also reveal that EBOV sequences from the two documented introductions into Mali (October and November 2014) belong to another larger cluster of Guinean viruses, denoted here as GUI-2. This phylogenetically distinct lineage is most closely related to the second cluster of Sierra Leone sequences (SLE-2), and could represent either a reintroduction from Sierra Leone or the continued diffusion in Guinea of strains related to those initially introduced to Sierra Leone. Finally, a third cluster of viruses (SLE-GUI-3) is found in Conakry, Forécariah, Dalaba and to a limited extent in Coyah (Fig. 2b), with multiple sequences falling within the third cluster4 of Sierra Leonean sequences. Such a phylogenetic structure suggests that there have been multiple migrations of EBOV into Guinea from Sierra Leone (although viral traffic from Guinea to Sierra Leone may also have occurred on occasion). An example of such cross-border virus traffic is a documented case that initiated a transmission chain in June 2014 in Conakry5, as well as transmission chains in Dalaba (260 km from Conakry), each of which is directly linked to different travellers from Sierra Leone. Although the numbers are small, the decreasing proportion of these sequences (matching the third cluster of Sierra Leone sequences) along the road from the Sierra Leonean border towards Conakry via Forécariah, and deeper inland in Coyah, might reflect the major transmission route of these viruses (Fig. 2b).
The area constituted by the urban setting of Conakry and the neighbouring prefectures harbours extensive EBOV genetic diversity, characterized by multiple co-circulating viral lineages. For example, all three lineages defined above (GUI-1 to SLE-GUI-3) co-circulated in Conakry during September and October 2014 (Figs 1 and 2c). Although early concerns associated with the presence of Ebola virus disease in high population density settings such as Conakry did not result in increased viral transmission, the capital city of Guinea nevertheless represents an important regional travel hub and highlights the challenge of controlling Ebola virus disease in and near large urban centres. In addition, although it is clear that a number of the EBOV strains circulating in Guinea are also present in neighbouring Sierra Leone (lineage SLE-GUI-3), reflecting the continued mobility of individuals between these localities during the peak of the epidemic and in the face of outbreak control measures, Guinea is also characterized by a number of independently evolving viral lineages, such that the epidemics in these countries have generated localized genetic diversity. Despite case reports reaching very low values in Conakry at several points during the summer of 2014, the recurrent transmission of the three distinct lineages in this locality is another indication of the challenges of controlling Ebola virus disease in large urban centres with highly mobile populations.
A total of 207 single nucleotide polymorphisms (SNPs) (51 non-synonymous, including 24 novel, 88 synonymous and 68 intergenic), have been fixed in individual patients within the sample of viruses analysed here. In contrast to the situation early in Sierra Leone, the viruses sampled from Guinea harbour numerous non-synonymous mutations which define lineages (Fig. 2a). Notably in GP, in which mutations could affect the efficacy of vaccines or antibody treatments, a C7025T (Pro→Ser) substitution in part defines GUI-1, and belongs to the heavily glycosylated mucin-like domain. Although O-glycosylation does involve the attachment of N-acetylglucosamine (GlcNac) to a serine (and/or threonine) residue, the sialylation pattern of this disordered domain appears to vary with the cellular environment. Two mutations (A6357G (Asn→Asp), in GP1 domain II and G7476A (Gly→Asp) in GP1 carboxy terminus) co-occur in a later branch of this cluster, whereas C7256T (His→Tyr), again in the mucin-like domain, is observed in another branch. We also observed one change in a glycosylation site (A6726G (Thr→Ala)) in a sub-cluster of sequences in SLE-GUI-3. Surprisingly, a mutation in the highly conserved interferon inhibitory domain of VP35 (C4116T) introduces a phenylalanine, characteristic of Sudan EBOV, but never previously observed in EBOV-Zaire. Another mutation in VP35, G3151A (Arg→Lys), lies in the sequence targeted by AVI-7539, a phosphorodiamidate morpholino oligomer (PMO)-based therapeutic candidate6. Studies of the phenotypic consequences of such mutations on viral components directly interacting with the host immune response could provide key insights into their epidemic potential, and also inform the therapeutic options currently considered for deployment7,8.
