
Abstract
The origins of neural systems remain unresolved. In contrast to other basal metazoans, ctenophores (comb jellies) have both complex nervous and mesoderm-derived muscular systems. These holoplanktonic predators also have sophisticated ciliated locomotion, behaviour and distinct development. Here we present the draft genome of Pleurobrachia bachei, Pacific sea gooseberry, together with ten other ctenophore transcriptomes, and show that they are remarkably distinct from other animal genomes in their content of neurogenic, immune and developmental genes. Our integrative analyses place Ctenophora as the earliest lineage within Metazoa. This hypothesis is supported by comparative analysis of multiple gene families, including the apparent absence of HOX genes, canonical microRNA machinery, and reduced immune complement in ctenophores. Although two distinct nervous systems are well recognized in ctenophores, many bilaterian neuron-specific genes and genes of ‘classical’ neurotransmitter pathways either are absent or, if present, are not expressed in neurons. Our metabolomic and physiological data are consistent with the hypothesis that ctenophore neural systems, and possibly muscle specification, evolved independently from those in other animals.
Similar content being viewed by others

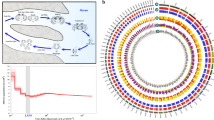
Main
Approximately 150 recognized species of comb jellies form a clade of pre-bilaterian animals1,2,3 (Fig. 1f) with an elusive genealogy, possibly tracing their ancestry to the Ediacaran biota4,5. We selected the Pacific sea gooseberry, Pleurobrachia bachei (A. Agassiz, 1860; Figs 1a, 2, Extended Data Fig. 1, Supplementary Data 1 and Supplementary Videos 1 & 2) as a model ctenophore because of preserved traits thought to be ancestral for this lineage (for example, cydippid larva and tentacles). Three next-generation sequencing platforms (454, Illumina and Ion Torrent) were used to obtain >700-fold coverage (Supplementary Tables 1 and 2) of Pleurobrachia’s genome, and about 2,000-fold coverage of the transcriptome representing all major organs and developmental stages (Supplementary Tables 3 and 4). Consequently, the draft assembly was 156,146,497 base pairs (bp) with 19,523 predicted protein-coding genes (Supplementary Tables 5–7). About 90% of these predicted genes are expressed in at least one tissue or developmental stage (Supplementary Methods) and 44% of Pleurobrachia genes have orthologues in other animals (Supplementary Tables 7 and 8). More than 300 families of transposable elements constitute at least 8.5% of the genome (Supplementary Table 9 and Supplementary Data 2) with numerous examples of diversification of some ancient transposable element classes (for example, transposases and reverse transcriptases). Approximately 1.0% of the genome is methylated. Pleurobrachia also uses DNA demethylation during development, with both 5-hydroxymethyl cytosine (5-hmC) and its synthetic enzyme TET6 (Extended Data Fig. 2). The obtained genome and transcriptome data provide rich resources (http://neurobase.rc.ufl.edu/Pleurobrachia) for investigating both animal phylogeny and evolution of animal innovations including nervous systems2,3,7,8,9.

a, The sea gooseberry, Pleurobrachia bachei (Supplementary Fig. 1), was selected as a target for genome sequencing due to preservation of traits ancestral for this lineage and because in situ hybridization/immunolabelling is possible. b–e, Major ctenophore innovations. b, Nervous system revealed by tyrosinated α-tubulin immunolabelling (scale bar, 60 μm). c, Scanning electron microscopy (SEM) imaging of nerve net in a tentacle pocket (scale bar, 20 μm). d, Locomotory ciliated combs (SEM; scale bar, 100 μm). e, Glue-secreting cells (colloblasts) in tentacles (SEM; scale bar, 20 μm). f, Relationships among major animal clades with choanoflagellates sister to all Metazoa.

Cydippid (Euplokamis, Pleurobrachia, Dryodora and Mertensiidae) and lobate (Mnemiopsis and Bolinopsis) ctenophores were polyphyletic, suggesting independent loss of both cydippid larval stage and tentacle apparatus as well as independent development of bilateral symmetry in benthic ctenophores, Vallicula and Coeloplana (Supplementary Data 4).
Ctenophore phylogeny
Although relationships among basal animal lineages are controversial1,10,11,12,13,14,15,16, our analyses (Supplementary Data 4) with Ctenophora represented by Pleurobrachia and Mnemiopsis suggest the placement of Ctenophora as the most basal animal lineage (Fig. 1 and Extended Data Fig. 3). Porifera was recovered as sister taxon to remaining metazoans (bootstrap support = 100%) with Cnidaria sister to Bilateria (bootstrap support = 100%, Fig. 1f). Shimodaira–Hasegawa tests17 (corresponding to Extended Data Fig. 3a–c with 586 gene matrix) rejected both Eumetazoa (sponges sister group to all other metazoans) and Coelenterata (Cnidaria+Ctenophora). Placement of Ctenophora at the base of Metazoa also provides the most parsimonious explanation for the pattern of global gene gain/loss seen across major animal clades (Fig. 3a and Supplementary Table 14a, b). Transcriptome data from ten additional ctenophores (Supplementary Table 13) with stricter criteria for orthology inference (Supplementary Methods 7) also placed ctenophores basal, albeit with less support (Extended Data Fig. 3d). When the most conserved set of genes was considered (Supplementary Methods 7.5 and Supplementary Data 4.3), the topology was unresolved. Weak support is probably due to under-representation of comparable transcriptomes from sponges and large protein divergence. Nevertheless, Shimodaira–Hasegawa tests based on expanded ctenophore sampling (with a reduced 114 gene matrix due to lack of other ctenophore and sponge genomes; Supplementary Methods 7.2) also rejected Coelenterata but not Eumetazoa. Notably, relationships within Ctenophora were strongly supported (Fig. 2). Both cydippid and lobate ctenophores, previously viewed as monophyletic clades, were recovered polyphyletic, indicating independent loss of both the cydippid larval stage and tentacle apparatus. Interestingly, Platyctenida was the second basal-most branch in the Ctenophore clade, suggesting that their benthic and bilaterial nature are secondarily derived.

