
Abstract
The reference human genome sequence set the stage for studies of genetic variation and its association with human disease, but epigenomic studies lack a similar reference. To address this need, the NIH Roadmap Epigenomics Consortium generated the largest collection so far of human epigenomes for primary cells and tissues. Here we describe the integrative analysis of 111 reference human epigenomes generated as part of the programme, profiled for histone modification patterns, DNA accessibility, DNA methylation and RNA expression. We establish global maps of regulatory elements, define regulatory modules of coordinated activity, and their likely activators and repressors. We show that disease- and trait-associated genetic variants are enriched in tissue-specific epigenomic marks, revealing biologically relevant cell types for diverse human traits, and providing a resource for interpreting the molecular basis of human disease. Our results demonstrate the central role of epigenomic information for understanding gene regulation, cellular differentiation and human disease.
Similar content being viewed by others

Main

While the primary sequence of the human genome is largely preserved in all human cell types, the epigenomic landscape of each cell can vary considerably, contributing to distinct gene expression programs and biological functions1,2,3,4. Epigenomic information, such as covalent histone modifications, DNA accessibility and DNA methylation can be interrogated in each cell and tissue type using high-throughput molecular assays2,5,6,7,8. The resulting maps have been instrumental for annotating cis-regulatory elements and other non-exonic genomic features with characteristic epigenomic signatures9,10, and for dissecting gene regulatory programs in development and disease7,9,11,12,13,14. Despite these technological advances, we still lack a systematic understanding of how the epigenomic landscape contributes to cellular circuitry, lineage specification, and the onset and progression of human disease.
To facilitate and spearhead these efforts, the NIH Roadmap Epigenomics Program was established with the goal of elucidating how epigenetic processes contribute to human biology and disease. One of the major components of this programme consists of the Reference Epigenome Mapping Centers (REMCs)15, which systematically characterized the epigenomic landscapes of representative primary human tissues and cells. We used a diversity of assays, including chromatin immunoprecipitation (ChIP)9,10,16,17, DNA digestion by DNase I (DNase)7,18, bisulfite treatment1,2,19,20, methylated DNA immunoprecipitation (MeDIP)21, methylation-sensitive restriction enzyme digestion (MRE)22, and RNA profiling8, each followed by massively parallel short-read sequencing (-seq). The resulting data sets were assembled into publicly accessible websites and databases, which serve as a broadly useful resource for the scientific and biomedical community. Here we report the integrative analysis of 111 reference epigenomes (Fig. 1 and Extended Data Fig. 1a–d), which we analyse jointly with an additional 16 epigenomes previously reported by the Encyclopedia of DNA Elements (ENCODE) project9,23.

Primary tissues and cell types representative of all major lineages in the human body were profiled, including multiple brain, heart, muscle, gastrointestinal tract, adipose, skin and reproductive samples, as well as immune lineages, ES cells and iPS cells, and differentiated lineages derived from ES cells. Box colours match groups shown in Fig. 2b. Epigenome identifiers (EIDs, Fig. 2c) for each sample are shown in Extended Data Fig. 1.
We integrate information about histone marks, DNA methylation, DNA accessibility and RNA expression to infer high-resolution maps of regulatory elements annotated jointly across a total of 127 reference epigenomes spanning diverse cell and tissue types. We use these annotations to recognize epigenome differences that arise during lineage specification and cellular differentiation, to recognize modules of regulatory regions with coordinated activity across cell types, and to identify key regulators of these modules based on motif enrichments and regulator expression. In addition, we study the role of regulatory regions in human disease by relating our epigenomic annotations to genetic variants associated with common traits and disorders. These analyses demonstrate the importance and wide applicability of our data resource, and lead to important insights into epigenomics, differentiation and disease. Specific highlights of our findings are given below.
• Histone mark combinations show distinct levels of DNA methylation and accessibility, and predict differences in RNA expression levels that are not reflected in either accessibility or methylation.
• Megabase-scale regions with distinct epigenomic signatures show strong differences in activity, gene density and nuclear lamina associations, suggesting distinct chromosomal domains.
• Approximately 5% of each reference epigenome shows enhancer and promoter signatures, which are twofold enriched for evolutionarily conserved non-exonic elements on average.
• Epigenomic data sets can be imputed at high resolution from existing data, completing missing marks in additional cell types, and providing a more robust signal even for observed data sets.
• Dynamics of epigenomic marks in their relevant chromatin states allow a data-driven approach to learn biologically meaningful relationships between cell types, tissues and lineages.
• Enhancers with coordinated activity patterns across tissues are enriched for common gene functions and human phenotypes, suggesting that they represent coordinately regulated modules.
• Regulatory motifs are enriched in tissue-specific enhancers, enhancer modules and DNA accessibility footprints, providing an important resource for gene-regulatory studies.
• Genetic variants associated with diverse traits show epigenomic enrichments in trait-relevant tissues, providing an important resource for understanding the molecular basis of human disease.
Reference epigenome mapping across tissues and cell types
The REMCs generated a total of 2,805 genome-wide data sets, including 1,821 histone modification data sets, 360 DNA accessibility data sets, 277 DNA methylation data sets, and 166 RNA-seq data sets, encompassing a total of 150.21 billion mapped sequencing reads corresponding to 3,174-fold coverage of the human genome.
Here, we focus on a subset of 1,936 data sets (Fig. 2) comprising 111 reference epigenomes (Fig. 2a–d), which we define as having a core set of five histone modification marks (Fig. 2e). The five marks consist of: histone H3 lysine 4 trimethylation (H3K4me3), associated with promoter regions10,24; H3 lysine 4 monomethylation (H3K4me1), associated with enhancer regions10; H3 lysine 36 trimethylation (H3K36me3), associated with transcribed regions; H3 lysine 27 trimethylation (H3K27me3), associated with Polycomb repression25; and H3 lysine 9 trimethylation (H3K9me3), associated with heterochromatin regions26. Selected epigenomes also contain a subset of additional epigenomic marks, including: acetylation marks H3K27ac and H3K9ac, associated with increased activation of enhancer and promoter regions27,28,29 (Fig. 2f); DNase hypersensitivity7,18, denoting regions of accessible chromatin commonly associated with regulator binding (Fig. 2g); DNA methylation, typically associated with repressed regulatory regions or active gene transcripts4,30 and profiled using whole-genome bisulfite sequencing (WGBS)19, reduced-representation bisulfite sequencing (RRBS)20, and mCRF-combined31 methylation-sensitive restriction enzyme (MRE)22 and immunoprecipitation based21 assays (Fig. 2h); and RNA expression levels8, measured using RNA-seq and gene expression microarrays (Fig. 2i). Our definition of 111 reference epigenomes is very similar to that used by the International Human Epigenome Consortium (IHEC), which required RNA-seq, WGBS and H3K27ac that are only available in a subset of epigenomes here. Lastly, an additional 16 histone modification marks on average were profiled across 7 deeply covered cell types (Fig. 2j).

List of 127 epigenomes including 111 by the Roadmap Epigenomics program (E001–E113) and 16 by ENCODE (E114–E129). See Supplementary Table 1 for a full list of names and quality scores. a–d, Tissue and cell types grouped by type of biological material (a), anatomical location (b), reference epigenome identifier (EID, c) and abbreviated name (d). PB, peripheral blood. ENCODE 2012 reference epigenomes are shown separately. e–g, Normalized strand cross-correlation quality scores (NSC)37 for the core set of five histone marks (e), additional acetylation marks (f) and DNase-seq (g). h, Methylation data by WGBS (red), RRBS (blue) and mCRF (green). A total of 104 methylation data sets available in 95 distinct reference epigenomes. i, Gene expression data using RNA-seq (brown) and microarray expression (yellow). j, A total of 26 epigenomes contain 184 additional histone modification marks. k, Sixty highest-quality epigenomes (purple) were used for training the core chromatin state model, which was then applied to the full set of epigenomes (purple and orange).
We jointly processed and analysed our 111 reference epigenomes with 16 additional epigenomes from ENCODE9,23. We generated genome-wide normalized coverage tracks, peaks and broad enriched domains for ChIP-seq and DNase-seq7,32, normalized gene expression values for RNA-seq33, and fractional methylation levels for each CpG site31,34,35. We computed several quality control measures (Fig. 2 and Supplementary Table 1) including the number of distinct uniquely mapped reads; the fraction of mapped reads overlapping areas of enrichment18,36; genome-wide strand cross-correlation37 (Fig. 2e–g); inter-replicate correlation; multidimensional scaling of data sets from different production centres (Supplementary Fig. 1); correlation across pairs of data sets (Extended Data Fig. 1e); consistency between assays carried out in multiple mapping centres (Supplementary Table 2); read mapping quality for bisulfite-treated reads38,39; and agreement with imputed data40. Outlier data sets were flagged, removed or replaced, and lower-coverage data sets were combined where possible (see Methods).
The resulting data sets provide global views of the epigenomic landscape in a wide range of human cell and tissue types (Fig. 3), including the largest and most diverse collection to date of chromatin state annotations (Fig. 3a); some of the deepest surveys of individual cell types using diverse epigenomic assays (with 21–31 distinct epigenomic marks for seven deeply profiled epigenomes; Fig. 3b); and some of the broadest surveys of individual epigenomic marks across multiple cell types (Fig. 3c). These data sets enable genome-wide epigenomic analyses across multiple dimensions (Fig. 3d). All data sets, standards and protocols are publicly available from web portals, linked from the main consortium homepage http://www.roadmapepigenomics.org, and also at http://compbio.mit.edu/roadmap.

a, Chromatin state annotations across 127 reference epigenomes (rows, Fig. 2) in a ∼3.5-Mb region on chromosome 9. Promoters are primarily constitutive (red vertical lines), while enhancers are highly dynamic (dispersed yellow regions). b, Signal tracks for IMR90 showing RNA-seq, a total of 28 histone modification marks, whole-genome bisulfite DNA methylation, DNA accessibility, digital genomic footprints (DGF), input DNA and chromatin conformation information72. c, Individual epigenomic marks across all epigenomes in which they are available. d, Relationship of figure panels highlights data set dimensions.
Chromatin states, DNA methylation and DNA accessibility
As a foundation for integrative analysis, we used a common set of combinatorial chromatin states41 across all 111 epigenomes, plus 16 additional epigenomes generated by the ENCODE project (127 epigenomes in total), using the core set of five histone modification marks that were common to all. We trained a 15-state model (Fig. 4a, b and Supplementary Table 3a) consisting of 8 active states and 7 repressed states (Fig. 4c) that were recurrently recovered (Extended Data Fig. 2a), and showed distinct levels of DNA methylation (Fig. 4d), DNA accessibility (Fig. 4e), regulator binding (Extended Data Fig. 2b and Supplementary Fig. 2) and evolutionary conservation (Fig. 4f and Supplementary Fig. 3). The active states (associated with expressed genes) consist of active transcription start site (TSS) proximal promoter states (TssA, TssAFlnk), a transcribed state at the 5′ and 3′ end of genes showing both promoter and enhancer signatures (TxFlnk), actively transcribed states (Tx, TxWk), enhancer states (Enh, EnhG), and a state associated with zinc finger protein genes (ZNF/Rpts). The inactive states consist of constitutive heterochromatin (Het), bivalent regulatory states (TssBiv, BivFlnk, EnhBiv), repressed Polycomb states (ReprPC, ReprPCWk), and a quiescent state (Quies), which covered on average 68% of each reference epigenome. Enhancer and promoter states covered approximately 5% of each reference epigenome on average, and showed enrichment for evolutionarily conserved non-exonic regions42.

a, Chromatin state definitions, abbreviations and histone mark probabilities. b, Average genome coverage. Genomic annotation enrichments in H1-ES cells. c, Active and inactive gene enrichments in H1-ES cells (see Extended Data Fig. 2b for GM12878). d, DNA methylation. e, DNA accessibility. d, e, Whiskers show 1.5× interquartile range. Circles are individual outliers. f, Average overlap fold enrichment for GERP evolutionarily conserved non-exonic nucleotides. Bars denote standard deviation. g, DNA methylation (WGBS) density (colour, ln scale) across cell types. Red = max ln(density + 1). Left column indicates tissue groupings; a full list is shown in Extended Data Fig. 4f. h, DNA methylation levels (left) and transcription factor enrichment (right) during ES cell differentiation50,51,52,53. i, Chromatin mark changes during cardiac muscle differentiation. Heat map = average normalized mark signal in Enh. C2 cluster enrichment55, with all clusters shown in http://compbio.mit.edu/roadmap.
To capture the greater complexity afforded by additional marks, we trained additional chromatin state models in subsets of cell types. In the subset of 98 reference epigenomes that also included H3K27ac data, we also learned an 18-state model (Extended Data Fig. 2c and Supplementary Table 3b), enabling us to distinguish enhancer states containing strong H3K27ac signal (EnhA1, EnhA2), which showed higher DNA accessibility (Extended Data Fig. 3a), lower methylation (Extended Data Fig. 3b) and higher transcription factor binding (Extended Data Fig. 2c) than enhancers lacking H3K27ac. In a subset of 7 epigenomes with an average of 24 epigenomic marks, we learned separate 50-state chromatin state models based on all the available histone marks and DNA accessibility in each epigenome (Supplementary Fig. 4), which additionally distinguished: a DNase state with distinct transcription factor binding enrichments (Supplementary Fig. 4f), including for mediator/cohesin components43 (even though CTCF was not included as an input track to learn the model) and repressor NRSF; transcribed states showing H3K79me1 and H3K79me2 and associated with the 5′ ends of genes and introns; and a large number of putative regulatory and neighbouring regions showing diverse acetylation marks even in the absence of the H3K4 methylation signatures characteristic of enhancer and promoter regions.
We used chromatin states to study the relationship between histone modification patterns, RNA expression levels, DNA methylation and DNA accessibility. Consistent with previous studies19,23,44,45, we found low DNA methylation and high accessibility in promoter states, high DNA methylation and low accessibility in transcribed states, and intermediate DNA methylation and accessibility in enhancer states (Fig. 4d, e and Extended Data Fig. 3a, b). These differences in methylation level were stronger for higher-expression genes than for lower-expression genes, leading to a more pronounced DNA methylation profile (Extended Data Fig. 3c, Supplementary Fig. 5 and Supplementary Table 4f). Genes proximal to H3K27ac-marked enhancers show significantly higher expression levels (Extended Data Fig. 3d), and conversely, higher-expression genes were significantly more likely to be neighbouring H3K27ac-containing enhancers (Extended Data Fig. 3e).
