
Abstract
T-cell acute lymphoblastic leukaemia (T-ALL) is a haematological malignancy with a dismal overall prognosis, including a relapse rate of up to 25%, mainly because of the lack of non-cytotoxic targeted therapy options. Drugs that target the function of key epigenetic factors have been approved in the context of haematopoietic disorders1, and mutations that affect chromatin modulators in a variety of leukaemias have recently been identified2,3; however, ‘epigenetic’ drugs are not currently used for T-ALL treatment. Recently, we described that the polycomb repressive complex 2 (PRC2) has a tumour-suppressor role in T-ALL4. Here we delineated the role of the histone 3 lysine 27 (H3K27) demethylases JMJD3 and UTX in T-ALL. We show that JMJD3 is essential for the initiation and maintenance of T-ALL, as it controls important oncogenic gene targets by modulating H3K27 methylation. By contrast, we found that UTX functions as a tumour suppressor and is frequently genetically inactivated in T-ALL. Moreover, we demonstrated that the small molecule inhibitor GSKJ4 (ref. 5) affects T-ALL growth, by targeting JMJD3 activity. These findings show that two proteins with a similar enzymatic function can have opposing roles in the context of the same disease, paving the way for treating haematopoietic malignancies with a new category of epigenetic inhibitors.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Change history
23 October 2014
A minor change was made to the author list.
References
Baylin, S. B. & Jones, P. A. A decade of exploring the cancer epigenome: biological and translational implications. Nature Rev. Cancer 11, 726–734 (2011)
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012)
Jankowska, A. M. et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 118, 3932–3941 (2011)
Ntziachristos, P. et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nature Med. 18, 298–303 (2012)
Kruidenier, L. et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012)
Simon, C. et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 26, 651–656 (2012)
Hubner, M. R. & Spector, D. L. Role of H3K27 demethylases Jmjd3 and UTX in transcriptional regulation. Cold Spring Harb. Symp. Quant. Biol. 75, 43–49 (2010)
Kooistra, S. M. & Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nature Rev. Mol. Cell Biol. 13, 297–311 (2012)
Morales Torres, C., Laugesen, A. & Helin, K. Utx is required for proper induction of ectoderm and mesoderm during differentiation of embryonic stem cells. PLoS ONE 8, e60020 (2013)
Wang, C. et al. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proc. Natl Acad. Sci. USA 109, 15324–15329 (2012)
Mansour, A. A. et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488, 409–413 (2012)
De Santa, F. et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094 (2007)
Agger, K. et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 (2009)
Barradas, M. et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 23, 1177–1182 (2009)
Jepsen, K. et al. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 450, 415–419 (2007)
Sen, G. L., Webster, D. E., Barragan, D. I., Chang, H. Y. & Khavari, P. A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 22, 1865–1870 (2008)
Zhao, W. et al. Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell 152, 1037–1050 (2013)
Wang, J. K. et al. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 24, 327–332 (2010)
Thieme, S. et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 121, 2462–2473 (2013)
van Haaften, G. et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nature Genet. 41, 521–523 (2009)
Mar, B. G. et al. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia 26, 1881–1883 (2012)
Weng, A. P. et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004)
Espinosa, L. et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell 18, 268–281 (2010)
Dik, W. A. et al. New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J. Exp. Med. 201, 1715–1723 (2005)
Van Vlierberghe, P. et al. ETV6 mutations in early immature human T cell leukemias. J. Exp. Med. 208, 2571–2579 (2011)
Valk, P. J. et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 350, 1617–1628 (2004)
Wang, H. et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl Acad. Sci. USA 108, 14908–14913 (2011)
Welstead, G. G. et al. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc. Natl Acad. Sci. USA 109, 13004–13009 (2012)
Satoh, T. et al. The Jmjd3–Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nature Immunol. 11, 936–944 (2010)
Palomero, T. et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to γ-secretase inhibitors. Leukemia 20, 1279–1287 (2006)
Sharma, V. M. et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 26, 8022–8031 (2006)