a, Predicted scope of gene loss (blue numbers; for example, −4,952 in Placozoa) from the common metazoan ancestor. Red and green numbers indicate genes shared between bilaterians and ctenophores (7,771), as well as between ctenophores and other eukaryotic lineages sister to animals, respectively. Text on tree indicates emergence of complex animal traits and gene families. Deu, deuterostomes; Pro, protostomes. b, Uniquely shared and lineage-specific genes among basal metazoans. Values under species names indicate number of genes (bold) without any recognized homologues (e-value is 10−4) versus the total number of predicted gene models in relevant species (Supplementary Table 14b).
A highly reduced complement of animal-specific genes is a feature shared for the entire ctenophore lineage (Fig. 3 and Supplementary Table 15). HOX genes involved in anterio-posterior patterning of body axes and present in all metazoans are absent in ctenophores and sponges18 (Supplementary Tables 17 and 18). Likewise, canonical microRNA machinery (that is, Drosha and Pasha, Supplementary Table 19) is lacking in Pleurobrachia and other ctenophores. Using small RNA sequencing from Pleurobrachia, Bolinopsis and Beroe, we were unable to experimentally detect microRNAs (Supplementary Data 5.4). Pleurobrachia also lacks major elements that initiate innate immunity such as pattern recognition receptors (Toll-like, Nod-like, RIG-like, Ig-Toll-IL-1R domain) and immune mediators, MyD88 and RHD transcription factors, that are present in bilaterians, cnidarians and, in divergent forms, in sponges19,20 (Supplementary Table 20).
Key bilaterian myogenic/mesoderm-specification genes are absent in Pleurobrachia’s genome and transcriptomes of ten other ctenophores (Supplementary Table 35). These data indicate that muscles21 and, possibly, mesoderm evolved independently in Ctenophora to control the hydroskeleton, body shape and food capture. Thus, ctenophores might have independently developed complex phenotypes and tissue organization, raising questions about the nature of ctenophore-specific traits such as their unique development, combs, tentacles, aboral/apical organs and nervous systems.
Ctenophore innovations
To assess the genomic bases of ctenophore-specific innovations, we performed RNA-seq profiling of the major developmental stages (Fig. 4a, b) as well as adult organs and identified genes differentially expressed in these structures. Many Pleurobrachia genes that have no homologues in other species are specifically expressed and most abundant during the 4- to 32-cell cleavage stages as well as in tentacles, combs and the aboral organ (Fig. 4b and Extended Data Fig. 4). Thus, structures that are known as ctenophore innovations (Fig. 1d, e) have the largest complement of highly expressed Pleurobrachia/ctenophore-specific genes. These data suggest extensive gene gain in cell lineages associated with early segregation of developmental potential leading to ctenophore-specific traits in structures controlling feeding, locomotion and integrative functions; a finding consistent with proposed ‘orphan’ genes contributing to variation in early development and evolution of novelties22,23.

a, Main developmental stages in Pleurobrachia from eggs to cleavage (2–64 cells), gastrulation (1–3 h) and formation of cydippid larvae (∼24 h). b, Hierarchical clustering of approximately 400 ctenophore-specific genes differentially expressed among different development stages and adult structures as revealed by RNA-seq experiments. Colour index: black indicates highest level of expression, followed by purple, red then down to white indicating no expression. Most of these ctenophore-specific genes are primarily expressed during 4–32-cell stages (asterisks). The red circle indicates a subset of novel genes uniquely expressed in combs, tentacles and the aboral organ (AO). These genes lack recognized homologues in other organisms. c, Diversity and differential expression of RNA-editing genes in Pleurobrachia development and adult tissues (RNA-seq). ADAR1 has the highest expression level in early cleavage stages whereas ADAR2 and ADAR3 and ADAT1 and ADAT2 are most abundant in the combs. d, Morphological appearance of neurons during the third day of development (the top inset, neuronal cell bodies are stained with tyrosinated α-tubulin antibodies, red arrows) correlates with abundant expression of multiple iGluR receptors, suggesting that glutamate has an important role as an intercellular messenger. Muscles formed well before neuronal differentiation at the end of the first day of development (the bottom insert, phalloidin staining, yellow arrow); white arrow points to the embryonic mouth with hundreds of cilia inside. In c and d expression levels of RNA-editing or iGluR genes are shown as a normalized frequency of sequence reads for a given transcript from all RNA-seq data for each developmental stage (Supplementary Methods). Scale bars in d: 15 μm (top); 40 μm (bottom).
