
Abstract
West Africa is currently witnessing the most extensive Ebola virus (EBOV) outbreak so far recorded1,2,3. Until now, there have been 27,013 reported cases and 11,134 deaths. The origin of the virus is thought to have been a zoonotic transmission from a bat to a two-year-old boy in December 2013 (ref. 2). From this index case the virus was spread by human-to-human contact throughout Guinea, Sierra Leone and Liberia. However, the origin of the particular virus in each country and time of transmission is not known and currently relies on epidemiological analysis, which may be unreliable owing to the difficulties of obtaining patient information. Here we trace the genetic evolution of EBOV in the current outbreak that has resulted in multiple lineages. Deep sequencing of 179 patient samples processed by the European Mobile Laboratory, the first diagnostics unit to be deployed to the epicentre of the outbreak in Guinea, reveals an epidemiological and evolutionary history of the epidemic from March 2014 to January 2015. Analysis of EBOV genome evolution has also benefited from a similar sequencing effort of patient samples from Sierra Leone. Our results confirm that the EBOV from Guinea moved into Sierra Leone, most likely in April or early May. The viruses of the Guinea/Sierra Leone lineage mixed around June/July 2014. Viral sequences covering August, September and October 2014 indicate that this lineage evolved independently within Guinea. These data can be used in conjunction with epidemiological information to test retrospectively the effectiveness of control measures, and provides an unprecedented window into the evolution of an ongoing viral haemorrhagic fever outbreak.
Similar content being viewed by others


Main
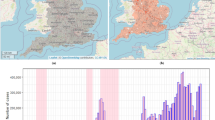
We used a deep sequencing approach to gain insight into the evolution of Ebola virus (EBOV) in Guinea from the ongoing West African outbreak. This was an approach based on analysis pipelines developed for a guinea-pig model of EBOV infection and Hendra virus infection of human and bat cells4,5. Here we use this approach to derive consensus EBOV genomes from individual patient samples that can be used to study viral genome evolution during the course of the outbreak. Viral genomes were derived primarily from blood samples that had been taken from patients in Guinea and sent to the European Mobile Laboratory (EMLab), deployed by the World Health Organisation within the Médecins Sans Frontières Ebola Treatment Centre Guéckédou in March 2014 to aid the diagnostic effort. With the permission of Guinean authorities a biobank of samples was assembled which had known provenance of EBOV infection. Linked to each sample were the following data: patient location (to district level), sample collection date, disease onset and outcome. The collection dates were a median of 4 days after the date of onset of symptoms. Baseline data was cleaned, formatted and imported into the Geographic Information System, ESRI ArcGIS. Statistical tools were used to generate tabular output and to join the numeric case data with the district level boundaries of Guinea, Liberia and Sierra Leone (district geometries freely available from http://www.gadm.org/) (Fig. 1a).

a, Geographical location of patient samples. The origin of the sequenced samples (one sample per patient) from Guinea, Sierra Leone, and Liberia processed by EMLab Guéckédou are plotted as numbers of cases by district. EMLab data are overlaid on an Ebola outbreak distribution map where cumulative cases are plotted as a heat map (low (yellow) to high (brown)) of confirmed cases from March 2014 to January 2015. Case data sourced from World Health Organization (WHO) Ebola response situation reports (http://apps.who.int/ebola/en/ebola-situation-reports); Geographic Information Systems (GIS) data sourced from Environmental Systems Research Institute (ESRI) and Database of Global Administrative Areas (GADM; http://www.gadm.org/). b, Sequence depth per nucleotide position. The number of reads for each nucleotide position was plotted across the full length of the virus genome for each of the 179 virus isolates we analysed. In red is shown the uniformity of the depth across individual genomes, although the median number of reads per nucleotide position had a variation spanning over four log10 units. c, Linear regression of the log10 median sequence depth of each virus isolate versus the Ct value of the viral load as determined by qRT–PCR. Red dots indicate samples taken from patients who went on to survive EBOV infection and grey shaded dots are from patients who records suggest died from EBOV infection.
The viral genome sequence was derived from RNA sequencing analysis of the patient samples with no pre-amplification of the viral genome. In general we selected a range of samples from both males and females of different ages and a fair representation of sequences for each month (Extended Data Fig. 1), and with Ct values less than 20 for EBOV RNA. In this selected patient cohort, with a relatively high viral load, there was approximately 80% mortality. The read depth mapping to the EBOV genome varied between samples and regions in the genome (Fig. 1b) and in general the number of sequence reads obtained for each genome correlated with the amount of viral load as determined by quantitative reverse-transcription PCR (qRT–PCR) (Fig. 1c).