Chromatin states sometimes captured differences in RNA expression that are missed by DNA methylation or accessibility. For example, TxFlnk, Enh, TssBiv and BivFlnk states show similar distributions of DNA accessibility but widely differing enrichments for expressed genes (Fig. 4c, d). Enh and ReprPC states show intermediate DNA methylation, but very different distributions of DNA accessibility and different enrichments for expressed genes (Fig. 4c–e). Lack of DNA methylation, typically associated with de-repression, is associated with both the active TssA promoter state and the bivalent TssBiv and BivFlnk states. Bivalent states TssBiv and BivFlnk also show overall lower DNA methylation and higher DNA accessibility than enhancer states Enh and EnhG, and binding by both activating and repressive regulatory factors (Extended Data Fig. 2b). These results also held for alternative methylation measurement platforms (Extended Data Fig. 4a–c), and for the 18-state chromatin state model (Extended Data Fig. 4d, e). Overall, these results highlight the complex relationship between DNA methylation, DNA accessibility and RNA transcription and the value of interpreting DNA methylation and DNA accessibility in the context of integrated chromatin states that better distinguish active and repressed regions.
Given the intermediate methylation levels of tissue-specific enhancer regions, we directly annotated intermediate methylation regions, based on 25 complementary DNA methylation assays of MeDIP31,46 and MRE-seq22,39 from 9 reference epigenomes47. This resulted in more than 18,000 intermediate methylation regions, showing 57% CpG methylation on average, that are strongly enriched in genes, enhancer chromatin states (EnhBiv, EnhG, Enh) and evolutionarily conserved regions. Intermediate methylation was associated with intermediate levels of active histone modifications and DNase I hypersensitivity. Near TSSs, intermediate methylation correlated with intermediate gene expression, and in exons it was associated with an intermediate level of exon inclusion47. Intermediate methylation signatures were equally strong within tissue samples, peripheral blood and purified cell types, suggesting that intermediate methylation is not simply reflecting differential methylation between cell types, but probably reflects a stable state of cell-to-cell variability within a population of cells of the same type.
Epigenomic differences during lineage specification
We next studied the relationship between DNA methylation dynamics and histone modifications across 95 epigenomes with methylation data, extending previous studies that focused on individual lineages19,48,49,50. We found that the distribution of methylation levels for CpGs in some chromatin states varied significantly across tissue and cell type (Fig. 4g, Extended Data Fig. 4f and Supplementary Table 4a). For example, TssAFlnk states were largely unmethylated in terminally differentiated cells and tissues, but frequently methylated for several pluripotent and embryonic-stem-cell-derived cells (Bonferroni-corrected F-test P < 0.01); Enh and EnhG states were highly methylated in pluripotent cells, but showed a broader distribution of intermediate methylation in differentiated cells and tissues (P < 0.01); EnhBiv states were unmethylated in most primary cells and tissues, but showed a broader distribution of methylation levels in pluripotent cells, possibly reflecting cell-to-cell heterogeneity (P < 0.01); the repressed state ReprPC showed varying methylation levels among epigenomes; and the Het state showed high levels of methylation in almost all epigenomes.
We also studied DNA methylation changes in three different systems. First, we studied DNA methylation changes during embryonic stem (ES) cell differentiation50,51. We identified regions that lost methylation (differentially methylated regions (DMRs), Supplementary Table 4c) upon differentiation of ES cells (E003) to mesodermal (E013), endodermal (E011) and ectodermal (E012) lineages (Fig. 4h). Each lineage showed a largely distinct set of ∼2,200–4,400 DMRs that are enriched for distinct transcription factor binding events (Fig. 4h, right column)52, consistent with their distinct developmental regulation. Upon further differentiation, ectodermal DMRs remained hypomethylated in three neural progenitor populations53, despite the usage of distinct human ES cell (hESC) lines, and mesodermal and endodermal DMRs remained highly methylated (Fig. 4h), highlighting the lineage-specific nature of changes in DNA methylation during early differentiation50,54.
Second, we studied DNA methylation changes associated with breast epithelia differentiation45. Ectoderm to breast epithelia differentiation was dominated by DNA methylation loss (1.3M CpGs lost methylation compared with 0.2M gained), consistent with other primary somatic cell types51. By distinguishing luminal versus myoepithelial cells by flow sorting, and comparing a set of DMRs (Supplementary Table 4d) defined specifically in epithelial lineages45, we found differences in nearest-gene enrichments55 (mammary gland epithelium development versus actin filament bundle, respectively) and differences in motif density (luminal DMRs show greater motif density for 51 transcription factors and lower density for 0 transcription factors). Proximal DMRs were highly associated with increased transcription, consistent with regulatory element de-repression associated with DNA methylation loss.
Third, we asked whether tissue environment or developmental origin is the primary driving factor in DNA methylation differences observed in more differentiated cell types56 using epigenomes from skin cell types (keratinocytes E057/058, melanocytes E059/E061 and fibroblasts E055/056) that share a common tissue environment but possess distinct embryonic origins (surface ectoderm, neural crest and mesoderm, respectively). We found that despite the shared tissue environment, these three cell types displayed lower overlap in their DNA methylation and histone modification signatures, and instead were more similar to other cell types with a shared developmental origin. Using a set of DMRs (Supplementary Table 4e) defined specifically in the skin cell types56, keratinocytes shared 1,392 (18%) of DMRs with surface ectoderm-derived breast cell types (hypergeometric P value <10−6), and 97% of these were hypomethylated. These shared DMRs were enriched for regulatory elements and cell-type-relevant genes, suggesting a common gene-regulatory network and shared signalling pathways and structural components56. These results suggest that common developmental origin can be a primary determinant of global DNA methylation patterns, and sometimes supersedes the immediate tissue environment in which they are found.
We also examined coordinated changes in chromatin marks associated with cellular differentiation57. We found that enhancers showing coordinated differences in multiple marks were enriched near genes showing common tissue-specific expression, and common knockout phenotypes based on their mouse orthologues. For example, enhancers that showed higher H3K27ac and H3K4me3 (Fig. 4i, cluster C2) in left ventricle (E095) relative to their ES cells (E003) and mesendodermal (E004) precursor lineages were enriched for heart ventricle expression and cardiac and muscle phenotypes in their mouse orthologues.
Most variable states and distinct chromosomal domains
We next sought to characterize the overall variability of each chromatin state across the full range of cell and tissue types. We first evaluated the observed consistency of each chromatin state at any given genomic position across all 127 epigenomes (Fig. 5a). We found that H3K4me1-associated states (including TxFlnk, EnhG, EnhBiv and Enh) are the most tissue specific, with 90% of instances present in at most 5–10 epigenomes, followed by bivalent promoters (TssBiv) and repressed states (ReprPC, Het). In contrast, active promoters (TssA) and transcribed states (Tx, TxWk) were highly constitutive, with 90% of regions marked in as many as 60–75 epigenomes. Quiescent regions were the most constitutive, with 90% consistently marked in most of the 127 epigenomes. These results held in the 18-state chromatin state model (Extended Data Fig. 5a), and in the subset of highest-quality epigenomes (Supplementary Fig. 6a, b).

a, Chromatin state variability, based on genome coverage fraction consistently labelled with each state. b, Relative chromatin state frequency for each reference epigenome. c, Chromatin state switching log10 relative frequency (inter-cell type versus inter-replicate). d, Clustering of 2-Mb intervals (columns) based on relative chromatin state frequency (fold enrichment), averaged across reference epigenomes. LaminB1 occupancy profiled in ES cells. Red lines show cluster average.
Adjusting for the overall coverage and variability of each state, we then studied differences in the relative fraction of the genome annotated to each chromatin state between cell types (Fig. 5b, Extended Data Fig. 5b and Supplementary Fig. 6c–e). Haematopoietic stem cells and immune cells show a consistent and previously unrecognized depletion of active and bivalent promoters (TssA, TssBiv) and weakly transcribed states (TxWk), which may be related to their capacity to generate sub-lineages and enter quiescence (reversible G0 phase). ES cells and induced pluripotent stem cells (iPS cells) show enrichment of TssBiv, consistent with previous studies58, and a depletion of ReprPCWk (defined by weak H3K27me3), possibly due to restriction of H3K27me3-establishing Polycomb proteins to promoter regions. Notably, IMR90 fetal lung fibroblasts, which were previously used as a somatic reference cell type59, are in fact a strong outlier in multiple ways, showing higher levels of Het, ReprPC and EnhG, and a depletion of Quies chromatin states.
We next studied the relative frequency with which different chromatin states switch to other states across different tissues and cell types (Fig. 5c), relative to switching in samples of the same tissue or cell type (Supplementary Fig. 7a, b). This revealed a relative switching enrichment between active states and repressed states, consistent with activation and repression of regulatory regions. The only exception was significant switching between transcribed states and active promoter and enhancer states, possibly due to alternative usage of promoters22 and enhancers60 embedded within transcribed elements. These chromatin state switching properties were also found in the 18-state model incorporating H3K27ac marks (Extended Data Fig. 5c) and in the subset of 16 ENCODE reference epigenomes using both models (Supplementary Fig. 7c, d). We found that enhancers and promoters maintained their identity, except for a small subset of regions switching between enhancer signatures and promoter signatures61. Luciferase assays showed that these regions indeed possess both enhancer and promoter activity61, consistent with their epigenomic marks.
While chromatin states were defined at nucleosome resolution (200 bp), we also studied the overall co-occurrence of chromatin states across tissues at a larger resolution (2 Mb) to recognize higher-order properties (Fig. 5d). This analysis revealed that 2-Mb segments rich in active enhancers are constrained to approximately 40% of the genome (clusters c1–c6), with the remainder marked predominantly by inactive regions (c7–c11), consistent with the identification of two large chromatin conformation compartments12,62. However, both compartments can be further subdivided by their chromatin state composition: inactive regions separate into predominantly quiescent (40%, c9, c11), heterochromatic (10%, c10), or bivalent (10%, c7, c8) marked regions; and active regions separate into regions rich in multiple marks (c3 and c6, showing a large diversity of active, ReprPC and bivalent states), enhancer and weakly transcribed regions (c5), and regions of intermediate activity (c1, c2, c4). These subdivisions were based on average state density across a large diversity of cell types and showed strong differences in gene density, CpG island occupancy, lamina association63,64 and cytogenetic bands (Fig. 5d and Extended Data Fig. 5d), suggesting that they represent stable chromosomal features.
Relationships between marks and lineages
We next studied the relationship between tissues and cell types, based on the similarity of diverse histone modification marks evaluated in their relevant chromatin states. Hierarchical clustering of our 111 reference epigenomes using H3K4me1 signal in Enh (Fig. 6a) showed consistent grouping of biologically similar cell and tissue types, including ES cells, iPS cells, T cells, B cells, adult brain, fetal brain, digestive, smooth muscle and heart. We also found several initially surprising but biologically meaningful groupings: fetal brain and germinal matrix samples clustered with neural stem cells rather than adult brain, consistent with fetal neural stem-cell proliferation; many ES-derived cells clustered with ES cells and iPS cells rather than the corresponding tissues, suggesting that those are still closer to pluripotent states than corresponding somatic states; adult and fetal thymus samples clustered with T cells rather than other tissues, consistent with roles in T-cell maturation and immunity. Several marks successfully recovered biologically meaningful groups when evaluated in their relevant chromatin states (Supplementary Fig. 8), including H3K4me3 in TssA, H3K27me3 in ReprPC, and H3K36me3 in Tx, suggesting that the signal of each mark in relevant chromatin states is highly indicative of cell type and tissue identity. These alternative clusterings also showed some differences; for example, H3K4me3 in TssA states grouped several fetal samples together with each other, in a cluster neighbouring ES cells and iPS cells, rather than in separate tissue groups.

a, Hierarchical epigenome clustering using H3K4me1 signal in Enh states. Numbers indicate bootstrap support scores over 1,000 samplings. b, c, Multidimensional scaling (MDS) plot of cell type relationships based on similarity in H3K4me1 signal in Enh states (b) and H3K27me3 signal in ReprPC states (c). First four dimensions are shown as dim1 versus dim2 and dim3 versus dim4.
We applied this approach to compare the Roadmap Epigenomics reference epigenomes with the 16 ENCODE 2012 samples with broad mark coverage (Extended Data Fig. 6). We found that H3K4me1 signal in enhancer chromatin states correctly groups primary cells from similar tissues across the two projects, emphasizing the robustness of our annotations and signal tracks across projects (Extended Data Fig. 6a). For example, NHEK epidermal keratinocytes group with other keratinocytes, HMEC mammary epithelial cells group with other skin cells, and obsteoblasts and HSMM skeletal muscle myoblasts group with bone marrow. Some cancer cell lines also grouped with corresponding primary tissues, including HepG2 hepatocellular carcinoma with liver tissue, NHLF primary lung fibroblasts with the IMR90 lung fibroblast cell line, and Dnd41 T-cell leukaemia with thymus, while in other cases cancerous cell lines grouped together, for example, HeLa-S3 cervical carcinoma with A549 lung carcinoma. H3K27me3 signal in Polycomb-repressed states grouped five immortalized cell lines together (Extended Data Fig. 6c), despite their T-cell, lung, cervical, leukaemia and hepatocellular origins12,65. The larger trees spanning ENCODE 2012 and Roadmap Epigenomics also highlighted the large number of lineages not previously covered by reference epigenomes, including brain, muscle, smooth muscle, heart, mucosa, digestive tract and fetal tissues.
To understand the relationship among different tissue/cell samples beyond the constraints of a tree representation, we also studied the full similarity matrix of each mark in relevant chromatin states (Supplementary Fig. 9) and also visualized the principal dimensions of epigenomic variation using multidimensional scaling (MDS) analysis (Supplementary Fig. 10). The pairwise similarity matrices of different marks were most effective in distinguishing different subsets of the samples, with H3K4me1 in Enh primarily capturing immune cell similarities, and H3K27me3 in ReprPC capturing pluripotent cell similarities (Supplementary Fig. 9). In the MDS analysis, the first four dimensions of variation for most marks separated major sample groups (Extended Data Fig. 7a–i), with some subtle differences between marks. For example, pluripotent cells and immune cells were two strong outliers in the first two dimensions of H3K4me1 variation in Enh (Fig. 6b), but H3K27me3 in ReprPC showed more uniform spreading of reference epigenomes (Fig. 6c), consistent with the coverage distributions of immune and pluripotent cells for the corresponding chromatin states (Fig. 5b). For most marks, the first five dimensions captured most of the variance, with additional dimensions capturing at most 4–6% for each mark (Extended Data Fig. 7).