Examples of known metazoan gene families that are considerably expanded in Ctenophora (Supplementary Data 5 and Supplementary Table 16) include collagens, RNA-editing enzymes and RNA-binding proteins (Supplementary Data 5). Pleurobrachia’s genome encodes the highest number of RNA-editing enzymes (ADAR1–4, ADAT1–3, CDA1–2) reported so far among metazoans24,25 (Supplementary Data 5.5), possibly acting as the generalized mechanism generating post-transcriptional diversity and ctenophore-specific traits in locomotory and integrative structures (combs and aboral organ). Matching the expansion of RNA regulatory mechanisms, Pleurobrachia has more RNA-binding proteins (especially RRM/ELAV, KH and NOVAs26,27, Supplementary Table 21) than any basal metazoan or choanoflagellate examined. Dozens of RNA-binding proteins are selectively expressed and abundant during 8–64-cell stages (Supplementary Table 31), and might contribute to sequestration of RNAs and segregation in developmental potential leading to early cell-fate specification.
Phenotypic complexity positively correlates with presence and high differential expression of 92 homeodomain Pleurobrachia genes (Supplementary Data 5.2 and Supplementary Table 17)—76 genes have been reported in Mnemiopsis18, whereas the Amphimedon homeodomain complement consists of only 32 genes. In contrast, some developmental pathways are either absent (Hedgehog, JAK/STAT) or have reduced representation in ctenophores (TGF-β, Wnt, Notch). Surprisingly, most Wnt genes are weakly expressed during Pleurobrachia development, whereas the ctenophore-specific subtype WntX is primarily restricted to adult neuroid elements such as polar fields, aboral organ and tentacular conductive tracts (Extended Data Fig. 5e), suggesting a distinct molecular make-up of neural systems.
Parallel evolution of neural organization
Extensive parallel evolution of neural organization in ctenophores is the most evident. Compared to other animals with nervous systems, many genes controlling neuronal fate and patterning (for example, neurogenins, NeuroD, Achaete-scute, REST, HOX, Otx) are absent in the ctenophores that we sampled. Orthologues of pre- and postsynaptic genes also have limited representation (Supplementary Table 34), and they lack components (for example, neuroligin) critical for synaptic function in other eumetazoans.
Notably, our combined molecular, ultrasensitive metabolomic, immunohistochemical and pharmacological data strongly suggest that ctenophores do not use serotonin, acetylcholine, dopamine, noradrenaline, adrenaline, octopamine, histamine or glycine as intercellular messengers (Extended Data Figs 6 and 7g, Supplementary Data 5.8 and Supplementary Tables 22–26). Lack of ionotropic receptors for these molecules in ctenophores is consistent with this conclusion (Supplementary Table 26a). Most genes involved in the synthesis of neurotransmitters are absent in non-metazoan opisthokonts Monosiga and Capsaspora, suggesting that they are cnidarian/bilaterian innovations.
We next investigated the identity of the ctenophore transmitters. Physiological and pharmacological tests suggest that l-glutamate is a candidate neuromuscular transmitter in Pleurobrachia (Fig. 5b, Extended Data Fig. 7), able to induce rapid inward currents and raise intracellular Ca2+ concentration in muscle cells causing muscle contractions at nanomolar concentrations (10−7 M). In contrast, all other classical neurotransmitters were ineffective even in concentrations up to 5 × 10−3 M whereas d-glutamate as well as l-/d-aspartate have significantly less affinity in these assays (Fig. 5b).

a, Two neural nets in Pleurobrachia as revealed by tyrosinated α-tubulin immunostaining. Top image shows subepithelial net with concentrations of neuronal elements in the polar fields and ciliated furrows, known as structures involved in sensory and motor functions, respectively (blue arrow in right insert indicates location of a neuronal somata with individual neurites marked by red arrows). The bottom image shows neurons of the mesogloeal net (arrows are neuronal somata; arrowheads are neuronal processes). Note that phalloidin (a muscle marker) did not stain these cells. Scale bar, 120 μm (top); 10 μm (bottom images). b, l-glutamate (10−7–10−3 M) induced action potentials in muscle cells, whereas other transmitter candidates were ineffective even at concentrations up to 5 mM. Typical responses of ctenophore muscle cells to local pulses of a transmitter application were recorded both as individual action potentials (whole-cell current-clamp mode) and video contractions from a single muscle cell. The graph shows normalized responses from the same muscle cell, indicating that l-glutamate is the most potential excitatory molecule compared to d-glutamate or l-/d-aspartate (Supplementary Methods). ACh, acetylcholine. c, Key molecular innovations underlying neural organization in ctenophores. Bars indicate the presence or relative expansions of selected gene families in all basal metazoan lineages from the inferred urmetazoan ancestor. The data suggest that sponges and placozoans never developed neural systems, or, unlikely assuming the presence of neuronal organization in the urmetazoan ancestor, sponges and placozoans lost their nervous systems. Either hypothesis points towards extensive parallel evolution of neural systems in ctenophores versus the Bilateria+Cnidaria clade. d, The aboral organ has the greatest diversity and highest expression levels of 12 gap junction proteins, suggesting unmatched expansion of electrical signalling in this complex integrative organ—an analogue of an elementary brain in ctenophores. Expression of different innexins is shown as a summation of normalized frequencies of respective sequencing reads in RNA-seq data obtained from each developmental stage and adult tissues (y axis is ‘expression frequency’ or ‘expression’; see Supplementary Methods).