King, B. et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 153, 1552–1566 (2013)
Armstrong, F. et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 113, 1730–1740 (2009)
Dickins, R. A. et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nature Genet. 37, 1289–1295 (2005)
Zhong, S. et al. High-throughput Illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 940–949 (2011)
Mullighan, C. G. Single nucleotide polymorphism microarray analysis of genetic alterations in cancer. Methods Mol. Biol. 730, 235–258 (2011)
Mullighan, C. G. et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–239 (2011)
Zhang, J. et al. SNPdetector: a software tool for sensitive and accurate SNP detection. PLOS Comput. Biol. 1, e53 (2005)
Chen, K. et al. PolyScan: an automatic indel and SNP detection approach to the analysis of human resequencing data. Genome Res. 17, 659–666 (2007)
Marks, D. I. et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 114, 5136–5145 (2009)
Verhaak, R. G. et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 94, 131–134 (2009)
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005)
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009)
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010)
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008)
Li, Q., Brown, J. B., Huang, H. & Bickel, P. J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 5, 1752–1779 (2011)
Tsirigos, A., Haiminen, N., Bilal, E. & Utro, F. GenomicTools: a computational platform for developing high-throughput analytics in genomics. Bioinformatics 28, 282–283 (2012)
Acknowledgements
We thank the members of the Aifantis laboratory and J. Siegle for discussions throughout the duration of the project; S. Shen for discussions on the analysis of sequencing data; GlaxoSmithKline for the GSKJ4 and GSKJ5 inhibitory compounds; A. Heguy and the NYU Genome Technology Center (supported in part by National Institutes of Health (NIH)/National Cancer Institute (NCI) grant P30 CA016087-30) for assistance with sequencing experiments; the NYU Flow Cytometry facility (supported in part by NIH/NCI grant 5P30CA16087-31) for cell sorting; the NYU Histology Core (5P30CA16087-31) and the NYU mouse facility (NYU Cancer Institute Center Grant 5P30CA16087-31); G. Natoli for providing the anti-JMJD3 antibody; J. Zhang for help with the analysis of the mutation data; and I. Rigo for technical support. I.A. was supported by the NIH (grants 1RO1CA133379, 1RO1CA105129, 1RO1CA149655, 5RO1CA173636 and 5RO1CA169784). J.N. was supported by the Damon Runyon Cancer Research Foundation. B.K. was supported by the NYU Cell and Molecular Biology Training Program. P.N. was supported by fellowships from Lady Tata Memorial Trust for leukaemia and the American Society of Hematology and NIH/NCI (K99CA188293). T.T. is supported by the NIH training grant 5 T32 CA009161-37. P.V.V. was supported by the Research Foundation Flanders and an Odysseus type II grant. Moreover, this study was supported by an NIH grant (R37-HD04502) to R.J., the ECOG tumour bank, an NIH grant (R01CA120196) to A.A.F.; NCI grants (U24 CA114737 and U10 CA21115) to E.P. and the Stand Up To Cancer Innovative Research Award (A.A.F.). I.A. was also supported by the William Lawrence and Blanche Hughes Foundation, The Leukemia & Lymphoma Society, the Ralph S. French Charitable Foundation Trust, The Chemotherapy Foundation, The V Foundation for Cancer Research and the St. Baldrick’s Foundation. I.A. is a Howard Hughes Medical Institute Early Career Scientist. A.T. carried out part of this work while at the Computational Biology Center, IBM Research, Yorktown Heights, New York.
Author information
Authors and Affiliations
Contributions
I.A. and P.N. designed the experiments and wrote the manuscript. P.N. performed most of the experiments. A.T. designed and performed the analysis of genome-wide data and wrote the manuscript. T.T., E.L., A.S., J.M., B.K., S.B. and J.N. performed experiments and contributed ideas. G.T. provided materials and tips related to the study. A.A.F. and L.X. designed and performed xenograft luciferase experiments and helped with ideas and concepts. P.V.V., E.P., M.S.T., J.M.R. and A.A.F. performed and analysed the mutational studies in adult T-ALL. G.G.W., R.J., T.S. and S.A. provided mouse tissues and helped with ideas and concepts. L.K. and R.P. helped with guidance on the biology and use of GSKJ inhibitors and with manuscript preparation. L.H., J.B. and C.G.M. performed and analysed the mutational studies in paediatric T-ALL.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 JMJD3 is induced through activation of the NF-κB pathway in a NOTCH1-dependent mode in T-ALL and binds to NOTCH1 target genes.