Imputation and completion of epigenomic data sets
We exploited the strong relationships between marks and lineages for epigenomic signal imputation to complete missing marks across remaining tissues, and to complement observed data sets with more robust predictions based on multiple data sets40. We predicted epigenomic signal tracks at 25-nucleotide resolution for histone marks, DNA accessibility, and RNA-seq data set and at single-base for CpG methylation, by exploiting correlations between multiple marks in the same cell type, and the same mark across multiple cell types.
We predict signal tracks for 34 epigenomic marks in 127 epigenomes, corresponding to 4,315 imputed genome-wide data sets, of which 3,193 (74%) are only available as imputed data. Imputed tracks showed high correlation with observed data, provided stronger and more consistent aggregate statistics relative to gene and TSS annotations, revealed lower-quality observed data sets in cases of disagreement between imputed and observed data, and captured cell type relationships and lineage-restricted information40.
We also used 12 imputed epigenomic marks to learn a 25-state chromatin state model jointly across all 127 reference epigenomes, which distinguished multiple subtypes of enhancer and promoter regions across the complete set of reference epigenomes, including several active, weak and transcribed enhancer states, and both upstream and downstream promoter regions, providing an important reference annotation for studies of gene regulation and human disease40.
Enhancer modules and their putative regulators
We next exploited the dynamics of epigenomic modifications at cis-regulatory elements to gain insights into gene regulation. We focused on 2.3M regions (12.6% of the genome) showing DNA accessibility in any reference epigenome and regulatory (promoter or enhancer) chromatin states, considering enhancer-only, promoter-only, or enhancer–promoter alternating states separately (Supplementary Fig. 11). We clustered enhancer-only elements (Enh, EnhBiv, EnhG) into 226 enhancer modules of coordinated activity (Fig. 7a), promoter-only elements into 82 promoter modules (Supplementary Fig. 11a) and promoter/enhancer ‘dyadic’ elements into 129 modules (Supplementary Fig. 11b), enabling us to distinguish ubiquitously active, lineage-restricted and tissue-specific modules for each group. Focusing on the enhancer-only clusters, we found that the neighbouring genes of enhancers in the same module showed significant enrichment for common functions66 (Fig. 7b and Supplementary Fig. 11c, d), common genotype–phenotype associations67 (Fig. 7c), and common expression in their mouse orthologues (Supplementary Fig. 12), each annotation type showing strong consistency with the known biology of the corresponding tissues. For example, stem-cell enhancers are enriched near developmental patterning genes, immune cell enhancers near immune response genes, and brain enhancers near learning and memory genes (Fig. 7b). Sub-clustering of individual modules continued to reveal distinct enrichment patterns of individual sub-modules (Supplementary Fig. 11e), suggesting increased diversity of regulatory processes beyond the 226 modules used here.

a, Enhancer modules by activity-based clustering of 2.3 million DNase-accessible regions classified as Enh, EnhG or EnhBiv (colour) across 111 reference epigenomes. Vertical lines separate 226 modules. Broadly active enhancers are shown first. Module IDs are shown in Supplementary Fig. 11c. b, c, Proximal gene enrichments55 for each module using gene ontology (GO) biological process (b) and human phenotypes (c). Rectangles pinpoint enrichments for selected modules. Representative gene set names (left) were selected using bag-of-words enrichment.
The genome sequence of enhancers in the same module showed substantial enrichment for sequence motifs68 associated with diverse transcription factors (Supplementary Fig. 13a). We found 84 significantly enriched motifs in 101 modules (Extended Data Fig. 8), indicating that enhancer modules likely represent co-regulated sets, and proposing candidate upstream regulators for nearly half of all modules. Direct application of the same approach and thresholds to the putative regulatory regions annotated in each of the 111 reference epigenomes led to significant enrichment for only 10 enriched motifs in 15 reference epigenomes (Supplementary Fig. 13b, c) of which 8 are blood samples, and focusing on the regions unique to each of the 17 tissue groups (Fig. 2b) only led to 19 enriched motifs in 10 tissue groups (Supplementary Fig. 13d, e), emphasizing the importance of studying regulatory motif enrichments at the level of enhancer modules.
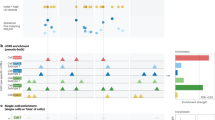
We next sought to distinguish likely activator and repressor motifs, by identifying regulators with expression patterns across cell/tissue types that show a strong (positive or negative) correlation with the activity of enhancers in the corresponding modules9. We focused on the 40 most strongly expression-correlated regulators (Extended Data Fig. 9a), and used the module-level motif enrichments to link each regulator to the cell/tissue types that define each module (Fig. 8). We found that many of the inferred links correspond to known regulatory relationships, including OCT4 (also known as POU5F1) in pluripotent cells, HNF1B and HNF4A1 in liver and other digestive tissues, RFX4 in neurosphere and neuronal cells, and MEF2D in muscle. The most enriched regulators showed primarily positive correlations, suggesting that they function as transcriptional activators, while a subset of factors showed a negative correlation, with the motif showing enhancer depletion in the lineages where the corresponding factor is expressed, suggesting a repressive role. For example, REST (also known as NRSF), a known repressor of neuronal lineages, showed lowest expression in neuronal tissues, where its motif was most enriched in enhancers, and a similar signature was found for ZBTB1B, a known repressor of myogenesis and brain development.

Module-level regulatory motif enrichment (Supplementary Fig. 11) and correlation between regulator expression and module activity patterns (Extended Data Fig. 8a) are used to link regulators (boxes) to their likely target tissue and cell types (circles). Edge weight represents motif enrichment in the reference epigenomes of highest module activity.
Regulatory motifs predicted to be drivers of enhancer activity patterns showed significant enrichment in tissue-specific high-resolution (6–40 bp) DNase digital genomic footprints (DGF)69 in matching cell types (Extended Data Fig. 9b and Supplementary Table 5b), providing DNA accessibility evidence that the motifs are indeed bound in these cell types. In addition, they showed positional bias relative to both the centre of DGF locations and relative to their boundaries (Extended Data Fig. 10), a property not found for shuffled motifs70. These positional biases were highly tissue- and cell-type-specific for most activating factors (Extended Data Fig. 9c), including POU5F1 in iPS cells, MEF2D in heart, HNF1B in gastrointestinal tissues, BHLH in brain, SPI1 in immune cells, and MEF2 in heart and muscle, in each case matching the tissues that showed the highest enrichment. In contrast, for repressive factors and CTCF, positional biases were found in large numbers of tissues, even when the motifs were not enriched in active enhancers. For example, REST (NRSF) was positionally biased in DGF sites in nearly all tissues except brain (Extended Data Fig. 9c), even though it was only enriched in active enhancers in brain (Extended Data Fig. 9a), consistent with widespread repressive binding in non-brain tissues.
Overall, these enhancer modules, motif enrichments and regulatory predictions provide an unbiased map that can help guide studies of candidate master regulators for fetal and adult lineage establishment and cell-type identity.
Impact of DNA sequence and genetic variation
We next studied the impact of primary DNA sequence on the epigenomic landscape, across genomic regions and between the two alleles of a given individual. First, we evaluated whether histone modifications and DNA methylation can be predicted by the underlying DNA sequence using DNA motifs for transcription factors expressed in ES cells and four ES-derived cell types. Using the area under the receiver operating curve (AUROC), we found between 71% predictive power for H3K4me1 peaks and 98% for H3K4me3 peaks (average of 85% across six marks and methylation-depleted regions)71. The most predictive motifs were those of factors associated with specific histone modifications or specific cell types, and were found within peak regions enriched for chromatin marks and at their boundaries. As an example of a boundary enrichment, H3K4me3 peaks were flanked by motifs consisting of a continuous stretch of A and T followed by a G and C, which may have a role in nucleosome positioning or recruiting promoter-associated transcription factors, such as nuclear receptors. Enhancer and promoter-predictive motifs were enriched in high-resolution DNase hypersensitive sites (Supplementary Table 5a), suggesting that they correspond to transcription-factor-bound sequences.
Second, we studied how sequence variants between the two alleles of the same individual can lead to allelic biases in histone modifications, DNA methylation and transcript levels. We reconstructed chromosome-spanning haplotypes for ES cells, four ES-cell-derived cell lines72 and 20 tissue samples61, and we resolved allele-specific activity and structure for each. We found widespread allelic bias in both transcript levels and epigenomic marks for each epigenome. For example, 24% of all testable genes that contain exonic variants demonstrate allelic transcription in one or more ES cell or ES-cell-derived cell lineages, and the majority of these genes also exhibit allelic epigenomic modifications in promoters (71%) and Hi-C-linked enhancers (69%)72. Similarly, as many as 11% of the testable enhancers display allelic bias in histone modification H3K27ac in the 20 tissue samples with allele-resolved transcription and chromatin states61. Allelic histone acetylation at enhancers is highly specific to individual genotypes, and often occurs near sequence variants that alter transcription factor binding, suggesting cis-acting sequence drivers for at least a subset of these regions61,72.
Trait-associated variants enrich in tissue-specific marks
We next used our tissue-specific epigenomic data sets to study the regulatory annotation enrichments of phenotype-associated variants from genome-wide association studies (GWAS) of diverse traits and disorders. Previous studies showed that disease-associated variants are enriched in specific regulatory chromatin states9, evolutionarily conserved elements73, histone marks74 and accessible regions14. We expanded these analyses using the diversity of primary tissues surveyed by our epigenomic maps, applied to a compendium of disease-associated variants from the NHGRI GWAS catalogue75. We intersected the set of variants identified in each curated study with peaks of H3K4me1, H3K4me3, H3K36me3, H3K27me3 and H3K9me3 across each of the 127 epigenomes, and H3K27ac, H3K9ac and DNase when available (Extended Data Figs 11, 12 and Supplementary Table 6), and we searched for significant enrichment in their overlap relative to what would be expected given the NHGRI GWAS catalogue as background (see Methods).
For enhancer-associated H3K4me1 peaks, we found 58 studies (Fig. 9a and Extended Data Fig. 11a) with significant enrichments in at least one tissue at 2% false discovery rate (FDR) (hypergeometric P < 10–3.9). Upon manual curation, the enriched cell types were consistent with our current understanding of disease-relevant tissues for the vast majority of cases. For example, diverse immune traits were enriched in immune cell enhancers, including rheumatoid arthritis, coeliac disease, type 1 diabetes, systemic lupus erythematosus, chronic lymphocytic leukaemia, allergy, multiple sclerosis, and Graves’ disease76,77,78,79,80,81,82. A large number of metabolic trait variants are enriched in liver enhancer marks, including LDL, HDL, total cholesterol, lipid metabolism phenotypes, and metabolite levels83,84. Fasting glucose was most enriched for pancreatic islet enhancer marks and insulin-like growth factors in placenta, consistent with their endocrine regulatory roles85,86. Several cardiac traits were enriched in heart tissue enhancers, including the PR heart repolarization interval, blood pressure and aortic root size. Interestingly, inflammatory bowel disease and ulcerative colitis variants show enrichment in both immune and gastrointestinal enhancer marks, suggesting that dysregulation of both organs may underlie disease predisposition. Both attention deficit hyperactivity disorder and adiponectin levels were enriched in brain regions, consistent with causal roles in brain dysregulation87,88. In contrast, late-onset Alzheimer’s disease variants were enriched in immune cell enhancers, rather than brain, consistent with recent evidence of a possible immune and inflammatory basis89,90,91.

Tissue-specific H3K4me1 peak enrichment significance (−log10 P value) for genetic variants associated with diverse traits. Circles denote reference epigenome (column) of most significant enrichment for SNPs reported by a given study (row), defined by trait and publication (PubMed identifier, PMID). Tissue (Abbrev) and P value (−log10) of most significant enrichment are shown. Only rows and columns containing a value meeting a FDR of 2% are shown (see Extended Data Figs 11 and 12 for full matrix for all studies showing at least 2% FDR).
For active enhancer-associated H3K27ac peaks (available in 98 cell types), we found a similar number of enriched studies (47 at 2% FDR, Extended Data Fig. 12b), but for promoter-associated H3K4me3 and H3K9ac peaks, we found only 25 and 18 enriched studies, respectively (Extended Data Fig. 12a, b), suggesting that enhancer-associated marks are more informative for tissue-specific disease enrichments than promoter-associated marks. For DNase peaks, we only found 9 enriched studies (Extended Data Fig. 12c), partly because they were only available in 53 reference epigenomes (restricting H3K4me1 to the same 53 resulted in 25 enriched studies, Supplementary Table 6), and possibly due to lack of distinction between enhancer and promoter regions. For transcription-associated H3K36me3, we found 15 enriched studies (Extended Data Fig. 12d), indicating that these help capture additional biologically meaningful variants outside annotated promoter and enhancer regions. In contrast, we found no enriched study for either Polycomb-associated H3K27me3 peaks or heterochromatin-associated H3K9me3 peaks (Extended Data Fig. 12e, f). These results indicate that enhancer-associated marks have the greatest ability to distinguish tissue-specific enrichments for regulatory regions, but promoter-, open-chromatin- and transcription-associated marks also have numerous significant enrichments, suggesting that disease variants affect a wide range of processes.
These results illustrate that the epigenomic annotations provided here across a broad range of primary tissues and cells will be of great utility for interpreting genetic changes associated with complex traits. We have made all these epigenomic annotations of GWAS regions publicly searchable and browsable through the Roadmap Epigenome Browser92 and an updated version of the HaploReg database93.
Discussion
The NIH Roadmap Epigenomics Program has been working to improve epigenomic assays, generate reference epigenomic maps, and use them to understand gene regulation, differentiation, reprogramming and human disease (see http://www.roadmapepigenomics.org/publications). This paper constitutes the first integrative analysis of all the reference epigenomes generated by the consortium, and represents an early component of the International Human Epigenome Consortium (http://ihec-epigenomes.org/), which seeks to extend such epigenomic maps to more than a thousand reference human epigenomes94.
In this paper, we use this resource to gain insights into the epigenomic landscape, its dynamics across cell types, tissues and development, and its regulatory circuitry. We find that combinations of histone modification marks are highly informative of the methylation and accessibility levels of different genomic regions, while the converse is not always true. Genomic regions vary greatly in their association with active marks, with approximately 5% of each epigenome marked by enhancer or promoter signatures on average, which show increased association with expressed genes, and increased evolutionary conservation, while two-thirds of each reference epigenome on average are quiescent, and enriched in gene-poor and nuclear-lamina-associated stably repressed regions. Even though promoter and transcription associated marks are less dynamic than enhancer mark, each mark recovers biologically meaningful cell-type groupings when evaluated in relevant chromatin states, allowing a data-driven approach to learn relationships between cell types, tissues and lineages. The coordinated activity patterns of enhancer regions enable us to cluster them into putative co-regulated modules, which are proximal to genes with common functions and phenotypes and enriched in regulatory motifs, enabling us to predict candidate upstream regulators.