Phylogenetic analysis revealed the dynamic nature of the epidemic and molecular change in the viral sequence (Fig. 2a). Several distinct lineages were identified, with an initial lineage A (Figs 2a, 3 and Extended Data Fig. 2) linked to early Guinean cases dating from March 2014 including the three original viruses published by Baize et al.2. A second lineage, B, emerged in May and June and comprises all the sequences from Gire et al.6 and the remainder of those described here. As the epidemic expanded, lineage A remained confined in Guinea from March to June 2014, except for one sequence from 18 July 2014. A single Liberian sequence from March 2014 grouped within this lineage. No further EBOV genomes that we sequenced from samples taken after July 2014 belonged to lineage A. This clade was likely to have been associated with the original outbreak in Guinea and was almost successfully contained in May 2014 by the interventions of the multi-agency response. Two clusters of Sierra Leone viruses described by Gire et al.6 (denoted by the authors as clusters SL1 and SL2), both of which contain later viruses from Guinea and Liberia, suggest continued spread across the border during this time. Early cases in SL1 and SL2 were both associated with a single funeral6, so it is possible that this event may have reignited the epidemic. Thereafter, lineage B spread into Guinea, Liberia and Sierra Leone. This lineage is associated with the large epidemics in these three countries and persisted into 2015. The spatiotemporal spread of these viruses based on the phylogenetic analysis presented in Figs 2a and 3 was summarized (Extended Data Fig. 3) and indicated how the virus may have spread between the neighbouring countries. There was no evidence from the data that increases or decreases in mortality were associated with any particular virus cluster (Extended Data Fig. 4).

a, Phylogenetic relatedness of EBOV isolates. Phylogenetic tree inferred using MrBayes11 for full-length EBOV genomes sequenced from 179 patient samples obtained between March 2014 and January 2015. Displayed is the majority consensus of 10,000 trees sampled from the posterior distribution with mean branch lengths. Posterior support is shown for selected key nodes. Twenty-two samples originated in Liberia and were collected between March and August 2014 and six samples from Sierra Leone were obtained in June and July 2014. In our analysis we also included published sequences, including the three early Guinean sequences2 and 78 sequences described by Gire et al.6. A number of lineages predominantly circulating in Guinea are denoted as GN1–4 along with a uniquely Sierra Leone lineage (SL3) recognised in Gire et al.6. b, EBOV nucleotide sequence divergence from root of the phylogeny in Fig. 2a plotted against time of collection of each virus. The date of the first documented case near Meliandou in eastern Guinea is indicated by the red triangle.

Shown is a maximum clade credibility tree constructed from 10,000 trees sampled from the posterior distribution with mean node ages. Clades described in Gire et al.6 are identified here (SL1, SL2 and SL3) as well as a number of lineages predominantly circulating in Guinea and posterior probability support is given for these. For certain key node ages, 95% credible intervals are shown by horizontal bars.
The Bayesian time-scaled phylogenetic analysis estimated an average rate of evolution over the genome of 1.42 × 10−3 substitutions per site per year with 95% credible intervals of 1.22 × 10−3 and 1.62 × 10−3. Details of the model assumptions are given in the Methods section. This rate is lower than that initially described for the West African outbreak by Gire et al.6 but still higher than the long-term, between-outbreak rate of 0.8 × 10−3 estimated using viruses back to the 1976 Yambuku outbreak6. This apparent drop in rate of evolution between these two studies is consistent with the explanation provided by Gire et al.6 that the short sampling interval (March to June) provided insufficient time for the action of purifying selection. However, the much longer sampling interval in the present study may simply be providing a more precise estimate of the rate. It should be noted, however, that the between-outbreak rate will exclusively reflect transmission and evolution that has occurred in the non-human reservoir species, so may not be directly comparable to the rate within a human outbreak. We observed no evidence of a change in evolutionary rate over the course of the epidemic with the accumulation of genetic change having a linear relationship with time (Fig. 2b), confirming that the apparent decline in rate between the two studies is an observational phenomenon7 rather than a change in the virus.
The estimate of the date of the most recent common ancestor of the sampled viruses is mid-January 2014 (95% credible intervals 12 December 2013, 18 February 2014). Although this is an estimate of first transmission event that resulted in more than one lineage in our sample, this provides an upper bound on the date of emergence of the virus into the human population. This date estimate is consistent with the epidemiological tracing of the first suspected cases to December 20132.