The proposed role of glutamate as a signal molecule in ctenophores is supported by an unprecedented diversity of ionotropic glutamate receptors (iGluRs; Extended Data Fig. 7a, b and Supplementary Table 27)—far exceeding the number of genes encoding iGluRs in other basal metazoans28. iGluRs might have undergone a substantial adaptive radiation in Ctenophora as evidenced by unique exon/intron organization for many subtypes, and ctenophoran iGluRs form a distinct clade within the gene tree. Notably, during development, Pleurobrachia’s neurons are formed 2 days after the initial muscle formation, and first neurogenesis events correlate with co-expression of all iGlu receptors in hatching larvae (Fig. 4d). All cloned iGluRs also show remarkable cell-type-specific distribution with predominant expression in tentacles, followed by combs and the aboral organ, revealing well-developed glutamate signalling in adults (Extended Data Fig. 7b). Additionally, Pleurobrachia contains more genes for glutamate synthesis (8 glutaminases) and transport (8 sialins) than any other metazoan investigated29,30. Although we detected GABA (γ-aminobutyric acid, Supplementary Tables 22–24) and immunohistochemically identified its localization in muscles, lack of pharmacological effects of GABA on Pleurobrachia behaviour and major motor systems, such as cilia, muscle and colloblasts, suggest that GABA is a by-product of glutamate metabolism by l-glutamic acid decarboxylase.
The first nervous systems are suggested to be primarily peptidergic in nature7. Although we did not find any previously identified neuropeptide homologue, secretory peptide prohormone processing genes (Supplementary Table 31) are present. We predicted 72 novel putative prohormones in Pleurobrachia and found >50 homologues in other sequenced ctenophores (Extended Data Fig. 8 and Supplementary Tables 28 and 32). Functions of these prohormone-derived peptides could include cell-to-cell signalling, toxins, involvement in innate immunity, or a combination of these functions. Several ctenophore-specific precursors are expressed in polarized cells around the mouth, tentacles and polar fields, indicative of a signalling role (Extended Data Fig. 8b). Secreted products of these prohormones may be natural ligands for >100 orphan neuropeptide-like G-protein-coupled receptors31 identified in Pleurobrachia (Supplementary Table 26b). A second example of neuropeptide receptor candidates is amiloride-sensitive sodium channels (ASIC), which are also known to be regulated by different classes of short peptides and protons32. Pleurobrachia’s genome has 29 genes encoding ASICs—more than any organism sequenced so far—and expression of most correlated with developmental appearance of neurons (Supplementary Table 31). ASIC expression is most abundant in tentacles, combs and aboral organs—structures enriched in neural elements and under complex synaptic control.
Moreover, ctenophores evolved an enormous diversity of electrical synapses (absent in Nematostella, Amphimedon and Trichoplax) with 12 gap junction proteins (pannexins/innexins33 but not chordate-specific connexins) found in Pleurobrachia. All pannexins/innexins have their highest expression in the aboral organ followed by tentacles and combs (Fig. 5d). Non-metazoans lack pannexin orthologues, suggesting that these are metazoan innovations with profound expansion of this family in ctenophores. The aboral organ, combs and tentacles have a relatively large diversity of ion channels (Extended Data Fig. 9b), confirming complex regulation of excitability in these structures. However, the overall complement of voltage-gated ion channels in ctenophores is reduced compared to other eumetazoans34 (Extended Data Fig. 9a).
Our genome-wide survey also indicates that some bilaterian and cnidarian pan-neural markers are present (for example, three elav and musashi genes), but they are not expressed in neurons; a finding consistent with early divergence and extreme parallel evolution of neural systems in this lineage (Extended Data Figs 5 and 9b).
Discussion
Figure 5c summarizes key molecular innovations underlying neural organization in ctenophores. Evidently, with a markedly different molecular and genomic make-up, ctenophores have achieved complex phenotypic plasticity and tissue organization. Thus, ctenophores might represent remarkable examples of convergent evolution including the emergence of neuro-muscular organization from the metazoan common ancestor without differentiated nervous system or bona fide neurons (Extended Data Fig. 10b and Supplementary Data 15). The alternative ‘single-origin hypothesis’, where the common ancestor of all metazoans had a nervous system with complex molecular and transmitter organization including all classical cnidarian/bilaterian transmitters and neurogenic genes (Extended Data Fig. 10a), is a less parsimonious scenario. This hypothesis implies that ctenophores, despite being active predators, underwent massive loss of neuronal and signalling toolkits and then replaced them with novel neurogenic and signalling molecules and receptors.