We also demonstrate the usefulness of the resulting regulatory annotations for interpreting human genetic variation and disease. In an unbiased sampling across the GWAS catalogue, we find that genetic variants associated with complex traits are highly enriched in epigenomic annotations of trait-relevant tissues, providing insights on the likely relevant cell types underlying genome-wide significant loci. The GWAS enrichments in our analysis were strongest for enhancer-associated marks, consistent with their highly tissue-specific nature. However, promoter-associated and transcription-associated marks were also enriched, implicating several gene-regulatory levels as underlying genetic variants associated with complex traits. These results suggest that our data sets will be valuable in the study of human disease, as several companion papers explore in the context of autoimmune disorders95,96, Alzheimer’s disease91,97,98 and cancer99,100.
Overall, our epigenomic data sets, regulatory annotations and integrative analyses have resulted in the most comprehensive map of the human epigenomic landscape so far across the largest collection of primary cells and tissues. We expect that this map will be of broad use to the scientific and biomedical communities, for studies of genome interpretation, gene regulation, cellular differentiation, genome evolution, genetic variation and human disease.
Methods
No statistical methods were used to predetermine sample size.
Data matrix, primary analysis and processing quality control
All genome-wide maps of histone modifications, DNA accessibility, DNA methylation and RNA expression are freely available online. Links for raw sequencing data deposited at the Short Read Archive or dbGAP are available at http://www.ncbi.nlm.nih.gov/geo/roadmap/epigenomics/. All primary processed data (including mapped reads) for profiling experiments are contained within Release 9 of the Human Epigenome Atlas (http://www.epigenomeatlas.org). Complete metadata associated with each data set in this collection is archived at GEO and describes samples, assays, data processing details and quality metrics collected for each profiling experiment.
Release 9 of the compendium contains uniformly pre-processed and mapped data from multiple profiling experiments (technical and biological replicates from multiple individuals and/or data sets from multiple centres). To reduce redundancy, improve data quality and achieve uniformity required for our integrative analyses, experiments were subjected to additional processing to obtain comprehensive data for 111 consolidated epigenomes (see sections below for additional details). Numeric epigenome identifiers (EIDs; for example, E001) and mnemonics for epigenome names were assigned for each of the consolidated epigenomes. Supplementary Table 1 (QCSummary sheet) summarizes the mapping of the individual Release 9 samples to the consolidated epigenome IDs. Key metadata such as age, sex, anatomy, epigenome class (see Supplementary Table 1, EpigenomeClassSummary sheet), ethnicity and solid/liquid status were summarized for the consolidated epigenomes. Data sets corresponding to 16 cell lines from the ENCODE project (with epigenome IDs ranging from E114 to E129) were also used in the integrative analyses23. All data sets from the 127 consolidated epigenomes were subjected to processing filters to ensure uniformity in terms of read-length-based mappability and sequencing depth as described below.
Each of the 127 epigenomes included consolidated ChIP-seq data sets for a core set of histone modifications—H3K4me1, H3K4me3, H3K27me3, H3K36me3, H3K9me3—as well as a corresponding whole-cell extract sequenced control. Ninety-eight epigenomes and sixty-two epigenomes had consolidated H3K27ac and H3K9ac histone ChIP-seq data sets, respectively. A smaller subset of epigenomes had ChIP-seq data sets for additional histone marks, giving a total of 1,319 consolidated data sets (Supplementary Table 1, QCSummary sheet). 53 epigenomes had DNA accessibility (DNase-seq) data sets. Fifty-six epigenomes had mRNA-seq gene expression data. For the 127 consolidated epigenomes, a total of 104 DNA methylation data sets across 95 epigenomes involved either bisulfite treatment (WGBS or RRBS assays) or a combination of MeDIP-seq and MRE-seq assays. In addition to the 1,936 data sets analysed here across 111 reference epigenomes, the NIH Roadmap Epigenomics Project has generated an additional 869 genome-wide data sets, linked from GEO, the Human Epigenome Atlas, and NCBI, and also publicly and freely available.
RNA-seq uniform processing and quantification for consolidated epigenomes
We uniformly reprocessed mRNA-seq data sets from 56 reference epigenomes that had RNA-seq data. For RNA-seq analysis, after library construction45, we aligned 75-bp-long or 100-bp-long reads using the BWA aligner, and generated read coverage profiles separately for positive and negative strand strand-specific libraries. We used several QC metrics for the RNA-seq library, including intron–exon ratio, intergenic reads fraction, strand specificity (for stranded RNA-seq protocols), 3′-5′ bias, GC bias and RPKM discovery rate (Supplementary Table 1, RNaseqQCSummary sheet). We quantified exon and gene expression using a modified RPKM measure8, whereby we used the total number of reads aligned into coding exons for the normalization factor in RPKM calculations, and excluded reads from the mitochondrial genome, reads falling into genes coding for ribosomal proteins, and reads falling into top 0.5% expressed exons. RPKM for a gene was calculated using the total number of reads aligned into all merged exons for a gene normalized by total exonic length. The resulting files contain RPKM values for all annotated exons and coding and non-coding genes (excluding ribosomal genes), as well as introns (Gencode V10 annotations were used). We also report the coordinates of all significant intergenic RNA-seq contigs not overlapping the annotated genes.
ChIP-seq and DNase-seq uniform reprocessing for consolidated epigenomes
Read mapping. Sequenced data sets from the Release 9 of the Epigenome Atlas involved mapping a total of 150.21 billion sequencing reads onto hg19 assembly of the human genome using the PASH read mapper34. These read mappings were used (except for RNA-seq data sets, which were mapped as described above) for constructing the 111 consolidated epigenomes. Only uniquely mapping reads were retained and multiply-mapping reads were filtered out. BED files containing the mapped reads were obtained from http://www.epigenomeatlas.org. Alignment parameters for each assay type and experiment are specified in the associated publicly accessible Release 9 metadata archived at GEO. For the ENCODE data sets, BAM files containing mapped reads were downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/. Only uniquely mapping reads were retained and multiply mapping reads were discarded.
Mappability filtering, pooling and subsampling. The raw Release 9 read alignment files contain reads that are pre-extended to 200 bp. However, there were significant differences in the original read lengths across the Release 9 raw data sets reflecting differences between centres and changes of sequencing technology during the course of the project (36 bp, 50 bp, 76 bp and 100 bp). To avoid artificial differences due to mappability, for each consolidated data set the raw mapped reads were uniformly truncated to 36 bp and then refiltered using a 36-bp custom mappability track to only retain reads that map to positions (taking strand into account) at which the corresponding 36-mers starting at those positions are unique in the genome. Filtered data sets were then merged across technical/biological replicates, and where necessary to obtain a single consolidated sample for every histone mark or DNase-seq in each standardized epigenome. Supplementary Table 1 summarizes the mapping of the individual Release 9 primary data sample files to the consolidated data files corresponding to the 127 consolidated reference epigenomes.
To avoid artificial differences in signal strength due to differences in sequencing depth, all consolidated histone mark data sets (except the additional histone marks the seven deeply profiled epigenomes, Fig. 2j) were uniformly subsampled to a maximum depth of 30 million reads (the median read depth over all consolidated samples). For the seven deeply profiled reference epigenomes (Fig. 2j), histone mark data sets were subsampled to a maximum of 45 million reads (median depth). The consolidated DNase-seq data sets were subsampled to a maximum depth of 50 million reads (median depth). These uniformly subsampled data sets were then used for all further processing steps (peak calling, signal coverage tracks, chromatin states).
Peak calling. For the histone ChIP-seq data, the MACSv2.0.10 peak caller was used to compare ChIP-seq signal to a corresponding whole-cell extract (WCE) sequenced control to identify narrow regions of enrichment (peaks) that pass a Poisson P value threshold 0.01, broad domains that pass a broad-peak Poisson P value of 0.1 and gapped peaks which are broad domains (P < 0.1) that include at least one narrow peak (P < 0.01) (https://github.com/taoliu/MACS/)32. Fragment lengths for each data set were pre-estimated using strand cross-correlation analysis and the SPP peak caller package (https://code.google.com/p/phantompeakqualtools/)37 and these fragment length estimates were explicitly used as parameters in the MACS2 program (–shift-size = fragment_length/2).
For DNase-seq data, we used two methods to identify DNase I accessible sites. First, the Hotspot algorithm was used to identify fixed-size (150 bp) DNase hypersensitive sites, and more general-sized regions of DNA accessibility (hotspots) using an FDR of 0.01 (http://www.uwencode.org/proj/hotspot)104. MACSv2.0.10 was also used to call narrow peaks using the same settings specified above for the histone mark narrow peak calling.
Narrow peaks and broad domains were also generated for the unconsolidated, 36-bp mappability filtered histone mark ChIP-seq and DNase-seq Release 9 data sets using MACSv2.0.10 with the same settings as specified above.
Genome-wide signal coverage tracks. We used the signal processing engine of the MACSv2.0.10 peak caller to generate genome-wide signal coverage tracks. Whole-cell extract was used as a control for signal normalization for the histone ChIP-seq coverage. Each DNase-seq data set was normalized using simulated background data sets generated by uniformly distributing equivalent number of reads across the mappable genome. We generated two types of tracks that use different statistics based on a Poisson background model to represent per-base signal scores. Briefly, reads are extended in the 5′ to 3′ direction by the estimated fragment length. At each base, the observed counts of ChIP-seq/DNase I-seq extended reads overlapping the base are compared to corresponding dynamic expected background counts (λlocal) estimated from the control data set. λlocal is defined as max(λBG, λ1k, λ5k, λ10k) where λBG is the expected counts per base assuming a uniform distribution of control reads across all mappable bases in the genome and λ1k, λ5k, λ10k are expected counts estimated from the 1 kb, 5 kb and 10 kb window centred at the base. λlocal is adjusted for the ratio of the sequencing depth of ChIP-seq/DNase-seq data set relative to the control data set. The two types of signal score statistics computed per base are as follows.
(1) Fold-enrichment ratio of ChIP-seq or DNase counts relative to expected background counts λlocal. These scores provide a direct measure of the effect size of enrichment at any base in the genome.
(2) Negative log10 of the Poisson P-value of ChIP-seq or DNase counts relative to expected background counts λlocal. These signal confidence scores provide a measure of statistical significance of the observed enrichment.
The −log10(P value) scores provide a convenient way to threshold signal (for example, 2 corresponds to a P value threshold of 1 × 10−2), similar to what is used in identifying enriched regions (peak calling). We recommend using the signal confidence score tracks for visualization. A universal threshold of 2 provides good separation between signal and noise. Both types of signal tracks were also generated for the unconsolidated data sets using the same parameter settings described above.
Quality control. For the primary Release 9 data sets, data quality enrichment scores were computed as the fraction of the uniquely mapped reads overlapping with areas of enrichment. Several methods were employed to select signal enrichment regions. The SPOT quality score was computed based on regions identified with the HotSpot peak caller104; the FindPeaks quality score was inferred based on peak calls made using the FindPeaks36 software; finally, a Poisson metric was derived by modelling the read distribution in genome-tiling 1,000-bp windows with a Poisson distribution and selecting as enriched regions windows with P < 0.05. All the quality scores in Release 9 are in agreement, with strong pairwise correlation (Pearson correlation >0.9). Concordance between centres was confirmed and data analysis pipeline was validated at the outset of the project using data sets for the H1 cell line. The same pipeline was subsequently used to produce Release 9 data. ChIP-seq data for six histone modifications (H3K4me3, H3K27me3, H3K9ac, H3K9me3, H3K36me3 and H3K4me1) were independently generated for the H1 cell line by three REMCs (Broad, UCSD, UCSF-UBC). To quantify concordance, the reads from each experiment were mapped (Level 1 data), read density tracks (Level 2 data) were generated using the EDACC’s primary data processing pipeline, and finally Pearson correlation coefficients were computed between each pair of experiments, as well as between experiments and H1 input acting as a control for background correlation between signals (Supplementary Table 2). The methylome processing pipeline was characterized experimentally on four independent samples38,39.
For the uniformly reprocessed and consolidated ChIP-seq and DNase-seq data sets, strand cross-correlation measures were used to estimate signal-to-noise ratios (https://code.google.com/p/phantompeakqualtools/)37. Data sets for each mark were rank-ordered based on the normalized strand cross-correlation coefficient (NSC) and flagged if the scores were significantly below the median value or in the range of NSC values for WCE extract controls. Consolidated data sets with extremely low sequencing depth (<10M reads) were also flagged. Each standardized epigenome was then manually assigned a subjective quality flag of 1 (high), 0 (medium) or −1 (low), based on the number of flagged data sets it contained. The SPOT, FindPeaks and Poisson quality scores were also recomputed for the consolidated data sets. We observed high correlations of the NSC scores with the SPOT (Pearson correlation of 0.7) and FindPeaks scores (Pearson correlation of 0.65). All QC measures are provided in Supplementary Table 1 (Sheets QCSummary and AdditionalQCScores).
To identify potential antibody cross-reactivity or mislabelling issues, a pairwise correlation heat map (Extended Data Fig. 1e) was computed across all consolidated data sets for H3K4me1, H3K4me3, H3K36me3, H3K27me3, H3K9me3, H3K27ac, H3K9ac and DNase. We computed the Pearson correlation between all pairs of the signal tracks based on signal in chromosomes 1–22 and chromosome X. We used the signal confidence score tracks (−log10(Poisson P value)) where we first computed the average signal scores within each consecutive 25-bp interval. To order the experiments in the heat map we defined the distance between two pairs of experiments as 1-correlation value and used a travelling salesman problem formulation105.
Methylation data cross-assay standardization and uniform processing for consolidated epigenomes
We used PASH38 alignments for the WGBS and RRBS read alignments. From the number of converted and unconverted reads at each individual CpG the total coverage and fractional methylation were reported. The data were uniformly post-processed and formatted into two matrices for each chromosome. One matrix contained read coverage information for each base (C and G) in every CpG (row) and for each reference epigenome (column). Another matrix similarly contained fractional methylation ranging from 0 to 1. For the locations where coverage was ≤3 we considered data as missing. For MeDIP/MRE methylation data we used the output of the mCRF tool31 that reports fractional methylation in the range from 0 to 1 and uses an internal BWA mapping. The mCRF results were combined in a single matrix per chromosome for all reference epigenomes where available.