a, Levels of p65 (RELA) protein in control T cells and T-ALL tumour cells. A representative sample from three mice is shown. b, Schematic representation of the Jmjd3 locus showing the p65 binding site (upper) and ChIP analysis for p65 binding to the Jmjd3 locus in mouse control T cells and T-ALL tumour cells, as well as T-ALL cells upon treatment with γ-secretase inhibitor (γSI), which affects NOTCH1 levels (centre). NOTCH1 binding to this region upon γSI treatment in T-ALL cells is also shown (right). c, Analysis of JMJD3 and HES1 messenger RNA levels upon γSI treatment of CUTLL1 cells. The average of three independent studies is shown. d, e, Expression levels of the JMJD3 transcript (d) and protein (e) upon treatment of human T-ALL lines (DND41 and CEM) with a NEMO binding domain (NBD) inhibitor of the NF-κB pathway. f, JMJD3 levels in T-ALL cells upon inhibition of the NF-κB pathway using a dominant negative form of IκBα (DN-IκBα). g, h, ChIP for NOTCH1 (g) and H3K27me3 (h) on the Hes1 promoter upon γSI treatment of mouse T-ALL cells. In d and f–h, the average of three studies is shown. In e, a representative example from three studies is shown. i, Genes correlated with selected human genes (including JMJD3 and NFKB1) were tested for enrichment in loss-of-H3K27me3 genes during the transition to T-ALL in the mouse model. j, Overlap of JMJD3 peaks with peaks of important activating (H3K4me3 and H3K4me1) and repressive (H3K27me3) epigenetic marks, as well as members of the NOTCH1 complex. The percentage of TSSs containing JMJD3 peaks was used as a conservative control and is an alternative to the much lower genome-wide JMJD3 occupancy. k, Genome-wide distribution of JMJD3 peaks in human T-ALL.
Extended Data Figure 2 JMJD3 is vital for T-ALL growth through participation in NOTCH1 transcriptional programs.
a, NOTCH1 interaction analyses for JMJD3, MAML1 and WDR5 proteins in 293T cells. Interaction with JMJD3 was confirmed in a reciprocal way (right-most lane, immunoprecipitation (IP) using an anti-haemagglutinin (HA) antibody). b, Expression of JMJD3 and WDR5 in 293T cells, followed by immunoprecipitation using the anti-HA antibody against HA–JMJD3. An anti-Flag antibody was used for the detection of both proteins. c, NOTCH1 interaction studies for JMJD3 and MAML1 proteins in mouse T-ALL cells expressing a Flag/Strep form of intracellular NOTCH1. StrepTactin beads were used for NOTCH1 precipitation in the absence of detectable intracellular NOTCH1, and different antibodies were used for the detection of JMJD3, MAML1, EZH2 and UTX. Extracts from green fluorescent protein (GFP)-expressing cells were used as negative control. All experiments were repeated three times (biological replicates), and a representative example is shown. d, mRNA expression of JMJD3 and UTX upon treatment with shRNA against JMJD3 or UTX. The expression after treatment of CEM cells with two shRNAs against JMJD3 and one shRNA against UTX and one control (Renilla) is shown. e, The effects on cell proliferation as measured by the loss of GFP-expressing shRNA. HL-60 is an acute promyelocytic leukaemia cell line (APL), which is a subtype of acute myeloid leukaemia (AML) and is used as control in this study. For both cell lines, the average results from three representative studies are shown. f, Annexin V staining upon shJMJD3 and shRenilla treatment of CUTLL1 cells (top) and HPB-ALL cells (bottom).
Extended Data Figure 3 JMJD3 binds to genes with important oncogenic functions and is vital for T-ALL growth.