These findings are of relevance for regenerative and synthetic biology in designing novel signalling pathways and systems. In this case, ctenophores and their genomes present matchless examples of ‘experiments’ in nature and the possible preservation of ancient molecular toolkits lost in other animal lineages.
Methods Summary
Genome sequencing and transcriptome profiling used high-throughput methods. Gene prediction (Augustus/Fgenesh) and annotations were complemented by metabolome and microchemical assays using capillary electrophoresis with laser-induced fluorescence and mass spectrometry detectors to validate the presence/absence of selected transmitters. RNA-seq, PCR with reverse transcription (RT–PCR), in situ hybridization and immunohistochemistry were used to characterize and localize gene expression in adults and embryos. Pharmacological and electrophysiological tests were performed on semi-intact preparations using freshly isolated ctenophore cells.
Online Methods
Source material
Animals (Pleurobrachia bachei, Euplokamis dunlapae, Dryodora glandiformis, Beroe abyssicola, Bolinopsis infundibulum and Mertensiid) were collected at Friday Harbour Laboratories (Pacific North-Western Coast of USA) and maintained in running seawater for up to 2 weeks. Other species were collected at the Atlantic coast of Florida and around Woods Hole, Massachusetts (Pleurobrachia pileus, Pleurobrachia sp., Mnemiopsis leidyi) as well as central Pacific (Palau, Hawaii, Coeloplana astericola, Vallicula multiformis). Animals were anaesthetized in 60% (volume/body weight) isotonic MgCl2 (337 mM). Specific tissues were surgically removed with sterile fine forceps and scissors and processed for DNA/RNA isolations as well as metabolomics or pharmacological/electrophysiological tests. Whole animals were used for all in situ hybridization and immunohistochemical tests as described35. Genomic DNA (gDNA) was isolated using Genomic-tip (QIAGEN) and total RNA was extracted using RNAqueous-Micro (Ambion/Life Technology) or RNAqueous according to manufacturers’ recommendations. Quality and quantity of gDNA was analysed on a Qubit2.0 Fluorometer (Life Technologies) and for RNA we used a 2100 Bioanalyzer (Agilent Technologies). For all details see Supplementary Methods sections 1.1–1.3.
Genome sequencing
All genomic sequence data for de novo assembly were generated on Roche 454 Titanium and Illumina Genome Analyzer IIx, HiSeq2000 and MiSeq instruments using both shotgun pair-end and mate-pair sequencing libraries with 3–9 kb inserts as summarized in Supplementary Tables 1 and 2. Shotgun sequencing was performed from a single individual. Owing to a limited amount of starting gDNA, mate pair libraries were constructed from 10–12 individuals. In total, the genome sequencing is composed of 132,015,600,107 bp or ∼132 Gb of data, which corresponds to 733–825× physical coverage of the Pleurobrachia genome (the size of the P. bachei genome is estimated to be ∼160–180 Mb); see Supplementary Methods sections 1.4–2.1.2.
Genome assemblies
The Pleurobrachia bachei draft genome was assembled using a custom approach designed to leverage the individual strengths of three popular de novo assembly packages and strategies: Velvet36, SOAPdenovo37, and pseudo-454 hybrid assembly with ABySS38. First, using filtered and corrected data, we performed individual assemblies from 454 and Illumina reads by the Newbler (Roche, Inc.) software. Then the merged/hybrid assembly was achieved using three individual assemblies (SOAPdenovo, Velvet and ABySS/Newbler as described in Supplementary Methods 2.2). Three gene model predictions were performed by Augustus39 and Fgenesh predictions with the Softberry Inc. Fgenesh++ pipeline40,41 to incorporate information from full-length cDNA alignments and similar proteins from the eukaryotic section of the NCBI NR database42. After initial gene predictions in each of the three sets of genomic scaffolds, we screened each set of gene models for internal redundancy with the BLASTP program from NCBI’s BLAST+ software suite43. A model was considered redundant if it: had 90% identity to other model; the alignment between the two models had a bit score of at least 100; and the model was shorter than the other model.
Scaffolds producing these gene models were pooled and then screened for prokaryotic contamination using UCSC’s BLAT software package44 to produce the draft genome assembly version 1.0 (statistics can be found in Supplementary Table 5 and Supplementary Methods 2).
Genome annotation
For annotation, gene models were uploaded to the In-VIGO BLAST interface, a blastp alignment of gene models was performed against the entirety of NCBI’s non-redundant protein database and the Swiss-Prot protein database, and subsequently annotated in terms of Gene Ontology and KEGG pathways as well as Pfam domain identification. Transposable elements (TEs) were identified using not only WU-BLAST and its implementation in CENSOR but also databases for all known classes, superfamilies and clades of TEs described in the literature and/or collected in Repbase45. Detected sequences have been clustered based on their pairwise identities by using BLASTclust. All autonomous non-LTR retrotransposons have been classified based on RTclass1 (ref. 46). To merge partially predicted, non-redundant gene models with assembled transcriptome data, a custom Java tool was developed. This Java tool extended partial gene model predictions based on using transcriptome sequences to bridge 5′ and 3′ fragments of partially predicted genes. Using this Java tool, analysis of alignments of non-redundant gene models to assembled Pleurobrachia transcriptomes resulted to 19,523 (Supplementary Table 30) gene models. These gene models were used to also identify their possible homologues in assembled transcriptomes from 10 other ctenophore species sequenced (Supplementary Tables 10 and 11). All genomic sequences were submitted to NCBI on SRA accession number Project SRP001155 (Supplementary Methods 3.1–3.2).