Chromatin state learning
To capture the significant combinatorial interactions between different chromatin marks in their spatial context (chromatin states) across 127 epigenomes, we used ChromHMM v.1.10106, which is based on a multivariate Hidden Markov Model.
‘Core’ 15-state model
A ChromHMM model applicable to all 127 epigenomes was learned by virtually concatenating consolidated data corresponding to the core set of five chromatin marks assayed in all epigenomes (H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3). The model was trained on 60 epigenomes with highest-quality data (Fig. 2k), which provided sufficient coverage of the different lineages and tissue types (Supplementary Table 1; Sheet QCSummary). The ChromHMM parameters used were as follows: reads were shifted in the 5′ to 3′ direction by 100 bp. For each consolidated ChIP-seq data set, read counts were computed in non-overlapping 200-bp bins across the entire genome. Each bin was discretized into two levels, 1 indicating enrichment and 0 indicating no enrichment. The binarization was performed by comparing ChIP-seq read counts to corresponding whole-cell extract control read counts within each bin and using a Poisson P value threshold of 1 × 10−4 (the default discretization threshold in ChromHMM). We trained several models in parallel mode with the number of states ranging from 10 states to 25 states. We decided to use a 15-state model (Fig. 4a–f and Extended Data Fig. 2b) for all further analyses since it captured all the key interactions between the chromatin marks, and because larger numbers of states did not capture sufficiently distinct interactions. The trained model was then used to compute the posterior probability of each state for each genomic bin in each reference epigenome. The regions were labelled using the state with the maximum posterior probability.
‘Expanded’ 18-state model
A second ‘expanded’ model applicable to 98 epigenomes that also have an H3K27ac ChIP-seq data set was learned by virtually concatenating consolidated data corresponding to the core set of five chromatin marks and H3K27ac. The model was trained on 40 high-quality epigenomes using the same parameters as those used for the primary model (Supplementary Table 1; Sheet QCSummary). We trained several models with the number of states ranging from 15 states to 25 states. An 18-state model was used for further analyses (Extended Data Fig. 2c) based on similar considerations.
State labels, interpretation and mnemonics
To assign biologically meaningful mnemonics to the states, we used the ChromHMM package to compute the overlap and neighbourhood enrichments of each state relative to various types of functional annotations (Fig. 4b, c, f and Extended Data Fig. 2b, c and Supplementary Fig. 2).
For any set of genomic coordinates representing a genomic feature and a given state, the fold enrichment of overlap is calculated as the ratio of ‘the joint probability of a region belonging to the state and the feature’ versus ‘the product of independent marginal probability of observing the state in the genome’ times ‘the probability of observing the feature’, namely the ratio between the (number of bases in state AND overlap feature)/(number of bases in genome) and the [(number of bases overlap feature)/(number of bases in genome) × (number of bases in state)/(number of bases in genome)]. The neighbourhood enrichment is computed for genomic bins around a set of single-base-pair anchor locations such as transcription start sites.
For the overlap enrichment plots in the figures, the enrichments for each genomic feature (column) across all states is normalized by subtracting the minimum value from the column and then dividing by the max of the column. So the values always range from 0 (white) to 1 (dark blue); that is, it’s a column-wise relative scale. For the neighbourhood positional enrichment plots, the normalization is done across all columns; that is, the minimum value over the entire matrix is subtracted from each value and divided by the maximum over the entire matrix.
The functional annotations used were as follows (all coordinates were relative to the hg19 version of the human genome): (1) CpG islands obtained from the UCSC table browser. (2) Exons, genes, introns, transcription start sites (TSSs) and transcription end sites (TESs), 2-kb windows around TSSs and 2-kb windows around TESs based on the GENCODEv10 annotation (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeGencodeV10/) restricted to GENCODE biotypes annotating long transcripts. (3) Expressed and non-expressed genes, their TSSs and TESs. Genes were classified into the expressed or non-expressed class based on their RNA-seq expression levels in the H1-ES cells (Fig. 4c) and GM12878 (Extended Data Fig. 2b) cell lines. A gaussian mixture model with two components was fit on expression levels of all genes to obtain thresholds for the two classes. (4) Zinc finger genes (obtained by searching the ENSEMBL annotation for genes with gene names starting with ZNF). (5) Transcription factor binding sites (TFBS) based on ENCODE ChIP-seq data in the H1-ES cell line. The uniformly processed transcription factor ChIP-seq peak locations were downloaded from the ENCODE repository: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeAwgTfbsUniform/. We also computed percentage transcription factor binding site coverage for state calls in the GM12878 and K562 cell lines using corresponding transcription factor ChIP-seq data from ENCODE which matched and supported the mnemonics and state interpretations obtained from the H1 cell line (Supplementary Fig. 2). (6) Conserved GERP elements based on 34 way placental mammalian alignments http://mendel.stanford.edu/SidowLab/downloads/gerp/ (Supplementary Fig. 3). (7) Enrichment for conserved GERP elements subtracting parts of the above-mentioned GERP elements that overlap exons.
Comparison to chromatin states learned on individual epigenomes
We also learned independent 15-state models individually on each of the 127 epigenomes using the core set of 5 marks and the same parameter settings as for the primary model. To compare the individual models to the joint 15-state primary model, we stacked the emission vectors for all states from all the models and hierarchically clustered them using Euclidean distance and Ward linkage (Extended Data Fig. 2a). The individual epigenome models consistently and repeatedly identified states that were also recovered by the joint model (Extended Data Fig. 2a). Two additional clusters which included states recovered by the independent models learned in individual cell types, but not recovered in the joint model, were HetWk, characterized by weak presence of H3K9me3, and Rpts, characterized by presence of H3K9me3 along with a diversity of other marks, which was enriched in a large number of repeat elements.
Expanded chromatin states using large numbers of histone marks
For each of the seven deeply profiled reference epigenomes (Fig. 2j) we independently learned chromatin states on observed data for all available histone modifications or variants, and DNase in the reference epigenome. The same binarization and model learning procedure was followed as for the core set of 5 marks. We chose to consistently focus on a larger set of 50-states to capture the additional state distinctions afforded by using additional marks (Supplementary Fig. 4). Enrichments for annotations, including some of those described above for the 15-state model, were computed using ChromHMM. The HiC domains were obtained from ref. 107; the lamina-associated domains are described below; conserved element sets were the hg19 lift-over from ref. 73; repetitive element definitions were from RepeatMasker.
Relationship between histone marks, methylation and DNase
The distribution of DNA methylation (per cent CpG methylation from WGBS data) and DNA accessibility (DNase-seq −log10(P value) signal confidence scores) was computed using regions belonging to each of the 15 chromatin states based on the core set of 5 marks and the 18 chromatin states from the expanded model across all reference epigenomes for which these data sets were available (Fig. 4d, e).
CpGs with a minimum read coverage of 5 were used to calculate the average methylation percentages within genomic regions labelled with each chromatin state from the 15-state primary model and 18 state expanded model. Only regions containing more than 3 CpGs with at most 200 bp between consecutive CpGs were used. Plots were generated using ggplot2 package for R (v.3.02). The average methylation levels for the chromatin states across DNA methylation platforms (WGBS, RRBS and mCRF) were analysed using Standard Least Square models in JMP (v.11.0; SAS Ins.). The model included the platforms (3 levels), chromatin states (15 levels) and the interactions (Extended Data Fig. 4).
Calling of lamina-associated domains
Genome-wide DamID binding data for human lamin B1 in SHEF-2 ES cells were obtained from GEO series GSE22428 (ref. 63). Lamina associated domains were determined using a similar method to the one described in ref. 64. First, hg18-based data coordinates were converted to hg19-based coordinates using UCSC’s liftOver tool. Data were smoothed using a running median filter with a window size of 5 probes, after which domains were detected by estimating border and domain positions and comparing these to domains defined on 100 randomized instances of the same data set. Parameters are chosen such that the false discovery rate (FDR) for detected domains is 1%.
Chromatin state variability
For each state s for the core 15-state joint model we computed the number of genomic bins that were labelled with that state in at least one epigenome (Gs). From among these bins we counted the number of bins (gs,i) that were labelled as being in state s in exactly i epigenomes (i = 1...127). We converted these counts to fractions (gs,i/Gs) and computed the cumulative fraction that is consistently labelled with the same chromatin state in at most N epigenomes (N = 1...127). States whose cumulative fractions rise faster than others represent those that are less constitutive (more variable). We repeated the same procedure restricted to 43 high-quality and non-redundant Roadmap epigenomes (using only 1 representative epigenome from those corresponding to ES cells, iPS lines or epigenomes for the same tissue type from different individuals and excluding ENCODE cell lines) (Supplementary Table 1, Sheet VariationAnalysis) (Supplementary Fig. 6a). Analogous analysis was performed on states from the 18-state expanded model (Extended Data 5a and Supplementary Fig. 6b).
The observed cumulative fractions of cell-type specificity are a function of the composition of cell types in the compendium and do depend to some extent on the variability of data quality for the different marks. For example, the enhancer mark (H3K4me1) does have a much better signal-to-noise ratio than the transcribed mark (H3K36me3). One might expect this to result in more spurious variation of states associated with the transcribed mark. However, contrary to this expectation, the cumulative fractions for states involving only the transcription mark (Tx and TxWk) and not the enhancer mark indicate that these states are in fact less variable and more constitutive across cell types. On the other hand, all states composed of the enhancer mark (H3K4me1), irrespective of whether they do (TxFlnk, EnhG) or do not (EnhBiv, Enh, BivFlnk, TssAFlnk) include the transcription mark (H3K36me3), are far more cell-type specific. These observations indicate that the increased variability of states is largely due to the enhancer mark (H3K4me1) than the transcribed mark (H3K36me3). As replicates are not available in all epigenomes, we did not correct for inter-replicate variation in this analysis, but in the state-switching analysis below we utilize samples from the same tissue as quasi-replicates.
Chromatin state switching
To avoid spurious switching due to differences in data quality, we restricted this analysis to chromatin states from the 43 high-quality and non-redundant Roadmap epigenomes (see above). Using the 15 state primary model, we computed the empirical switching frequency of any pair of states across all pairs of 43 epigenomes. For a given pair of states A and B, we counted the number of genomic bins that were labelled as (A,B) or (B,A) in all pairs of epigenomes. The switching frequency matrix (which is symmetric) was then row-normalized to convert the switching frequencies to switching probabilities. This is done to avoid a dependence on the total number of epigenomes. Also, the switching probabilities unlike switching frequencies are not dominated by states that are highly prevalent (for example, quiescent state). Supplementary Fig. 7b shows the empirical switching probabilities for all pairs of states across the 43 epigenomes. To differentiate between chromatin state dynamics across tissues (inter-tissue) relative to variation of states across individuals or replicates from the same tissue (intra-tissue), we also computed analogous switching frequencies by restricting to subgroups of epigenomes from the same tissue type (Supplementary Table 1, Sheet VariationAnalysis). The frequencies were added across all sub-groups and then row-normalized to switching probabilities. Supplementary Fig. 7a shows the intra-tissue switching probabilities. We then computed the relative enrichment of state switches as the log10 ratio of inter-tissue switching probability across the 43 epigenomes relative to the intra-tissue switching probabilities (Fig. 5c). We repeated this analysis on the 16 ENCODE cell lines and obtained similar conclusions regarding relative enrichment of state switches (Supplementary Fig. 7c). Analogous analyses were performed using the 18-state expanded model in Roadmap Epigenomics samples (Extended Data Fig. 5c) and ENCODE samples (Supplementary Fig. 7d).
Large-scale chromatin structure
To study large-scale chromatin structure we first calculated ChromHMM (15-state model) state frequencies identified in 200-bp genome-wide bins across 127 epigenomes. Then we averaged state frequencies over the 2-Mb genomic regions, thus defining vectors of length 1,458 for each state. The unsupervised clustering of a 15 × 1,458 matrix (using Pearson correlation as a similarity measure and complete linkage) revealed 11 distinct genomic clusters enriched in different subsets of chromatin states (Fig. 5d, top heat map). Clusters had different sizes, with the smallest one (c1) containing only 27 bins, while the largest cluster (c9), occupied predominantly by a ‘quiescent’ state for all epigenomes, had 377 bins. For each 2-Mb bin in each cluster we calculated average gene density, lamin B1 signal (see section 4 above) and overlap with different cytogenetic bands (Fig. 5d, bottom, which displays also average levels across each cluster). We also show chromosomal locations of the clusters as well as distributions of CpG island frequency across the 2-Mb bins in each cluster (Extended Data Fig. 5d).
DMR calls across reference epigenomes
As a general resource for epigenomic comparisons across all epigenomes for which DNA methylation data is available, we defined DMRs using the method of Lister et al.108, combining all differentially methylated sites (DMSs) within 250-bp of one another into a single DMR and excluded any DMR with less than 3 DMSs. For each DMR in each sample, we computed its average methylation level, weighted by the number of reads overlapping it109. This resulted in a methylation level matrix with rows of DMRs and columns of samples.
DMRs in hESC differentiation (Fig. 4h)
For analysing differentiation of hESCs in Fig. 4h, we used a second set of DMRs. We used a pairwise comparison strategy between ES cells and three in vitro derived cell types representative of the three germ layers (mesoderm, endoderm, ectoderm) and performed DMR calling as previously described53. Only DMRs losing more than 30% methylation compared to the ES cell state at a significance level of P ≤ 0.01 were retained. Subsequently, we computed weighted methylation levels for all three DMR sets across HUES64, mesoderm, endoderm and ectoderm as well as three consecutive stages of in vitro derived neural progenitors (please see accompanying paper53 for details on the cell types). Finally, we plotted the corresponding distribution using the R function vioplot in the vioplot package. In order to identify potential regulators associated with the loss of DNA methylation at these regions, we determined binding sites of a compendium of transcription factors profiled in distinct cell lines and types that overlapped with each set of hypomethylated DMRs51. Next, we determined a potential enrichment over a random genomic background by randomly sampling 100 equally sized sets of genomic regions, respecting the chromosomal and size distribution of the different DMR sets and determined their overlap with the same transcription factor binding site compendium to estimate a null distribution. Only transcription factors that showed fewer binding sites across the control regions in 99 of the cases were considered for further analysis. Next, we computed the average enrichment over background for each transcription factor with respect to the 100 sets of random control regions for each germ layer DMR and report this enrichment level in Fig. 4h right, where we capped the relative enrichment at 12.