Given the error-prone nature of EBOV genome replication we examined the potential amino acid variation in EBOV proteins from the start of our sample collection in March 2014 to January 2015. The location of amino acid changes on EBOV proteins and their relative representation in the 179 assembled genomes were compared to an isolate identified in March 2014 (ref. 2) (Fig. 4). While there is amino acid variation in all of the genomes sampled, there were very few changes in viral protein 30 (VP30), viral protein 40 (VP40) and viral protein 24 (VP24), and these changes are only in less than ∼2% of the genomes sampled. However, a single amino acid substitution in VP24 is associated with adaptation to a new host4,8, and this may be due to interactions with host-cell proteins9,10. While some of the variation may be attributed to a purely random molecular clock pattern, in GP, VP35, NP and L there are some amino acid variations that are present in over ∼15% of the genomes sampled. For example, in GP there is an A to V substitution in ∼70.5% of the genomes sampled compared to the reference genome. Implications of the mutations within GP in relation to immune escape of therapeutics and vaccines will need to be assessed in pseudotype neutralization assays using EBOV monoclonal antibodies and serum from people who have been vaccinated.

Shown is the frequency of all amino acid positions that had variability and the substitution that occurred with the first single letter position indicating the reference sequence and the second position showing the variation. The percentage frequency in the 179 genomes is shown on the y axis. GP, glycoprotein; NP, nucleoprotein; L, RNA polymerase; VP, viral protein.
Methods
No statistical methods were used to predetermine sample size. There was no randomization or blinding in selection of samples for sequencing.
Ethics statement
The National Committee of Ethics in Medical Research of Guinea approved the use of diagnostic leftover samples and corresponding patient data for this study (permit no. 11/CNERS/14). As the samples had been collected as part of the public health response to contain the outbreak in Guinea, informed consent was not obtained from patients.
Genome sequencing and consensus building
Viral genome sequence was derived from the RNA extracted for diagnostic purposes from blood samples in the field with no pre-amplification of the viral genome. These samples were processed by the EMLab and are detailed in Supplementary Table 1, which indicates sample name, geographical location, date of onset of symptoms, date sample was collected, and the Ct value of EBOV RNA at the date of test. The clinical status is also indicated as well as malaria co-infection where known. Extracted RNA was DNase treated with Turbo DNase (Ambion) using the rigorous protocol. RNA sequencing libraries were prepared from the resultant RNA using the Epicentre ScriptSeq v2 RNA-Seq Library Preparation Kit. Following 10–15 cycles of amplification, libraries were purified using AMPure XP beads. Each library was quantified using Qubit and the size distribution assessed using the Agilent 2100 Bioanalyzer. These final libraries were pooled in equimolar amounts using the Qubit and Bioanalyzer data with 9–10 libraries per pool. The quantity and quality of the pool was assessed by Bioanalyzer and subsequently by qPCR using the Illumina Library Quantification Kit from Kapa on a Roche Light Cycler LC480II according to manufacturer’s instructions. Each pool of libraries was sequenced on one lane of a HiSeq2500 at 2 × 125-bp paired-end sequencing with v4 chemistry.
The trimmed fastq files were first aligned to a copy of the human genome using Bowtie2 (ref. 12) and the unaligned reads were then mapped with Bowtie2 to a list of 3731 known viral genomes excluding EBOV genomes. The reads that were still unmapped were then aligned to the EBOV genome—either the prototype strain isolated in Zaire in 1976 (AF086833.2) or a strain isolated during the current outbreak (KJ660348.2). For this step we again used Bowtie2 and the resultant alignment files were filtered with samtools to remove unmapped reads and reads with a mapping quality score below 11, followed by filtering with markdup to remove PCR duplicates. The resultant BAM file was then analysed by Quasirecomb13 to generate a phred-weighted table of nucleotide frequencies which were parsed with a custom perl script to generate a consensus genome in fasta format. This consensus genome was then used as a reference genome to which we remapped the sequence reads which did not map to the human genome or other viruses in order to generate a second consensus. In this way we were able to manually determine if the reference genome used by Bowtie2 influenced the process of calling a consensus genome. In addition, we used FreeBayes to independently call and identify SNPs and indels. The pipeline is entirely open source and implemented in the Galaxy environment14, a Galaxy compatible workflow, novel scripts and XML wrappers needed for implementation in Galaxy are freely available and included in Supplementary Data File 1. Sequence alignment maps were manually inspected and curated over regions with consistent low coverage (for example, at the 5′ ends).