Transcriptome sequencing and annotation
Three sequencing technology platforms were used for transcriptome profiling (RNA-seq): Roche 454 Titanium, Illumina HiSeq2000 and Ion Proton/PGM (Ion Torrent, Life Technologies). RNA-seq was performed from all major embryonic and developmental stages (1 cell, 2 cells, 4 cells, 8 cells, 16 cells, 32 cells, 64 cells, early and later gastrula, 1 day and 3 day larvae), major adult tissues and organs (combs, mouth, tentacles, stomach, the aboral organ, body walls), and whole body of Pleurobrachia bachei. We developed a reduced representation sequencing protocol for the 454 and Ion Torrent sequencing platforms that can detect low abundance transcripts47. The method reduces the amount of sequencing and gives more accurate quantification and additional details of the procedure are reported elsewhere47,48. In summary, we have generated 499,699,347 reads or ∼47.9 Gb to achieve approximately 2,000× coverage of the Pleurobrachia transcriptome.
In addition, Illumina HiSeq sequencing was also performed with RNA extracted from the following ctenophore species: Euplokamis dunlapae, Coeloplana astericola, Vallicula multiformis, Pleurobrachia pileus, Pleurobrachia sp. (collected from the Middle Atlantic and later identified as a subspecies of P. pileus), Dryodora glandiformis, Beroe abyssicola, Mnemiopsis leidyi, Bolinopsis infundibulum and an undescribed species which belongs to the family Mertensiidae (Supplementary Table 3). Each sequencing project was individually assembled using the Trinity de novo assembly package49 and in selected cases using MIRA. Reads from developmental stages were also assembled using the CLCBio Genomics Workbench. Before each assembly, reads were quality trimmed and had adaptor contamination removed with cutadapt50. Full summaries of the transcriptome assemblies are presented in Supplementary Tables 4 and 10. Each transcriptome was mapped to the Pleurobrachia genome, and aligned to both NCBI’s non-redundant protein database (NR) and the UniProtKB/Swiss-Prot (SP) protein database. Gene Ontology51 and Kyoto Encyclopedia of Genes and Genomes52,53 (KEGG) terms were associated with each transcript. By first translating transcripts in all six reading frames, Pfam/SMART domains54 were assigned to each reference transcriptome.
Each reference transcriptome and its full set of annotation and expression data was uploaded to our transcriptome database http://moroz.hpc.ufl.edu/slimebase2/browse.php for downstream analysis and visualization55,56. The database is integrated with UCSC type genome browser. Via the genome project homepage (http://neurobase.rc.ufl.edu/Pleurobrachia) all data sets have direct download options. Quantification of gene expression profiling was performed on all transcriptional data as described in Supplementary Methods 4.4. Hierarchical clustering was performed by Spotfire agglomerative algorithm. All primary transcriptome data was submitted to NCBI on SRA accession number Project SRP000992. (See Supplementary Methods 4.1–4.2.3 for details.)
Phylogenetic analyses
To reconstruct basal metazoan phylogeny (see controversies in10,11,12,13,14,15,57), we conducted two sets of phylogenomic analysis using tools described elsewhere58. All analyses included new data from Pleurobrachia bachei and the sponges Sycon (Calcarea) and Aphrocallistes (Hexactinellida). For the first set of analyses, Ctenophora was represented by two species of Pleurobrachia and Mnemiopsis leidyi. Initial analyses included the taxa in Supplementary Table 12. For a subsequent analysis, sampling within Ctenophora was expanded to include ten additional taxa, each represented by a relatively deeply sequenced Illumina transcriptome (Supplementary Table 13). In order to reduce noise in the phylogenetic signal, we used strict criteria to exclude paralogues, highly derived sequences, mistranslated sequence regions, and ambiguously aligned positions in sequence alignments. Analyses were conducted in RAxML 7.2.7 (refs 59) using maximum likelihood (ML) with the CAT +WAG + F model. Topological robustness (that is, nodal support) for all ML analyses was assessed with 100 replicates of nonparametric bootstrapping. Details of phylogenomic analyses are presented in Supplementary Methods 7. Shimodaira–Hasegawa test17 was implemented in RAxML with the PROTGAMMAWAGF model17.
In order to examine evolution of single genes or gene families, alignments were performed with either ClustalX260,61,62 or Muscle63 then, if appropriate, either trimmed manually or trimmed using GBlocks64 to exclude ambiguously aligned positions. Once alignments were obtained, gene trees were reconstructed in MEGA 565 using ML with the Whelan and Goldman (WAG) model. The bootstrap consensus tree was inferred from 100 replicates. All positions containing gaps and missing data were eliminated. Pfam composition54, Gene Ontology51 and KEGG52,53 were used to further validate P. bachei orthologues. Analyses of gene gain and gene loss were performed using custom scripts as described elsewhere66 and in Supplementary Methods 7.