Additional DMR calls
For studying breast epithelia differentiation, DMRs were called from WGBS, requiring at least five aligned reads to call differentially methylated CpG, and at least three differentially methylated CpGs within a distance of 200 bp of each other45. For studying tissue environment versus developmental origin, DMRs were called from MeDIP and MRE data using the M&M algorithm56.
DNA methylation variation
For variation in methylation of each chromatin state across epigenomes (Fig. 4g and Extended Data Fig. 4f), we first excluded any contiguous chromatin state region containing less than three CpG sites. Then, the mean of the methylation level for all contained CpG sites was calculated for each region, and for each epigenome density values were calculated for these mean methylation values between 0% and 100%, with density values estimated over n = 1,000 points with a gaussian kernel, with the default ‘nrd0’ bandwidth from the R stats package density function. Finally, for each chromatin state, we plotted the ln(density + 1) for each epigenome as rows, with the colour scale set with white as the minimum ln(density + 1) value and red, green, or blue, for WGBS, mCRF and RRBS, respectively, set as the maximum ln(density + 1) value in the matrix. Rows were ordered by the epigenomic lineage and grouping ordering shown in Fig. 2a. In Extended Data Fig. 4f, epigenomes were first grouped by methylation platform, and then ordered by Fig. 2a within each platform. The chromatin state methylation profiles in the cell lines versus primary cells/tissue cells were analysed using a mixed model with repeated measures. Overall effect of the group (cell lines versus primary cells/tissue cells) was tested using epigenomes within group as the error term. Testing for group effect was performed for each of the 15 chromatin states, resulting in a Bonferroni correction on the P values for the 15 tests.
Identifying coordinated changes in chromatin marks during development
To identify patterns of coordinated changes of histone marks over enhancers during heart muscle development, we compared ES cells, mesendoderm cells, and left ventricle tissue57. We identified relevant enhancers as those that show changes in at least one histone mark between a specific cell type cluster (heart muscle in our case) and other cell types using LIMMA (Linear Model for Microarray Analysis). We applied FDR-corrected P value significance threshold of 0.05 to obtain cluster-specific enhancers. For each tissue type (heart muscle in our case) we then clustered the enhancers into five clusters (C1–C5) based on their multi-mark epigenomic profiles using the k-means algorithm implemented in the Spark tool (Fig. 4i). The tools used to generate Fig. 4i are integrated into the Epigenomic Toolset within the Genboree Workbench and are accessible for online use at http://www.genboree.org.
Clustering of epigenomes reveals common lineages and common properties
For each analysed mark, we calculated Pearson correlation values between all pairwise combinations of reference epigenomes using the mark’s signal confidence scores (–log10(Poisson P value)) within 200 bp of the genomic regions deemed relevant for that mark. Relevance of regions is determined by whether a region was called in a particular (mark-matched) chromatin state with posterior probability of >0.95 in any of the reference epigenomes. For H3K4me1, H3K27ac and H3K9ac we used state Enh; for H3K4me3 state TssA; for H3K27me3 state ReprPC; for H3K36me3 state Tx; and for H3K9me3 state Het, unless otherwise noted (all based on the 15-state core model).
The resulting correlation matrices were used as the basis for a distance matrix for complete-linkage hierarchical clustering, followed by optimal leaf ordering110. Bootstrap support values are derived from 1,000 random samplings with replacement from all regions considered for a particular mark and a bootstrap tree was estimated for each resampling. The bootstrap support for a branch corresponds to the fraction of bootstrapped trees that support the bipartition induced by the branch.
In parallel to this, all correlation matrices mentioned above were used to perform Multi-Dimensional Scaling analyses using R.
Delineation of DNase I-accessible regulatory regions
For each of the 39 Roadmap reference epigenomes with DNase data, peak positions are combined across reference epigenomes by defining peak island areas, defined by stacking all DNase peak positions across epigenomes, and considering the full width at half maximum (FWHM). Note that for this we are only considering peak locations, not intensities. The goal of this is to obtain an estimate of the area of open chromatin, not to quantify the level of ‘openness’, as these data are not available for all reference epigenomes. In cases when peak islands overlap, they are merged because it means that the original DNase peak area populations overlap at least for half of the epigenomes with DNase peaks in that area (given the FWHM approach). Peak island summits are defined as the median peak summit of all peak island member DNase peaks. This results in a total of 3,516,964 DNase enriched regions across epigenomes.
We then annotate each of the ∼3.5M DNase peaks with the chromatin states they overlap with in each of the 111 Roadmap reference epigenomes, using the core 15-state chromatin state model, and focusing on states TssA, TssAFlnk and TssBiv for promoters, and EnhG, Enh and EnhBiv for enhancers, and state BivFlnk (flanking bivalent Enh/Tss) for ambiguous regions. Out of these, ∼2.5M regions are called as either enhancer or promoter across any of the 111 Roadmap reference epigenomes. Note that because DNase data are not available for all Roadmap epigenomes, the set of regulatory regions defined may exclude DNase regions active in cell types for which DNase was not profiled (Fig. 2g). Although most regions are undisputedly called exclusively promoter or enhancer, there are 535,487 regions that needed further study to decide whether they should be called promoters, enhancers, or both (‘dyadic’). We arbitrate on these regions by first clustering them (using the methods in the following section) with an expected cluster size of 10,000 regions, and then for each cluster calculating (a) the mean posterior probabilities for promoter and enhancer calls separately, and (b) the mean number of reference epigenomes in which regions were called promoter or enhancer. Clusters of regions for which the differences in mean posterior probabilities (a) is smaller than 0.05, or for which the absolute log2 ratio of the number of epigenomes called as promoter or enhancer (b) is smaller than 0.05, are called true ‘dyadic’ regions, along with a small number of ‘ambiguous’ regions in state BivFlnk. Note that this particular clustering is only to arbitrate on these regions using group statistics instead of one-by-one; the final clusterings are described next. Overall, we define ∼2.3M putative enhancer regions (12.63% of genome), ∼80,000 promoter regions (1.44% of genome) and ∼130,000 dyadic regions (0.99% of genome), showing either promoter or enhancer signatures across epigenomes.
Clustering of DNase I-accessible regulatory regions to identify modules of coordinated activity
To cluster regulatory (that is, enhancer, promoter or dyadic) regions based on their activity patterns across all reference epigenomes, we expressed each region in terms of a binary vector of length n×s, where n is the number of reference epigenomes (111) and s is the number of chromatin states considered. For enhancers and promoters, s = 3, as both of these types of regions are made up of 3 chromatin states in the 15-state ChromHMM model (enhancers, EnhG, Enh and EnhBiv; promoters, TssA, TssAFlnk and TssBiv).
The thus obtained binary matrices are subsequently clustered using a variation of a k-centroid clustering algorithm111. Instead of Euclidean distance we use a Jaccard-index-based distance. This is done to be able to correctly cluster highly cell-type-restricted regions. From a computational point of view, we optimized the method to both deal with the size of the used data matrices and leverage their sparsity, to efficiently compute and update distances for matrices with sizes on the order of 106 × 103. The algorithm has been further modified to converge when less than 0.01% of cluster assignments change between iterations.
We selected the number of clusters k by tuning the expected number of regions within each cluster to be approximately 1,000 for promoter and dyadic regions, and approximately 10,000 for enhancer regions, given their much larger count (81,000, 129,000 and 2.3M for promoter, dyadic and enhancer, respectively). This results in a value of k = 233 for enhancer clusters (for ∼10k elements per cluster), and the algorithm converged on k = 226 non-empty clusters, which are used for subsequent analyses.
Clusters are visualized (Fig. 7a) by ‘diagonalizing’ when possible. First, ‘ubiquitous’ clusters (defined as having at least 50% of epigenomes with an enhancer/promoter density of >25%) are shown. Then, the remaining clusters are ordered according to which epigenome has the maximum enhancer density.
Enrichment analyses of proximity to gene members of a catalogue of gene sets (Gene Ontology (GO), Human Phenotype Ontology (HPO)) have been performed using the GREAT tool55. In particular, the GREAT web API was used to automatically submit region descriptions and retrieve results for subsequent parsing. We restricted ourselves to interpretation of results with an enrichment ratio of at least 2, and multiple hypothesis testing corrected P values <0.01 for both the binomial and the hypergeometric distribution based tests.
For visualization of a representative subset of enriched terms in Fig. 7b, c, we select representative terms for display (after diagonalizing the enrichment matrix by re-ordering the rows). We do this using a weighted bag-of-words approach to select highly enriched terms that contain many words that are over-represented in gene-set labels showing similar enrichment patterns. Briefly, sliding along the row names (gene-set terms) of the diagonalized enrichment matrices, we collect word counts and multiply these by integer-rounded –log10(q values) obtained from GREAT. We do this in sliding windows of size 33 for Fig. 7b (resulting in 35 terms) and size 16 for Fig. 7c (resulting in 15 terms). For each word in a window, these values are expressed relative to the same words across all row names, registering to what extent they are over-represented. Each gene-set term in the window is then assigned a score based on the mean over-representation of all words it consists of. Lastly, gene sets are co-ranked based on this mean over-representation and their GREAT significance. The best-ranked gene set label is selected as the representative label for that window. All terms are shown in Supplementary Fig. 11d and are available for download at http://compbio.mit.edu/roadmap.
Predicting regulators active in each tissue, cell type and lineage
We collected 1,772 known transcription factor recognition motifs (position weight matrices) from primarily large-scale databases68,112,113,114,115,116,117 and measured their enrichment in the enhancers for each enhancer module compared to the union of the 226 enhancer modules (as described in refs 9,68) using a 0.3 conservation-based confidence cutoff70,73. We clustered motifs using a 0.75 correlation cutoff resulting in 300 motif clusters68 and selected for each motif cluster the motif with the highest enrichment in any enhancer module for further analysis.
We computed an expression score for each enhancer module and transcription factor as the Pearson correlation between the transcription factor expression across cell types with expression data (quantile-normalized log(RPKM) with zeroes replaced by log(0.0005)) and the ‘centre’ of a module. For each enhancer module, its centre is defined as a vector of length 111, containing the fraction of regions in that module called as (any type of) enhancer in each of the 111 epigenomes analysed. This expression score is meant to act as the ‘expression’ of a transcription factor within a module of cell types. We then computed an expression-enrichment value for each transcription factor as the correlation of this expression score and the enrichment of the corresponding motif across enhancer modules. The top 40 motifs in terms of their absolute expression-enrichment correlation and the clusters with log2 enrichment or depletion of at least log2 = 1.5 for at least one motif are shown in Fig. 8 and Extended Data Fig. 8a (only one motif is shown in Fig. 8 for each factor).
We show all 84 motifs that were significantly enriched (log2 ≥ 1.5) in any enhancer modules, across the full set of 226 enhancer modules (Supplementary Fig. 13a) and in the 101 modules in which they were significantly enriched (Extended Data Fig. 8a). Similarly, we show all 10 enriched motifs across the full set of 111 individual reference epigenomes (Supplementary Fig. 13b) and specifically in the 15 enriched epigenomes (Supplementary Fig. 13c). Lastly, we show all 19 enriched motifs across the full set of 17 tissue groups (Supplementary Fig. 13d), and specifically within the 10 groups that showed significant enrichments (Supplementary Fig. 13e).
For visualization of regulator–cell type links (Fig. 8), we computed edge weights between each cell type and motif using these motif-module enrichments. For each motif and cell type, we computed the sum across all modules of the product of the log2 motif enrichment and the value of the cell type within the module centre (only consider the highly associated cell types by replacing values <0.7 with 0). We show all resulting edge weights of at least 1.5 and visualize the network using Cytoscape118.
Based on the same motif enrichment method mentioned above, we computed the motif enrichment in the tissue-specific Digital Genomic Footprinting (DGF) regions in each library. The tissue-specific DGF regions were identified by selecting the DGF region occurring in no more than 20 DGF libraries among 42 DGF libraries. To generate Extended Data Fig. 9b, we standardized the motif enrichment in each library into z-scores for each motif (row) and colour each DGF library (column) based on their tissue type.
DNA motif positional bias in digital genomic footprinting sites
We computed the positional enrichment of each driver motif (Extended Data 9c and 10) related to the digital genomic footprinting (DGF) sites in each cell type (Supplementary Table 5b). For each driver transcription factor motif, we generated two views corresponding to the motif position (the centre of the motif instance) relative to the centre of closest DGF site (centre view) and the motif position relative to the boundary of closest DGF site (boundary view). We only considered the motif instances with closest DGF site within 100 bp. For the centre view, we plotted the motif occurrence density versus the distance to the DGF centre for different cell types. For the boundary view, we considered the shortest distance between the centre of a motif instance and either side of DGF boundary, and gave a negative distance value for motif instances inside the DGF, and a positive distance value otherwise. Similar to the centre view, we plotted the motif density versus the derived distance value in the boundary view for each cell type.
To access the significance of the motif concentration within DGF in each cell type, we computed the DGF enrichment ratio as the ratio between the number of motif instances with distance less than 20 bp to the DGF centre and that number in the immediate flanking window, that is, the number of motif instances with distance to the DGF centre larger than 20 bp and smaller than 40 bp. As control, we randomly sampled the same number of motif instances from the shuffled versions of the given motif, and obtained the DGF enrichment ratio for the shuffled motif instances. The DGF enrichment ratio of the true motif is further converted to z-score by mean and standard deviation from the DGF enrichment ratios of shuffled motif from 1,000 times random sampling. Then the adjusted P value is further computed from z-score and Bonferroni correction for number of cell types.
Comparing DGF with DNA motifs that are predictive of epigenomic modification
The motifs that were predictive of epigenomic modifications71 were compared to DGF in Supplementary Table 5a. This was done in three cell types where both DGF and predictive motifs were available: ‘H1 BMP4 derived mesendoderm cultured cells’ (E004), ‘H1 BMP4 derived trophoblast cultured cells’ (E005), and ‘H1 derived mesenchymal stem cells’ (E006). The motifs that were predictive of the following seven inputs were considered: H3K27me3, H3K27ac, H3K9me3, H3K36me3, H3K4me1, H3K4me3 and DNA methylation valleys (DMV)11. To identify overlaps the predictive motifs were scanned against the modification peaks of the corresponding modification and the location of the best match between motif and sequence was recorded. Then we counted the number of times the locations of the best motif matches overlapped a DGF by at least 1 bp. These counts were compared to the number of overlaps identified randomly, which was calculated by comparing DGF to random locations within the modifications peaks. The reported random frequency was the average of 100 repeats. To calculate the fold enrichment we divided the observed frequency by the random frequency.