There has been some debate over the rate at which EBOV has evolved during the West African outbreak of EBOV, and what this may mean for the adaptive capacity of the virus, including changes in virulence9. Our estimates of the rate of nucleotide substitution for the combined Guinea and Mali and Sierra Leone data set under both strict and relaxed molecular clocks and using a variety of demographic and substitution models fall within the range of those obtained previously for EBOV4,9,10,11, with mean rates of between 0.87 × 10−3 to 0.91 × 10−3 nucleotide substitutions per site per year (range of credible intervals of 0.68 × 10−3 to 1.1 × 10−3 substitutions per site per year) (Extended Data Fig. 2). Essentially identical rates were observed when studying the Guinean viruses in isolation. However, these rates are lower than those observed during the early spread of the virus in Sierra Leone4. It is therefore possible that the rate estimate provided by ref. 4 represents a random fluctuation due to limited genetic variation within sequences from Sierra Leone sampled over a relatively short time-period, and/or has been elevated by the presence of transient deleterious mutations that have yet to be removed by purifying selection, as suggested by those authors4. Indeed, evolutionary rates in RNA viruses are known to have a strongly time-dependent quality, such that they are expected to be higher in the short-term than the long-term12. In addition, it is possible that differences in rate estimates in part reflect minor differences in substitution model parameters, the duration of intra-host virus evolution, as well as local epidemiological variation. More generally, it is difficult to translate relatively small differences in estimates of substitution rate, such as those obtained for EBOV in West Africa, into predictions on the future evolution of such key phenotypic traits as virulence, as the latter are more dependent on the nature of the selection pressures acting on the virus as well as the complex relationship between virulence and transmissibility. The data presented here indicates EBOV is able to generate and fix nucleotide and amino acid variation within co-circulating viral lineages on the time-scale of individual outbreaks, including the presence of country-specific lineages, and which may ultimately produce variants with important fitness differences.
Continued genomic surveillance is a strong complement to sometimes difficult local epidemiological investigations. We believe that the deployment of additional next-generation sequencing facilities in the West African surveillance network, thereby avoiding the logistical and regulatory13 hurdles associated with long-distance sample transportation, will positively contribute to the control of the current epidemic and help limit future outbreaks.
Methods
Ethics statement
This study has been evaluated and approved by the ethics committee of Guinea (ref: 35/CNERS/15), the Ebola research committee and the Institutional Review Board at Institut Pasteur. The Office of the Guinean Ethics and Scientific Review Committee granted a waiver to provide written consent to sequence and make publicly available viral sequences obtained from patient and contact samples collected during the Ebola virus disease outbreak in Guinea.
Sample collection and processing
Samples were collected from suspected Ebola cases hospitalized at an Ebola treatment centre in Conakry (Donka hospital) or from other regions of Guinea. EBOV detection was tested by quantitative RT–PCR using a Taqman assay with 5-FAM and 3-TAMRA probes on a portable Smart-Cycler TD. Each sample was run three times on three separate assays.
Carrier RNA and host ribosomal RNA depletion
Carrier RNA and host ribosomal RNA was depleted from RNA samples as described in ref. 14, using the NEBNext rRNA Depletion Kit (New England Biolabs).
cDNA synthesis, Nextera library construction and Illumina sequencing of EBOV samples
RNA from selective depletion was used for cDNA synthesis and Illumina library preparation as described previously4,14. Each individual sample was indexed with a unique dual barcode and libraries were pooled equally and sequenced on a HiSeq2500 (101-base-pair (bp) paired-end reads; Illumina) platform.
Demultiplexing of raw Illumina sequencing reads
Illumina Analysis Pipeline version 1.8 was used for image analysis, base calling, error estimation and demultiplexing.
Mapping of full-length EBOV genomes
Sequencing read pairs were obtained, from which low-quality bases and remaining adaptor/barcode sequences were removed. Reads were mapped to a 2014 EBOV genome (GenBank accession number: KM233070) using the CLC Genomics Assembly Cell v4.2 implemented in Galaxy15,16,17. All genomes generated here were annotated and manually inspected for accuracy, such as the presence of intact open reading frames, using Geneious v8 (ref. 18). Multiple sequence alignments across all EBOV from the 2014–2015 outbreak were generated by first aligning amino acid sequences using MUSCLE19, and then aligning the nucleotide sequence based on the amino acid alignment.