Phylogenetic analysis
Phylogenetic analysis comprised the 179 EBOV genomes from this study, 78 genomes from Sierra Leone6, three sequences from Guinea2 and two sampled from Mali15. The genomes were partitioned into four sets of sites—1st, 2nd and 3rd codon positions of the protein-coding regions and the non-coding intergenic regions—with each partition being assigned a generalized time reversible substitution model16, gamma distributed rate heterogeneity17 and a relative rate of evolution. This model was used to construct a Bayesian nucleotide divergence tree (Fig. 2) using MrBayes11 and a time-scaled phylogenetic analysis (Fig. 3) using BEAST18 with a log-normal distributed relaxed molecular clock19, and the ‘Skygrid’ non-parametric coalescent tree prior20. The alignments and control files for both analyses are available in Supplementary Data Files 2 and 3 and provide documentation of all model parameters.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The 179 consensus genome sequences described in this study have been assigned the GenBank accession numbers KR817067–KR817245. Further information is provided in Supplementary Table 1.
Change history
05 August 2015
Spelling of author M.D.F.-G. was corrected.
References
Schieffelin, J. S. et al. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N. Engl. J. Med. 371, 2092–2100 (2014)
Baize, S. et al. Emergence of Zaire Ebola virus disease in Guinea. N. Engl. J. Med. 371, 1418–1425 (2014)
Gatherer, D. The unprecedented scale of the West African Ebola virus disease outbreak is due to environmental and sociological factors, not special attributes of the currently circulating strain of the virus. Evid. Based Med. 20, 28 (2015)
Dowall, S. D. et al. Elucidating variations in the nucleotide sequence of Ebola virus associated with increasing pathogenicity. Genome Biol. 15, 540 (2014)
Wynne, J. W. et al. Proteomics informed by transcriptomics reveals Hendra virus sensitizes bat cells to TRAIL-mediated apoptosis. Genome Biol. 15, 532 (2014)
Gire, S. K. et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372 (2014)
Ho, S. Y., Phillips, M. J., Cooper, A. & Drummond, A. J. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22, 1561–1568 (2005)
Mateo, M. et al. VP24 is a molecular determinant of Ebola virus virulence in guinea pigs. J. Infect. Dis. 204 (Suppl 3). S1011–S1020 (2011)
García-Dorival, I. et al. Elucidation of the Ebola virus VP24 cellular interactome and disruption of virus biology through targeted inhibition of host-cell protein function. J. Proteome Res. 13, 5120–5135 (2014)
Basler, C. F. & Amarasinghe, G. K. Evasion of interferon responses by Ebola and Marburg viruses. J. Interferon Cytokine Res. 29, 511–520 (2009)
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012)
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012)
Topfer, A. et al. Probabilistic inference of viral quasispecies subject to recombination. J. Comput. Biol. 20, 113–123 (2013)
Goecks, J., Nekrutenko, A., Taylor, J. & Galaxy, T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86 (2010)
Hoenen, T. et al. Mutation rate and genotype variation of Ebola virus from Mali case sequences. Science (2015)
Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences in Lectures on Mathematics in the Life Sciences Vol. 17 (ed. Muira, R. M. ) Some Mathematical Questions in Biology: DNA Sequence Analysis (American Mathematical Society, 1986)
Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314 (1994)
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012)
Drummond, A. J., Ho, S. Y., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006)
Gill, M. S. et al. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol. 30, 713–724 (2013)
Acknowledgements
The authors would like to acknowledge that the EMLab response and the subsequent EBOV genome sequencing study would not have been possible without the extensive support from the many different agencies and organisations working in the West African EBOV disease outbreak region. EMLab worked with WHO, MSF and the Guinean authorities to tackle the outbreak in the Guéckédou area where the samples from this study were collected. We thank those who helped make this possible and the Guinean authorities for their decision to release the diagnostic samples to EMLab for shipment to Europe to undergo further analysis, including sequencing. We acknowledge Air France, Brussels Airlines and Virgin Airlines for transporting EMLab personnel and equipment in and out of West Africa during the outbreak period; World Courier for shipping our EBOV-positive samples out of Guinea to Europe; and the logistics support units and pilots and drivers of WHO/United Nations in West Africa for transporting our people and equipment throughout the region, and especially the drivers who made the 28 h round trip journey from Conakry to enable the EMLab unit to be established and resupplied in Guéckédou. We appreciate the work of the numerous European Embassies operating in West Africa who provided emergency support to our personnel at times of need. We thank M. Bull, J. Lewis, P. Payne and S. Leach from the Microbial Risk Assessment and Behavioural Science Team, Emergency Response Department, Public Health England; J. Tree from Public Health England for help with GenBank submission; and S. Price and I. Stewart for helping with the running of our software on BlueCrystal, University of Bristol. We thank the people of West Africa for their gratitude and optimism, and for their positive attitude to our presence that we encountered on the daily journey to the Ebola Treatment Centre in Guéckédou. We acknowledge the efforts of the late Dr Lamine Ouendeno, who was one of the first healthcare workers to die during the current EBVD outbreak. We also thank Isabel and Maurice Ouendeno for providing us with food and shelter whilst delivering our Ebola response duties. This work was carried out in the context of the project EVIDENT (Ebola virus disease: correlates of protection, determinants of outcome, and clinical management) that received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 666100 and in the context of service contract IFS/2011/272-372 funded by Directorate-General for International Cooperation and Development. The EMLab is a technical partner in the WHO Emerging and Dangerous Pathogens Laboratory Network (EDPLN), and the Global Outbreak Alert and Response Network (GOARN) and the deployments in West Africa have been coordinated and supported by the GOARN Operational Support Team at WHO/HQ.