Tissue-specific activity of disease-associated regions
We tested the enrichment of SNPs from individual genome-wide association studies (GWAS) for the gapped peak call sets for histone marks H3K4me1, H3K4me3, H3K36me3, H3K9me3, H3K27me3, H3K9ac and H3K27ac as well as the DNase peak call set based on MACS2 in each reference epigenome where available. The SNPs used were curated into the NHGRI GWAS catalogue75 and obtained through the UCSC Table Browser119 on 12 September 2014. We restricted the enrichment analysis to chromosomes 1–22 and chromosome X. We defined a study to be a unique combination of annotated trait and PubMed ID. To reduce dependencies between pairs of SNPs assigned to the same study, we pruned SNPs such that no two SNPs were within 1 Mb of each other on the same chromosome. The pruning procedure considered each SNP in ranked order of P value with the most significant coming first, and we retained a SNP if there was no already retained SNP on the same chromosome within 1 Mb. We computed hypergeometric P values for the enrichment of each pruned set of SNPs overlapping peak calls against the pruned GWAS catalogue as the background. We estimated separately for each mark a mapping from a P value to a false discovery rate across tests for all study and reference epigenome combinations by generating 100 randomized versions of the pruned GWAS catalogues, shuffling which SNPs were assigned to which study and computing the average fraction of reference epigenome–study combinations that reached that level of significance (in a continuous mapping of P values to FDR) using randomized catalogues divided by the number based on the actual GWAS catalogue.
References
Rivera, C. M. & Ren, B. Mapping human epigenomes. Cell 155, 39–55 (2013)
Zhou, V. W., Goren, A. & Bernstein, B. E. Charting histone modifications and the functional organization of mammalian genomes. Nature Rev. Genet. 12, 7–18 (2011)
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Rev. Genet. 13, 484–492 (2012)
Smith, Z. D. & Meissner, A. DNA methylation: roles in mammalian development. Nature Rev. Genet. 14, 204–220 (2013)
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev. Genet. 10, 57–63 (2009)
Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nature Rev. Genet. 10, 669–680 (2009)
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012)
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5, 621–628 (2008)
Ernst, J. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011)
Heintzman, N. D. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genet. 39, 311–318 (2007)
Xie, W. et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148 (2013)
Zhu, J. et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654 (2013)
Neph, S. et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90 (2012)
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012)
Bernstein, B. E. et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature Biotechnol. 28, 1045–1048 (2010)
Mikkelsen, T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007)
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007)
John, S. et al. Genome-scale mapping of DNase I hypersensitivity. Curr. Protoc. Mol. Biol. Ch. 27, Unit 21 27. (2013)
Lister, R. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009)
Meissner, A. et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877 (2005)
Weber, M. et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genet. 37, 853–862 (2005)
Maunakea, A. K. et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257 (2010)
ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012)
Bernstein, B. E. et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181 (2005)
Bonasio, R., Tu, S. & Reinberg, D. Molecular signals of epigenetic states. Science 330, 612–616 (2010)
Peters, A. H. et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12, 1577–1589 (2003)
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009)
Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011)
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010)
Cedar, H. & Bergman, Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature Rev. Genet. 10, 295–304 (2009)
Stevens, M. et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 23, 1541–1553 (2013)
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008)
Butterfield, Y. S. et al. JAGuaR: Junction Alignments to Genome for RNA-Seq Reads. PLoS ONE 9, e102398 (2014)
Coarfa, C. et al. Pash 3.0: A versatile software package for read mapping and integrative analysis of genomic and epigenomic variation using massively parallel DNA sequencing. BMC Bioinform. 11, 572 (2010)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Fejes, A. P. et al. FindPeaks 3.1: a tool for identifying areas of enrichment from massively parallel short-read sequencing technology. Bioinformatics 24, 1729–1730 (2008)
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012)
Kunde-Ramamoorthy, G. et al. Comparison and quantitative verification of mapping algorithms for whole-genome bisulfite sequencing. Nucleic Acids Res. 42, e43 (2014)
Harris, R. A. et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nature Biotechnol. 28, 1097–1105 (2010)
Ernst, J. & Kellis, M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nature Biotechnol http://dx.doi.org/10.1038/nbt.3157 (in the press)
Ernst, J. & Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nature Biotechnol. 28, 817–825 (2010)
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput. Biol. 6, e1001025 (2010)
Kagey, M. H. et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 (2010)
Stadler, M. B. et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 (2011)
Gascard, P. et al. Epigenetic and transcriptional determinants of the human breast. Nature Commun http://dx.doi.org/10.1038/ncomms7351 (in the press)
Mohn, F., Weber, M., Schubeler, D. & Roloff, T. C. Methylated DNA immunoprecipitation (MeDIP). Methods Mol. Biol. 507, 55–64 (2009)
Elliott, G. et al. Intermediate DNA methylation is a conserved signature of genome regulation. Nature Commun http://dx.doi.org/10.1038/ncomms7363 (in the press)
Ji, H. et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467, 338–342 (2010)
Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008)
Gifford, C. A. et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 153, 1149–1163 (2013)
Ziller, M. J. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481 (2013)
Tsankov, A. M. et al. Transcription factor binding dynamics during human ESC differentiation. Nature http://dx.doi.org/10.1038/nature14233 (this issue)
Ziller, M. J. et al. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature http://dx.doi.org/10.1038/nature13990 (this issue)
Xie, M. et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nature Genet. 45, 836–841 (2013)
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnol. 28, 495–501 (2010)
Lowdon, R. F. et al. Regulatory network decoded from epigenomes of surface ectoderm-derived cell types. Nat. Commun. 5, 5442 (2014)
Amin, V. et al. Epigenomic footprints across 111 reference epigenomes reveal tissue-specific epigenetic regulation of lincRNAs. Nature Commun http://dx.doi.org/10.1038/ncomms7370 (in the press)
Bernstein, B. E. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006)
Hawkins, R. D. et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491 (2010)
Varley, K. E. et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567 (2013)
Leung, D. et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature http://dx.doi.org/10.1038/nature14217 (this issue)
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009)
Meuleman, W. et al. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 23, 270–280 (2013)
Guelen, L. et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951 (2008)
Antequera, F., Boyes, J. & Bird, A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell 62, 503–514 (1990)
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet. 25, 25–29 (2000)
Kohler, S. et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 42, D966–D974 (2014)
Kheradpour, P. & Kellis, M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 42, 2976–2987 (2014)
Hesselberth, J. R. et al. Global mapping of protein–DNA interactions in vivo by digital genomic footprinting. Nature Methods 6, 283–289 (2009)
Kheradpour, P., Stark, A., Roy, S. & Kellis, M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 17, 1919–1931 (2007)
Whitaker, J. W., Chen, Z. & Wang, W. Predicting the human epigenome from DNA motifs. Nature Methods http://dx.doi.org/10.1038/nmeth.3065 (in the press)
Dixon, J. R. et al. Chromatin architecture reorganization during stem cell differentiation. Nature http://dx.doi.org/10.1038/nature14222 (this issue)
Lindblad-Toh, K. et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011)
Trynka, G. et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genet. 45, 124–130 (2013)
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014)
Franke, A. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nature Genet. 42, 1118–1125 (2010)
Cooper, J. D. et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nature Genet. 40, 1399–1401 (2008)
Berndt, S. I. et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nature Genet. 45, 868–876 (2013)
Stahl, E. A. et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nature Genet. 42, 508–514 (2010)
Barrett, J. C. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nature Genet. 41, 703–707 (2009)
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012)
Yang, W. et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am. J. Hum. Genet. 92, 41–51 (2013)
Musunuru, K. et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466, 714–719 (2010)
Willy, P. J. et al. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 9, 1033–1045 (1995)
Pasquali, L. et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nature Genet. 46, 136–143 (2014)
Dalcik, H. et al. Expression of insulin-like growth factor in the placenta of intrauterine growth-retarded human fetuses. Acta Histochem. 103, 195–207 (2001)
Lesch, K. P. et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J. Neural Transm. 115, 1573–1585 (2008)
Repunte-Canonigo, V. et al. A potential role for adiponectin receptor 2 (AdipoR2) in the regulation of alcohol intake. Brain Res. 1339, 11–17 (2010)
Sawcer, S. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011)
Heneka, M. T., Kummer, M. P. & Latz, E. Innate immune activation in neurodegenerative disease. Nature Rev. Immunol. 14, 463–477 (2014)
Gjoneska, E. et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature http://dx.doi.org/10.1038/nature14252 (this issue)
Zhou, X. et al. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nature Biotechnol http://dx.doi.org/10.1038/nbt.3158 (in the press)
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012)
Satterlee, J. S., Schubeler, D. & Ng, H. H. Tackling the epigenome: challenges and opportunities for collaboration. Nature Biotechnol. 28, 1039–1044 (2010)
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature http://dx.doi.org/10.1038/nature13835 (this issue)
Seumois, G. et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nature Immunol. 15, 777–788 (2014)
De Jager, P. L. et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nature Neurosci. 17, 1156–1163 (2014)
Lunnon, K. et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nature Neurosci. 17, 1164–1170 (2014)
Polak, P. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature http://dx.doi.org/10.1038/nature14221 (this issue)
Yao, L., Tak, Y. G., Berman, B. P. & Farnham, P. J. Functional annotation of colon cancer risk SNPs. Nat. Commun. 5, 5114 (2014)
Zhou, X. et al. The Human Epigenome Browser at Washington University. Nature Methods 8, 989–990 (2011)
Karolchik, D. et al. The UCSC Genome Browser Database. Nucleic Acids Res. 31, 51–54 (2003)
Chadwick, L. H. The NIH Roadmap Epigenomics Program data resource. Epigenomics 4, 317–324 (2012)
John, S. et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature Genet. 43, 264–268 (2011)
Ernst, J. & Kellis, M. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res. 23, 1142–1154 (2013)
Ernst, J. & Kellis, M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods 9, 215–216 (2012)
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012)
Lister, R. et al. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905 (2013)
Schultz, M. D., Schmitz, R. J. & Ecker, J. R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 28, 583–585 (2012)
Bar-Joseph, Z., Gifford, D. K. & Jaakkola, T. S. Fast optimal leaf ordering for hierarchical clustering. Bioinformatics 17 (suppl. 1). S22–S29 (2001)
Leisch, F. A toolbox for KK-centroids cluster analysis. Comput. Stat. Data Anal. 51, 526–544 (2006)
Matys, V. et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31, 374–378 (2003)
Sandelin, A., Alkema, W., Engstrom, P., Wasserman, W. W. & Lenhard, B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32, D91–D94 (2004)
Berger, M. F. et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature Biotechnol. 24, 1429–1435 (2006)
Berger, M. F. et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133, 1266–1276 (2008)
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013)
Badis, G. et al. Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723 (2009)
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003)
Karolchik, D. et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 (2004)
Acknowledgements
This work was supported by the NIH Common Fund as part of the NIH Roadmap Epigenomics Program through U01ES017155 (B.B. and A.M.), U01ES017154 (J.C. and M.M.), U01ES017166 (B.R.), U01ES017156 (J.S.), U01DA025956 (A.M. and A.B.), and by NHGRI through RC1HG005334, R01HG004037 and R01HG004037-S1 (M.K.), RO1NS078839 (L.-H.T.), and by NIH ES017166, NSFC 91019016 and NBRPC 2012CB316503 (M.Q.Z.). Sample procurement was supported by grants 5R24HD000836 (I.A.G.) for staged fetal tissues; P30AG10161, R01AG15819, R01AG17917 (D.A.B.) and U01AG46152 (P.L.D. and D.A.B.) for adult brain samples. This work was also supported by NIH fellowship grants F32HL110473 and K99HL119617 (S.L.), and NSF CAREER award 1254200 (J.E.). We acknowledge program leadership by members of the NIH Epigenomics Workgroup, especially J. S. Satterlee, F. L. Tyson, J. Rutter, K. A. McAllister, A. Haugen, C. Colvis (NCATS), J. Battey (NIDCD), L. Birnbaum (NIEHS) and N. Volkow (NIDA). We acknowledge feedback from our External Scientific Panel members M. Bartolomei, S. Baylin, S. Beck, A. Chakravarti, L. Jackson-Grusby, J. Lieb, S. Peckman, J. Quackenbush and S. Stice.
Author information
Authors and Affiliations
Consortia
Contributions
Details of author contributions are provided in the Roadmap Epigenomics Consortium list.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
All data sets and analysis results are available at http://compbio.mit.edu/roadmap/. Browsable views of all data sets (as shown in Fig. 3) are available from the WashU Epigenome Browser101 at http://epigenomegateway.wustl.edu/ and the UCSC Genome Browser102 at http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hubUrl=http://vizhub.wustl.edu/VizHub/RoadmapReleaseAll.txt. All primary data sets and protocols are available at REMC portal103 at http://www.roadmapepigenomics.org, GEO data sets at http://ncbi.nlm.nih.gov/geo/roadmap/epigenomics, and the Human Epigenome Atlas at http://epigenomeatlas.org. Epigenomic annotations and motif predictions are incorporated into HaploReg for mining GWAS at http://compbio.mit.edu/haploreg.
Extended data figures and tables
Extended Data Figure 1 Tissues and cell types of reference epigenomes.
Comprehensive listing of all 111 reference epigenomes generated by the consortium, along with epigenome identifiers (EIDs), including: (a) adult samples; (b) fetal samples; (c) ES cell, iPS cell and ES-cell-derived cells; and (d) primary cultures. Colours indicate the groupings of tissues and cell types (as in Fig. 2b, and throughout the manuscript). For five samples (adult osteoblasts, and fetal liver, spleen, gonad and spinal cord), no colour is present, indicating that these are not part of the 111 reference epigenomes (ENCODE 2012 samples, or not all five marks in the core set were present), but data sets from these samples are high quality and were sometimes used in companion paper analyses, and are publicly available. e, Assay correlations. Heat map of the pairwise experiment correlations for the core set of five histone modification marks (H3K4me1, H3K4me3, H3K36me3, H3K27me3, H3K9me3) across all 127 reference epigenomes, the two common acetylation marks (H3K27ac and H3K9ac), and DNA accessibility (DNase) across the reference epigenomes where they are available. Yellow indicates relatively higher correlation and blue lower correlation. Rows and columns were ordered computationally to maximize similarity of neighbouring rows and columns (see Methods). All experiments for H3K9me3, H3K27me3, H3K36me3, DNase and H3K4me1 are consistently ordered into distinct and contiguous groups. For H3K4me3, H3K9ac and H3K27ac, experiments group primarily based on the mark, but in some cases, the correlations and ordering appear more cell-type driven.