a, JMJD3 but not UTX genetic inactivation impairs the expression of important oncogenic genes. NOTCH1, MYC and MAZ, as well as JMJD3, expression levels are shown. shUTX treatment results in significant upregulation of JMJD3 compared with shRenilla (control)-treated cells. The average results from three studies are shown. b, Significant expression changes in NRARP transcript levels upon JMJD3 knockdown. c, ChIP for H3K27me3 on the NRARP locus. d, e, Binding of JMJD3 to the NOTCH1 (d) and MAZ (e) promoters upon shJMJD3 and shRenilla (control) treatment. The average results from three studies are shown. f, Numbers of upregulated and downregulated genes are shown for shJMJD3- and shUTX-treated cells compared with shRenilla-treated cells. g, Scatter plot showing the expression levels of important genes in shJMJD3- and shUTX-treated CUTLL1 T-ALL cells. Emphasis is given to the NOTCH1 pathway and apoptosis-related genes. This is a scatter plot representation of an expression analysis comparing three independent studies for shJMJD3 and two for shUTX. h, i, Scatter plots showing the expression levels of important genes in shJMJD3- and shRenilla-treated CCRF-CEM T-ALL cells (h) and in shUTX-treated CCRF-CEM T-ALL cells (i). CCRF-CEM cells exhibit increased NOTCH1 levels through mutations in the heterodimerization (HD) domain of NOTCH1 and in the NOTCH1-associated ligase FBXW7. Emphasis is given to the NOTCH1 pathway and apoptosis-related genes. This is a scatter plot representation of an expression analysis comparing two studies for shJMJD3, two for shUTX and two for shRenilla.
Extended Data Figure 4 In vivo studies of the role of JMJD3 in T-ALL using luciferase analysis of CEM-, P12- and CUTLL1-based xenograft models in immunocompromised (NRG) mouse recipients.
a, b, In vivo growth of CEM T-ALL cells in subcutaneous xenograft studies upon genomic ablation of JMJD3 and UTX (red and green circles denote shJMJD3-expressing cells (two different shRNAs); blue denotes shUTX-expressing cells; and black circles denote shRenilla-expressing cells). One million CEM cells were injected into the animals, and representative graphs from five mouse recipients and an image of a representative mouse on days 0 and 6 are shown (a). Representative graphs from five mouse recipients and the average luciferase intensity on days 0 and 6 are shown (b). c, Results for growth of CEM cells at different time points post transplantation in subcutaneous xenograft studies (n = 5). d, Comparison of in vivo cell growth in the subcutaneous model of shJMJD3-, shUTX- and shRenilla-expressing P12 cells (n = 5). One million P12 cells were injected into sublethally irradiated NRG (immunocompromised) recipients, and the mice were monitored every day for luciferase activity. Day 0 was the first day that a substantially detectable luciferase intensity was measured. The last day of the experiment was the day that either luciferase intensity reached saturation or the mice were euthanized for humanitarian reasons. Red and green circles denote shJMJD3-expressing cells (two different shRNAs, shJMJD3A and shJMJD3B); blue denotes shUTX-expressing cells; and black circles denote shRenilla-expressing cells. e, Monitoring the change in luciferase intensity over a period of seven days in the subcutaneous xenograft model using CUTLL1 T-ALL cells (n = 4). f, g, Intravenous xenograft studies using CUTLL1 cells injected into sublethally irradiated NRG (immunocompromised) recipients (n = 8 or 6, as indicated in the figure). In e–g, 0.5 ×106 CUTLL1 cells were transplanted, and the mice were monitored every day for luciferase activity.
Extended Data Figure 5 UTX is a tumour suppressor and is genetically inactivated in T-ALL but is dispensable for physiological T-cell development.