Author information
Authors and Affiliations
Contributions
M.W.C., S.G., J.A.H., D.A.M and N.M. designed the study. J.A.H., D.A.M., M.J.E., A.R., G.P., S.G. and M.W.C. wrote the manuscript. D.A.M., J.A.H., M.J.E., A.R., G.P., M.W.C., S.G., Y.H. and I.G.D. analysed the data. All other authors were involved either in sample collection, processing and/or logistical support and strategic oversight for the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Spatial and temporal location of patient samples. Geographical locations of sequenced samples are plotted by district as panels for each month of collection (March 2014–January 2015).
In brief, the number of samples obtained for each month was as follows: March 2014, 11; April 2014, 14; May 2014, 14; June 2014, 22; July 2014, 16; August 2014, 19; September 2014, 18; October 2014, 21; November 2014, 11; December 2014, 22; January 2015, 11. Total number of samples sequenced, 179.
Extended Data Figure 2 Enlarged view of phylogenetic tree presented in Fig. 3.
Posterior support shown where >0.5.
Extended Data Figure 3 Temporal spread of EBOV based on phylogenetic analyses in Figs 2a and 3.
Colour scheme is as follows: Guinea is red/blue (1st half/2nd half of 2014, respectively), Sierra Leone is grey-black, Liberia is green, Mali is brown. Lineage A (A) is associated with the initial focus of the outbreak (Guéckédou, Macenta and Kissidougou) in March 2014, expanded around this area and then declined around July 2014. From lineage A a second lineage (B) emerged in May/June 2014 and expanded into Sierra Leone (end of May 2014) and Liberia (small arrow). Lineage B continued to spread into Sierra Leone, Liberia, and further into Guinea (beyond the original focus into most districts of Guinea). EBOV disease entered Mali from Guinea via two separate routes (from the Beyla district (possibly originally from Kissidougou) in October 2014 and from the Siguiri district in November 2014).
Extended Data Figure 4 Survival rate amongst individuals with known EBOV sequences.
The total survival rate for the 179 sequenced virus isolates included in this study is presented, as is the survival rate for two sub-lineages, GN1 and GN2, as defined by phylogenetic inference in Figs 2a and 3. The sequences available for GN1 were collected during the period of March–July 2014 and the sequences available for GN2 were collected during the period of August 2014–January 2015. Red dots indicate survivors.
Supplementary information
Supplementary Data 1
This file contains the Galaxy compatible workflow, novel scripts and xml wrappers for implementation of the sequencing pipeline.(ZIP 7 kb)
Supplementary Data 2
This file contains the NEXUS file used for constructing the MrBayes divergence tree (Figure 2a). (TXT 4865 kb)
Supplementary Data 3
This file contains the BEAST XML file for the time-scaled phylogenetic analysis in Figure 3. (XML 4972 kb)
Supplementary Table 1
This table contains background patient sample information and GenBank accession numbers for the viral sequences described in this study. (XLSX 26 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Carroll, M., Matthews, D., Hiscox, J. et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 524, 97–101 (2015). https://doi.org/10.1038/nature14594
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14594
This article is cited by
-
Oligonucleotide usage in coronavirus genomes mimics that in exon regions in host genomes
Virology Journal (2023)
-
Analysis of an Ebola virus disease survivor whose host and viral markers were predictive of death indicates the effectiveness of medical countermeasures and supportive care
Genome Medicine (2021)
-
Electronic data collection, management and analysis tools used for outbreak response in low- and middle-income countries: a systematic review and stakeholder survey
BMC Public Health (2021)
-
Validation of multiplex PCR sequencing assay of SIV
Virology Journal (2021)
-
Ebola virus antibody decay–stimulation in a high proportion of survivors
Nature (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