Screening for recombinant sequences
To screen for potential recombination in the 2014–2015 EBOV sequences we used the RDP, GENECONV, MAXCHI, CHIMAERA, 3SEQ, BOOTSCAN and SISCAN methods as implemented in the RDP4 (ref. 20) software package with default settings. No recombinant sequences were identified, nor was there any evidence for phylogenetic incongruence among the sequences analysed here.
Phylogenetic tree inference
Phylogenetic trees on our total data set of 195 sequences, 18,959 bp alignment length, were estimated using the maximum likelihood (ML) procedure in RAxML v8, employing the GTR-Γ model of nucleotide substitution. Fifty instances were run to obtain the best tree, and statistical support for each node was calculated using the standard bootstrapping algorithm with 500 pseudoreplicates (Extended Data Fig. 1). A topologically equivalent ML tree was obtained using the GTR+Γ model available in PhyML21.
Analysis of evolutionary rates
We employed the Bayesian Markov Chain Monte Carlo (MCMC) method in BEAST v1.8 (ref. 22) to estimate the rate of EBOV evolution (nucleotide substitution) during the 2014–2015 epidemic. The date (day) for each individual sample was based on the time of diagnostic testing. Importantly, very similar estimates were obtained using a variety of substitution, coalescent and molecular clock models, namely (i) the HKY-Γ and GTR-Γ nucleotide substitution models (with four categories of the gamma distribution of among-site rate variation, Γ), (ii) constant population size, Bayesian SkyGrid, and exponential population tree priors, and (iii) strict and relaxed (uncorrelated lognormal) molecular clocks (Extended Data Fig. 2). In all cases the MCMC was run until convergence was (easily) achieved (Extended Data Fig. 2). A broadly similar substitution rate (0.99 × 10−3 substitutions per site per year), and relatively strong temporal structure (correlation coefficient = 0.87, R2 = 0.76), was obtained using a regression of root-to-tip genetic distance in the ML (PhyML) tree against sampling date using the Path-O-Gen program (http://tree.bio.ed.ac.uk/software/pathogen/). The posterior distribution of trees obtained from the BEAST analysis was also used to obtain the maximum clade credibility (MCC) tree for these sequences. For simplicity, we used the results of the HKY+Γ, constant population size, strict molecular clock analysis to create the MCC tree as this had the narrowest distribution (Extended Data Fig. 2). Prior to inference of the MCC tree, 10% of the runs were removed as burn-in.
Glycoprotein RNA editing
The RNA editing site of the GP gene consists of 7 U residues; co-transcriptional stuttering can result in transcripts with more or less A residues. The resulting frameshifts allow for the expression of distinct glycoproteins called sGP (7 A), GP (predominantly 8 A), and ssGP (predominantly 6 A). Deep sequencing revealed 8 U at ∼1% and 7 U at ∼99%, values similar to those described in ref. 4.