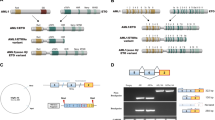
a, b, Study of lymphoid development in Utx−/Y compared with Utx+/+ (or Utx+/Y, data not shown) background mice. Flow cytometric analyses of CD4+ and CD8+ expression (a), and the relative proportions of CD4+CD8+ (double-positive) thymocytes across different genotypes (b) are shown. A representative example from three independent samples (biological replicates) is shown. c, Illustration of the transplantation scheme for the in vivo leukaemia studies. d, e, T-ALL progresses faster in the male Utx knockout background (Utx−/Y) than in the female wild-type background (Utx+/+) in recipients transplanted with NOTCH1-IC–GFP-expressing haematopoietic progenitors, as is demonstrated by the white blood cell counts in the peripheral blood (d), as well as the percentage of GFP+ leukaemic cells in the peripheral blood upon transplantation of wild-type progenitors (e) from female mice (Utx+/+) compared with the corresponding knockout cells (Utx−/Y). f, Survival study of the recipients of cells from male wild-type (Utx+/Y, n = 7) and knockout (Utx−/Y, n = 5) mice expressing NOTCH1-deltaE(ΔE)–GFP (an allele with weaker oncogenic action than NOTCH1-IC). g, h, Survival analysis of recipients upon transplantation of wild-type progenitors from female mice (Utx+/+) compared with the corresponding knockout cells (Utx−/Y) carrying NOTCH1-IC (g) or NOTCH1-ΔE (h). i, Quantitative PCR (qPCR) validation of the expression levels of one downregulated gene (Suz12) and one upregulated gene (Il7r) in Utx−/Y (compared with UTX+/Y) mice. The average results from three independent samples (studies) are presented. j, Targeted Sanger sequencing in paediatric T-ALL led to the identification of three cases with frameshift mutations. The positions of the mutations are indicated by dashed lines in the electropherograms. k, Identification of one in-frame deletion (p.Ala14_Ala17del, #1, top panel), one splice acceptor site (#2, second panel) and one missense mutation (#3, third panel) in adult T-ALL. Case #4 is an adult T-ALL case with wild-type UTX (control, bottom panel). Mutations are indicated by red characters. l, The levels of UTX in CUTLL1 T-ALL cells in the absence (−dox) or presence (+dox) of doxycycline. m, n, Apoptosis analysis through measuring annexin V staining using control LacZ-expressing and UTX-expressing CUTLL1 cells in the absence or presence of doxycycline. Representative plots (l, n), as well as the average results (l, m), from three independent experiments are shown.
Extended Data Figure 6 Physiological development of the haematopoietic system in the absence of JMJD3.
a, b, Targeting scheme for the generation of the Jmjd3−/− mouse (a) and PCR-based quantification of the wild-type and mutant transcripts (b) using a specific primer set for the 3′ end of Jmjd3 cDNA. c, d, Analysis of the fetal liver for lineage markers (c), as well as the bone marrow (d) of recipients for haematopoietic progenitors (the Lin−c-Kit+Sca1+ (LSK) population), for the Jmjd3+/+ and Jmjd3−/− genotypes. Representative plots from three independent experiments are shown. e–g, Analysis of major thymic subsets in Jmjd3+/+ (n = 7) and Jmjd3−/− (n = 7) mice. Schematic representation of the flow cytometric analysis performed (e). Relative proportions of the major cell populations in the thymi of Jmjd3+/+ and Jmjd3−/−mice (f). The mRNA expression of the Jmjd3 gene at different stages of thymic development (g). h, The expression of NOTCH1 target genes (such as Hes1, n = 7) in CD4+CD8+ (double positive) and CD4−CD8−CD25+ lymphocyte progenitor cells. Representative plots (e), as well as average results (g, h), from seven independent thymi are shown.
Extended Data Figure 7 JMJD3 is necessary for disease initiation in an animal model of T-ALL.
Initiation of the disease was studied by transplanting c-Kit+ haematopoietic progenitors. a, b, Blood analysis of the recipients for NOTCH1-IC–GFP leukaemic blasts (Jmjd3+/−, n = 8; Jmjd3−/−, n = 8; a) and white blood cells (WBCs, Jmjd3+/−, n = 4; Jmjd3−/−, n = 6; b). c–e, Comparison of the organ size (c), histochemistry (haematoxylin and eosin staining) (d) and flow-cytometry-based analysis (e) of the leukaemic cell infiltration of the spleen. f, Survival studies of recipients. Eight recipients for the Jmjd3+/− and eight for the Jmjd3−/− background were used in c–f.
Extended Data Figure 8 GSKJ4 inhibitor induces apoptosis and cell cycle arrest of T-ALL but not myeloid leukaemia or physiological LSK cells.