Extended Data Figure 2 Chromatin state model robustness and enrichments.
a, Chromatin state model robustness. Clustering of 15-state ‘core’ chromatin state model learned jointly across reference epigenomes (Fig. 4a) with chromatin state models learned independently in 111 reference epigenomes. We applied ChromHMM to learn a 15-state ChromHMM model using the five core marks in each of the 111 reference epigenomes generated by the Roadmap Epigenomics program, and clustered the resulting 1,680-state emission probability vectors (leaves of the tree) with the 15 states from the joint model (indicated by arrows). We found that the vast majority of states learned across cell types clustered into 15 clusters, corresponding to the joint model states, validating the robustness of chromatin states across cell types. This analysis revealed two new clusters (red crosses) which are not represented in the 15 states of the jointly learned model: ‘HetWk’, a cluster showing weak enrichment for H3K9me3; and ‘Rpts’, a cluster showing H3K9me3 along with a diversity of other marks, and enriched in specific types of repetitive elements (satellite repeats) in each cell type, which may be due to mapping artefacts. This joint clustering also revealed subtle variations in the relative frequency of presence of H3K4me1 in states TxFlnk, Enh and TssBiv, and H3K27me3 in state TssBiv. Overall, this analysis confirms that the 15-state chromatin state model based on the core set of five marks provides a robust framework for interpreting epigenomic complexity across tissues and cell types. b, Enrichments for 15-state model based on five histone modification marks. Top left: transcription factor binding site overlap enrichments of 15 states in H1-ES cells from the ‘core’ model for transcription factor binding sites (TFBS) based on ChIP-seq data in H1-ES cells. Transcription factor binding coverage for other cell types based on matched transcription factor ChIP-seq data are shown in Supplementary Fig. 2. Top right: enrichments for expressed and non-expressed genes in H1-ES cells and GM12878. Bottom: positional enrichments at the transcription start site (TSS) and transcription end site (TES) of expressed (expr.) and repressed (repr.) genes in H1-ES cells. Transition probabilities show frequency of co-occurrence of each pair of chromatin states in neighbouring 200-bp bins. c, Definition and enrichments for 18-state ‘expanded’ model that also includes H3K27ac associated with active enhancer and active promoter regions, but which was only available for 98 of the 127 reference epigenomes. Inclusion of H3K27ac distinguishes active enhancers and active promoters. Top: TFBS enrichments in H1-ES cells (E003) chromatin states using ENCODE transcription factor ChIP-seq data in H1-ES cells. Bottom: positional enrichments in H1-ES cells for genomic annotations, expressed and repressed genes, TSS and TES, and state transitions as in Extended Data Fig. 2b and Fig. 4a–c. Right: average fold-enrichment (colours bars) and standard deviation (black line) across 98 reference epigenomes (Supplementary Fig. 3d) for the fold enrichment for non-exonic genomic segments (GERP) in each chromatin state (rows) in the 18-state model. Excluding protein-coding exons (see Supplementary Fig. 3b versus Supplementary Fig. 3d), the TSS-proximal states show the highest levels of conservation, followed by EnhBiv and the three non-transcribed enhancer states. In contrast, Tx and TxWk elements are weakly depleted for conserved regions, and Znf/Rpts and Het are strongly depleted for conserved elements.
Extended Data Figure 3 Relationship between histone marks, DNA methylation, DNA accessibility and gene expression.
a, H3K27ac-marked ‘active’ enhancers show higher levels of DNA accessibility, based on enrichment of DNase-seq signal confidence scores (–log10(Poisson P value)) for elements in each chromatin state in our extended 18-state model that includes the core five histone modification marks and H3K27ac, similar to Fig. 4e. b, Level of whole-genome bisulfite methylation for all chromatin states in the 18-state model shows that H3K27ac-marked ‘active’ enhancers associated with H3K27ac in addition to H3K4me1 show lower methylation levels, consistent with higher regulatory activity. The whiskers in a and b show 1.5× interquartile range and the filled circles are individual outliers. c, DNA methylation levels for genes showing different expression levels. The depletion of DNA methylation in promoter regions, and the enrichment of DNA methylation in transcribed regions, are both more pronounced for highly expressed genes. The enrichment for high DNA methylation is more pronounced in the 3′ ends of the most highly expressed genes. d, Genes associated with active enhancer states have consistently significantly higher expression. ‘Active enhancer’ associated genes have at least one EnhA1 and/or EnhA2 ±20 kb from TSS (18-state model). ‘Weak-enhancer’ genes are associated with EnhG1, EnhG2, EnhWk, EnhBiv. Lowest expression have genes that are not associated with any enhancer. Plots with red markers show median expression of genes associated with ‘active’ enhancers, yellow markers ‘weak’ enhancers, and white markers no association with any enhancer state. e, Higher-expression genes show greater association with H3K27ac-marked ‘active’ enhancers. Highly expressed genes are consistently more frequently associated with H3K27ac-marked active enhancers (EnhA1 and EnhA2) across all cell types. Fraction of genes associated with H3K27ac-marked ‘active’ enhancers (red), H3K27ac-lacking ‘weak’ enhancers only (yellow), or no enhancers (white) for genes of varying expression levels in each cell type with RNA-seq data.
Extended Data Figure 4 Methylation relationship with chromatin state.
a–c, DNA methylation levels in 15-state model across technologies. We observed significant differences in the average methylation levels observed that were correlated with the different DNA methylation platforms used, but their relative relationships in average chromatin state methylation were conserved. Relative to WGBS (panel a, repeated from Fig. 4d for comparison purposes), RRBS (panel b) showed the lowest overall methylation levels (as expected given its CpG island enrichment), while mCRF showed the highest (panel c). This highlights the importance of recognizing and potentially correcting for DNA-methylation-platform-specific biases before performing integrative analyse. d, e, Distribution of DNA methylation levels measured using RRBS and mCRF in 18-state model (defined in Extended Data Fig. 2c). WGBS is shown in Extended Data Fig. 3b. The whiskers in a–e show 1.5× interquartile range and the filled circles are individual outliers. f, DNA methylation variation across cell types. Density plots denote distribution of DNA methylation levels from 0% to 100% for each chromatin state across the 95 reference epigenomes profiled for whole-genome bisulfite (WGBS, red), reduced representation bisulfite (RRBS, blue), or MeDIP/MRE (mCRF, green). The respective colour (red, blue, or green) was set to the maximum ln(density+1) value for each chromatin state and respective platform, with intermediate values coloured on a natural log scale. For each panel, the subset of reference epigenomes profiled using each technology are listed, using the colours, order, and abbreviations from Fig. 2.
Extended Data Figure 5 Chromatin state variability, switching and genomic coverage.
a, Variability level for 18-state model. Chromatin state variability (similar to Fig. 5a), quantified based on the fraction of the genomic coverage (y axis) of each state (colour) that is consistently labelled with that state in at most N (ranging from 1 to 98) reference epigenomes, using the 18-state model learned based on 6 chromatin marks, including H3K27ac. b, Chromatin state over- and under-representation for 18-state expanded model. c, Log-ratio (log10) of chromatin state switching probabilities for the 18-state expanded model across 34 high-quality, non-redundant epigenomes that have H3K27ac data, relative to intra-tissue switching probabilities across replicates or samples from multiple individuals. d, Chromatin state coverage grouped by epigenomic domains. Top: chromosome ‘painting’ of 11 clusters shown in Fig. 5d and discovered based on chromatin state co-occurrence at the 2-Mb scale across reference epigenomes. Bottom: enrichment of CpG islands in each cluster clearly showing higher CpG density ‘active’ clusters 3 and 6 comparing to passive clusters 9–11. Each box plot shows a distribution of CpG total occupancy in 2-Mb bins in each cluster (with box boundaries indicating 25th and 75th percentiles, the whiskers extend to the most extreme data points considered to not be outliers). Points are drawn as outliers if they are larger than Q3+1.5×(Q3-Q1) or smaller than Q1-1.5×(Q3-Q1), where Q1 and Q3 are the 25th and 75th percentiles, respectively.
Extended Data Figure 6 Hierarchical clustering of epigenomes using diverse marks.
a–e, Clustering of all 127 reference epigenomes, including ENCODE samples, using H3K4me1, H3K4me3, H3K27me3, H3K36me3 and H3K9me3 signal in Enh, TssA, ReprPC, Tx and Het chromatin states, respectively. All panels show hierarchical clustering with optimal leaf ordering. Colours indicate sample groups, as defined in Fig. 2. Numbers on internal nodes represent bootstrap support scores over 1,000 bootstrap samples.
Extended Data Figure 7 Multi-dimensional scaling (MDS) analysis.
a–i, MDS plots showing reference epigenome distances using similarity of different epigenomic marks in corresponding chromatin states. Reference epigenomes (dots) are coloured according to their group colouring defined in Fig. 2b. Thin lines connect same-group reference epigenomes. The first four axes of variation are shown in pairs. Marks are assessed in regions with relevant chromatin states (see Methods). j, Variance explained by each MDS dimension. The first five dimensions shown in Supplementary Fig. 10 (Fig. 6b, c) explain between 45% and 80% of the total epigenome-to-epigenome variance for all histone modification mark correlations, and additional dimensions explain less than 10%. Only a few components of H3K4me3 in TssA chromatin states explains a much larger fraction of the variance than other marks, possibly due to its stability across cell types.
Extended Data Figure 8 Regulatory motif analysis for modulesa.
Regulatory motifs enriched in enhancer modules. Enrichment (red) or depletion (blue) of regulatory motifs (rows) in the enhancer modules (columns) relative to shuffled control motifs. For each motif is shown the motif name, consensus logo, and correlation between regulator expression and module activity: positive correlation (orange) is indicative of activators, and negative correlation (purple) indicates a repressive role for the factor. Only clusters with log enrichment or depletion of at least 1.5-fold for one motif are shown. b, Average activity level of enhancers of each module in each reference epigenome (black, high; white, low). c, Total size of each enhancer module showing enrichment (in kb).
Extended Data Figure 9 Regulatory motif enrichment, DGF enrichment and positional bias for predicted driver motifs.
a, Regulatory motif enrichments for the 40 regulators showing the strongest absolute correlation between transcription factor expression and module activity. Of these, 36 were also recovered solely based on their motif enrichment scores (Extended Data Fig. 8), but 6 motifs showing significant and biologically relevant correlations were not discovered solely based on their motif enrichment (Esrra_4, Max_4, Mga_3, Nfatc1_3, Rest_2 and Tead3_1), illustrating the importance of studying motif enrichments in the context of transcription factor expression and enhancer activity patterns. b, Predicted driver regulatory motifs are enriched in high-resolution DNase footprints. Enrichment of predicted driver motif instances (Fig. 8 and Extended Data Fig. 9a) in 42 high-resolution (6–40 bp) DGF libraries from deeply sequenced DNase data sets69 shows consistent tissue preferences in matching cell types. For example, POU5F1 in iPS cells, HNF1B and HNF4A1 in digestive tissues, RFX4 in neural lineages, MFE2B in muscle. c, Matrix of significant positional bias across factors and cell types. For each DGF data set (columns), positional bias score (heat map) of predicted driver regulatory motifs (rows) found to be significantly enriched (Fig. 8 and Extended Data Fig. 9a) in enhancer modules (Fig. 7a).
Extended Data Figure 10 Positional biases of predicted driver motifs relative to high-resolution DNase footprint centres and boundaries.
a, Driver transcription factor motif instance logo, as in Fig. 8 and Extended Data Fig. 9a. b, Distribution of motif instances relative to the centre of the high-resolution DNase sites (DGF lengths range from 6 bp to 40 bp), each curve coloured according to the cell/tissue type (from Fig. 2 and Supplementary Table 5b). c, Distribution of shuffled motifs that match composition and number of conserved occurrences in the genome70,73. d, Positional bias relative to boundary of DGF region for true motifs, similar to b. e, Positional bias relative to boundary of DGF region for shuffled motifs, similar to c. f, Cell types showing significant positional bias after multiple testing correction, coloured according to Fig. 2 and Supplementary Table 5b.
Extended Data Figure 11 Epigenomic enrichments of genetic variants associated with diverse traits.
Tissue-specific enrichments for peaks of epigenomic marks for genetic variants associated with complex disease, expanding Fig. 9. Enrichments are shown for: a, H3K4me1 peaks (enhancers). This panel includes all the data shown in Fig. 9, but expands the enrichments shown to all reference epigenomes (columns) for studies (rows) that met the FDR = 0.02 threshold. b, H3K27ac peaks (active enhancers). a, b, Studies were defined by a set of SNPs annotated in the GWAS catalogue with the same combination of a publication (shown by the Pubmed ID) and trait. Epigenome with maximum enrichment, uncorrected −log10 P value and estimated FDR are indicated.
Extended Data Figure 12 Epigenomic enrichments of genetic variants associated with diverse traits.
Tissue-specific enrichments for peaks of epigenomic marks for genetic variants associated with complex disease, similar to Extended Data Fig. 11 except enrichments are shown for: a, H3K4me3 peaks (promoters); b, H3K9ac peaks (active promoters and active enhancers); c, DNase peaks (accessible regions); d, H3K36me3 peaks (transcribed regions); e, f, H3K27me3 peaks (Polycomb-repressed regions, e) and H3K9me3 peaks (heterochromatin regions, f) do not show any enrichments at the FDR = 0.02 threshold. As for Extended Data Fig. 11, studies were defined by a set of SNPs annotated in the GWAS catalogue with the same combination of a trait (far left column) and publication shown by the Pubmed ID (far right column), uncorrected P value (in -log10) and estimated FDR.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-13 and a guide to Supplementary Tables 1-6. See http://compbio.mit.edu/roadmap/ for additional information. (PDF 9329 kb)
Supplementary Information
This file contains Supplementary Tables 1-6. See Supplementary Information document for full table legends. (ZIP 1439 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Roadmap Epigenomics Consortium., Kundaje, A., Meuleman, W. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). https://doi.org/10.1038/nature14248
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14248
This article is cited by
-
The genetic architecture of youth anxiety: a study protocol
BMC Psychiatry (2024)
-
Quantifying the proportion of different cell types in the human cortex using DNA methylation profiles
BMC Biology (2024)
-
Causality-enriched epigenetic age uncouples damage and adaptation
Nature Aging (2024)
-
Decoding chromatin states by proteomic profiling of nucleosome readers
Nature (2024)
-
Advances in the multimodal analysis of the 3D chromatin structure and gene regulation
Experimental & Molecular Medicine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