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
References
The World Health Organization http://www.who.int/csr/don/archive/disease/ebola/en/ (2014)
Baize, S. et al. Emergence of Zaire Ebola virus disease in Guinea. N. Engl. J. Med. 371, 1418–1425 (2014)
Marí Saéz, A. et al. Investigating the zoonotic origin of the West African Ebola epidemic. EMBO Mol. Med. 7, 17–23 (2015)
Gire, S. K. et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372 (2014)
Faye, O. et al. Chains of transmission and control of Ebola virus disease in Conakry, Guinea, in 2014: an observational study. Lancet Infect. Dis. 15, 320–326 (2015)
Warren, T. K. et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nature Med. 16, 991–994 (2010)
Kugelman, J. R. et al. Evaluation of the potential impact of Ebola virus genomic drift on the efficacy of sequence-based candidate therapeutics. MBio 6, (2015)
Sayburn, A. WHO gives go ahead for experimental treatments to be used in Ebola outbreak. Br. Med. J. 349, g5161 (2014)
Hoenen, T. et al. Mutation rate and genotype variation of Ebola virus from Mali case sequences. Science 348, 117–119 (2015)
Duchêne, S., Holmes, E. C. & Ho, S. Y. Analyses of evolutionary dynamics in viruses are hindered by a time-dependent bias in rate estimates. Proc. R. Soc. B 281, 20140732 (2014)
Biek, R., Walsh, P. D., Leroy, E. M. & Real, L. A. Recent common ancestry of Ebola Zaire virus found in a bat reservoir. PLoS Pathog. 2, e90 (2006)
Carroll, S. A. et al. Molecular evolution of viruses of the family Filoviridae based on 97 whole-genome sequences. J. Virol. 87, 2608–2616 (2013)
Devaux, C. A. The hidden face of academic researches on classified highly pathogenic microorganisms. Infect. Genet. Evol. 29, 26–34 (2015)
Matranga, C. B. et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 15, 519 (2014)
Blankenberg, D. et al. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protocols Mol. Biol. (2010)
Giardine, B. et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 (2005)
Goecks, J., Nekrutenko, A. & Taylor, J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86 (2010)
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012)
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
Martin, D. P. et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463 (2010)
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010)
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012)
Acknowledgements
This study was supported by the Pasteur Ebola Task Force (PETF) and has also received funding from the French government’s Investissement d’Avenir programme, Laboratoire d’Excellence ‘Integrative Biology of Emerging Infectious Diseases’ (grant number ANR-10-LABX-62-IBEID) and Institut Pasteur de Dakar. We are grateful to all members of the PETF for their support, and in particular F. Rey and K. Victoir. High-throughput sequencing was performed on the Genomics Platform of Institut Pasteur, member of ‘France Génomique’ consortium (ANR10-INBS-09-08). We thank L. Ma for technical assistance and Institut Pasteur Clinical Research Department for their help with ethical approval procedures. E.C.H. is supported by an NHMRC Australia fellowship.
Author information
Authors and Affiliations
Contributions
E.S.-L., E.C.H. and A.A.S. designed the experiments. Ous.F., Our.F., L.K., N.M., S.K., G.F. and A.A.S. collected samples and epidemiological data. C.B.M. and P.C.S. provided protocols for sample processing and sequencing. E.S.-L., J.-M.T., L.D., C.B., M.V., V.C. and J.-C.M. performed the deep sequencing. E.S-L., E.C.H. and J.P.B. analysed the genomic sequence data. E.S-L. and E.C.H. wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Maximum likelihood phylogenetic tree of EBOV from the 2014–2015 outbreak in West Africa.
Published sequences from Sierra Leone are shown in blue, those from Mali in green, and those from Guinea in red. All horizontal branch lengths are scaled to the number of nucleotide substitutions per site. Bootstrap values are shown for key nodes. The tree is rooted according to the topology seen in the MCC tree (Fig. 1) under the assumption of a molecular clock, although the observation of three main lineages of EBOV in Guinea is robust to rooting position (including rooting on the oldest sequences from March 2014).
Extended Data Figure 2 Substitution rates and temporal signal.
a, Posterior distribution of nucleotide substitution rates (×10−4 substitutions per site per year) in the 195 sequence EBOV data set and using a range of substitution (HKY+Γ and GTR+Γ), demographic (constant population size, exponential population growth, Bayesian SkyGrid), and molecular clock (strict, relaxed lognormal (UCLN)) models. Note the extensive overlap among estimates under a range of models. b, Root-to-tip regression of genetic distance against day of sampling for the 195 sequence EBOV data set.
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Simon-Loriere, E., Faye, O., Faye, O. et al. Distinct lineages of Ebola virus in Guinea during the 2014 West African epidemic. Nature 524, 102–104 (2015). https://doi.org/10.1038/nature14612
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14612
This article is cited by
-
Ebola and Marburg Virus Infections in Resource-Rich Countries: Implications for Future Outbreaks
Current Infectious Disease Reports (2023)
-
The ability of single genes vs full genomes to resolve time and space in outbreak analysis
BMC Evolutionary Biology (2019)
-
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak
Nature Communications (2018)
-
Filovirus – Auslöser von hämorrhagischem Fieber
Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz (2018)
-
Mathematical models for devising the optimal Ebola virus disease eradication
Journal of Translational Medicine (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