a, Effect of GSKJ4 (at 2 μM concentration) on a panel of T-ALL and myeloid lines. The average results from three representative studies are shown. b–d, Effects on cell growth (b), apoptosis (c) and the cell cycle (d) in three primary T-ALL lines. The average results from three representative studies are shown. e, f, Measurement of apoptosis (e, n = 3) and cell cycle effects (f, representative study from three experiments) on CUTLL1 cells 72 h post treatment with the inhibitor. g, h, Apoptosis assays using annexin V staining of CEM cells (g) after a period of 72 h of treatment and measuring caspase 7/9 activity upon treatment of CUTLL1 T-ALL cells with GSKJ5 or GSKJ4 over a period of 24 h (h). i, Time course studies of annexin V (top) and cell cycle analysis (bottom) of CUTLL1 cells over a period of 72 h during GSKJ4 treatment according to the scheme on top of the figure. j, Expression of the wild-type and catalytic mutant of JMJD3 in T-ALL (CEM) cells. k, Cell growth analysis of T-ALL cells overexpressing wild-type JMJD3 or a catalytic mutant of JMJD3 upon GSKJ4 treatment over a period of 72 h. Average results from three independent experiments are shown. l, Cell growth of LSK cells upon treatment with the control (2 μM) and different concentrations of the inhibitor GSKJ4. m, Annexin V staining of THP-1 (AML) cells after a period of 72 h of GSKJ4 or GSKJ5 (control) treatment at 2 μM concentration. The average results from three independent experiments are shown. n, The mRNA levels are shown for three classical NOTCH1 targets (HEY1, NRARP and NOTCH1) over a period of 72 h during GSKJ4 treatment. The average results from three independent experiments are shown.
Extended Data Figure 9 GSKJ4 treatment leads to increased H3K27me3 levels on NOTCH1 target genes through specific inhibition of JMJD3 activity.
a–c, Analysis of the promoter area of HEY1 (a), NOTCH1 (b) and NRARP (c) for H3K27me3, H3K27me1, NOTCH1 and JMJD3 enrichment over a period of 24 h during GSKJ4 treatment. The average results from three independent experiments are shown. d, Analysis of the total protein extracts from CUTLL1 cells for JMJD3 and NOTCH1. e, Analysis of the chromatin fraction from CUTLL1 cells for the repressive mark H3K27me3, the activating marks H3K27me1 and H3K4me3, as well as total histone H3 levels. Representative plots from three independent experiments are shown. f, Snapshots of GSKJ4-associated H3K27me3 changes in major NOTCH1 and JMJD3 targets. g, ChIP-qPCR analyses for UTX binding to the NOTCH1 target genes HEY1, NRARP and NOTCH1. (RBBP6 was used as positive control). The average results from three independent experiments are shown.
Extended Data Figure 10 JMJD3 as a pivotal factor in NOTCH1-mediated oncogenic activation in T-cell leukaemia.
a, Schematic representation of the H3K27me3 writer (the polycomb complex, left) and eraser (JMJD3, right). EZH2 contains the catalytic subunit of the complex through its SET domain, whereas the EED subunit recognizes the H3K27me3 mark and aids in polycomb binding. JmjC domain activity is inhibited by the small molecule inhibitor GSKJ4. b, The main idea about the key role of JMJD3 in the NOTCH1 transcriptional complex. Before activation of the NOTCH1 signalling pathway, the promoters of classical NOTCH1 target genes are bound by RBP-Jκ, together with components of the co-repressor complexes and PRC2, leading to low gene expression. After the binding of NOTCH1 and its co-activator MAML1, the genes are activated through the recruitment of JMJD3 and the MLL complex, with simultaneous eviction of PRC2, which leads to the demethylation of H3K27me3 and the methylation of H3K4me3.
Supplementary information
Supplementary Information
This file contains Supplementary Text and References. (PDF 467 kb)
Rights and permissions
About this article

Cite this article
Ntziachristos, P., Tsirigos, A., Welstead, G. et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514, 513–517 (2014). https://doi.org/10.1038/nature13605
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13605
This article is cited by
-
Polycomb-mediated silencing of miR-8 is required for maintenance of intestinal stemness in Drosophila melanogaster
Nature Communications (2024)
-
KDM6B protects T-ALL cells from NOTCH1-induced oncogenic stress
Leukemia (2023)
-
UTX inactivation in germinal center B cells promotes the development of multiple myeloma with extramedullary disease
Leukemia (2023)
-
KDM6 demethylases integrate DNA repair gene regulation and loss of KDM6A sensitizes human acute myeloid leukemia to PARP and BCL2 inhibition
Leukemia (2023)
-
Therapeutic potential of inhibiting histone 3 lysine 27 demethylases: a review of the literature
Clinical Epigenetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