Analysis of DNA methylation
ELIZA-based colorimetric assays (Epigenteck) were performed to quantify both global 5-mC and 5-hmC methylation in the P. bachei genome. A total of 6 individual P. bachei and three rat (positive control) were used (Supplementary Methods 1.2). Three biological and technical replicates were performed for every sample. Absolute quantification of 5-mC and 2-hmC were determined and date is reported as a mean ± s.e.m. (Supplementary Methods 8).
Molecular cloning, in situ hybridization and immunohistochemistry
Methods were similar as reported elsewhere35,47,48,67 with some modifications (Supplementary Methods 9–11).
Scanning electron microscopy
Animals were fixed in 2.5% glutaraldehyde in 0.2 M phosphate-buffered saline (pH 7.6) for 3–4 h at room temperature, and washed. For secondary fixation, we used 2% osmium tetroxide in 1.25% sodium bicarbonate for 2–3 h at room temperature. After dehydration in ethanol, samples were placed for drying in Samdri-790 Critical Point Drying. After drying the samples were coated on Sputter Coater. SEM observations and recordings were done on NeuScope JCM-5000 microscope (Supplementary Methods 12).
Electrophysiological methods, calcium imaging and pharmacological assays
Patch electrodes for extracellular and whole-cell recordings were pulled from borosilicate capillary (P-87, Sutter Instruments). All currents were recorded using an Axopatch or 200B amplifier controlled by a Digidata 1322A and pClamp 9.2. Action potentials (APs, spikes) were recorded in track mode using cell-attached loose-patch configuration. Whole-cell currents were recorded in voltage-clamp mode at a holding potential of −70 mV. Neurotransmitter candidate (see Supplementary Method 15) application for both extracellular AP and whole cell recordings were performed with a rapid solution changer, RSC-160 (Bio-Logic-Science Instruments, France). Data were analysed with Clampfit 9.0 (Molecular Devices) in combination with SigmaPlot 10.0. Videomicroscopy and time-lapse series were acquired with QImaging EXi CCD camera using DIC mode of Nikon Eclipse 2000 inverted microscope. Calcium imaging was performed on isolated ctenophore muscle cells using an Olympus IX-71inverted microscope equipped with a cooled CCD camera (ORCA R2, Hamamatsu). Cells were injected with calcium-sensitive probe (Fluo-4, ∼5 μM) through a patch pipette. Fluorescence imaging was performed under the control of Imaging Workbench 6 software. Stored time series image stacks were analysed off-line using Imaging Workbench 6, Clampfit 10.3, SigmaPlot 10/11 or exported as TIFF files into ImageJ 1.42. Pharmacological tests and behavioural assays with video recording were performed on intact animals in 5–40 l aquaria or on semi-intact preparations in a Sylgard-coated Petri dish with free cilia beating and muscle contractions. To monitor and quantify cilia movements we used glass microelectrodes filled with 2 M potassium acetate with resistances of 5–20 MΩ with electrical signals recorded by A-M System amplifiers (Neuroprobe 1600) and Gould Recorder (WindoGraf 980).
Determination of the presence of classical neurotransmitters by capillary electrophoresis (CE)
Two CE separation techniques were used to analyse tissue extracts for the presence of a number of neurotransmitters (Supplementary Tables 22 and 23; Supplementary Methods 17). While both methods used CE separations, complimentary detection methods, laser-induced native fluorescence (LINF)68 and electrospray ionization mass spectrometry (ESI-MS)69,70 were used to ensure broad coverage and low detection limits for the specific analytes of interest. Whole bodies of small animals as well as individual organs and tissues were removed, rinsed with ultrapure water and analytes were extracted using 49.5/49.5/1, methanol (LC-MS grade)/water/glacial acetic acid (99%) by volume, homogenized, centrifuged and supernatant was removed and frozen at −80 °C until analysis. The CE-LINF instrument used ultraviolet excitation at 264 nm and the native fluorescence emission was collected and recorded using a UV-enhanced CCD array (Spec-10; 2KBUV/LN; Princeton Instruments). CE separations were performed by hydrodynamic injection of 10 nl of sample and using 25 mM citric acid (pH 2.5, applied voltage +30 kV) or 50 mM borate (pH 9.5, applied voltage +21 kV). Analytes were identified based on comparison of both the migration time and fluorescence spectrum to that of standard mixtures of analytes. CE-ESI-MS analysis was performed using a Bruker Microtof or a Maxis (Bruker Daltonics) mass spectrometer for detection. All separations were performed using 1% formic acid in water as the electrolyte and applied voltage of +30 kV. Sheath liquid was 0.1% formic acid in 50/50 methanol/water. Samples were hydrodynamically injected for a total volume of ∼6 nl. Mass spectra were collected and recorded at a rate of 2 Hz with calibration performed using sodium formate clusters. Analytes were identified based on comparison of both the CE migration time and mass match to that of standard mixtures of analytes.
Accession codes
Primary accessions
BioProject
GenBank/EMBL/DDBJ
Sequence Read Archive
Change history
04 June 2014
Amendments have been made to the Acknowledgements section and the Author Information section.
References
Nielsen, C. Animal Evolution: Interrelationships of the Living Phyla 402 (Oxford Univ. Press, 2012)
Hernandez-Nicaise, M.-L. in Microscopic Anatomy of Invertebrates: Placozoa, Porifera, Cnidaria, and Ctenophora Vol. 2 (eds Harrison, F. W. & Westfall, J. A. ) 359–418 (Wiley, 1991)
Tamm, S. L. in Electrical Conduction and Behavior in “Simple” Invertebrates 266–358 (Clarendon, 1982)
Tang, F., Bengtson, S., Wang, Y., Wang, X. L. & Yin, C. Y. Eoandromeda and the origin of Ctenophora. Evol. Dev. 13, 408–414 (2011)
Dzik, J. Possible ctenophoran affinities of the Precambrian “sea-pen” Rangea. J. Morphol. 252, 315–334 (2002)
Pastor, W. A., Aravind, L. & Rao, A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nature Rev. Mol. Cell Biol. 14, 341–356 (2013)
Moroz, L. L. On the independent origins of complex brains and neurons. Brain Behav. Evol. 74, 177–190 (2009)
Moroz, L. L. Phylogenomics meets neuroscience: how many times might complex brains have evolved? Acta Biol. Hung. 63 (suppl. 2). 3–19 (2012)
Pennisi, E. Nervous system may have evolved twice. Science 339, 391 (2013)
Nosenko, T. et al. Deep metazoan phylogeny: When different genes tell different stories. Mol. Phylogenet. Evol. 67, 223–233 (2013)
Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008)
Hejnol, A. et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. R. Soc. Lond. B 276, 4261–4270 (2009)
Philippe, H. et al. Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 19, 706–712 (2009)
Pick, K. S. et al. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol. Biol. Evol. 27, 1983–1987 (2010)
Schierwater, B. et al. Concatenated analysis sheds light on early metazoan evolution and fuels a modern “Urmetazoon” hypothesis. PLoS Biol. 7, e1000020 (2009)
Kohn, A. B. et al. Rapid evolution of the compact and unusual mitochondrial genome in the ctenophore, Pleurobrachia bachei. Mol. Phylogenet. Evol. 63, 203–207 (2012)
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 (1999)
Ryan, J. F., Pang, K., Mullikin, J. C., Martindale, M. Q. & Baxevanis, A. D. The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. Evodevo 1, 9 (2010)
Lange, C. et al. Defining the origins of the NOD-like receptor system at the base of animal evolution. Mol. Biol. Evol. 28, 1687–1702 (2011)
Leulier, F. & Lemaitre, B. Toll-like receptors–taking an evolutionary approach. Nature Rev. Genet. 9, 165–178 (2008)
Steinmetz, P. R. et al. Independent evolution of striated muscles in cnidarians and bilaterians. Nature 487, 231–234 (2012)
Chen, S., Krinsky, B. H. & Long, M. New genes as drivers of phenotypic evolution. Nature Rev. Genet. 14, 645–660 (2013)
Tautz, D. & Domazet-Loso, T. The evolutionary origin of orphan genes. Nature Rev. Genet. 12, 692–702 (2011)
Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 79, 321–349 (2010)
Savva, Y. A., Rieder, L. E. & Reenan, R. A. The ADAR protein family. Genome Biol. 13, 252 (2012)
Yano, M., Hayakawa-Yano, Y., Mele, A. & Darnell, R. B. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron 66, 848–858 (2010)
Zhang, C. et al. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329, 439–443 (2010)
Traynelis, S. F. et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 (2010)
Omote, H., Miyaji, T., Juge, N. & Moriyama, Y. Vesicular neurotransmitter transporter: bioenergetics and regulation of glutamate transport. Biochemistry 50, 5558–5565 (2011)
El Mestikawy, S., Wallen-Mackenzie, A., Fortin, G. M., Descarries, L. & Trudeau, L. E. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nature Rev. Neurosci. 12, 204–216 (2011)
Palczewski, K. & Orban, T. From atomic structures to neuronal functions of G protein-coupled receptors. Annu. Rev. Neurosci. 36, 139–164 (2013)
Krishtal, O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 26, 477–483 (2003)
Panchin, Y. V. Evolution of gap junction proteins–the pannexin alternative. J. Exp. Biol. 208, 1415–1419 (2005)
Liebeskind, B. J., Hillis, D. M. & Zakon, H. H. Evolution of sodium channels predates the origin of nervous systems in animals. Proc. Natl Acad. Sci. USA 108, 9154–9159 (2011)
Moroz, L. L. & Kohn, A. B. Analysis of gene expression in neurons and synapses by multi-color in situ hybridization. Methods Mol. Biol. (in the press).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008)
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 1–6 (2012)
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009)
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008)
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000)
Solovyev, V. in Handbook of Statistical Genetics (eds Balding, D. J., Bishop, M. & Cannings, C. ) 97–159 (John Wiley & Sons, 2007)
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 37, D5–D15 (2009)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–4002 (1997)
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002)
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005)
Kapitonov, V. V., Tempel, S. & Jurka, J. Simple and fast classification of non-LTR retrotransposons based on phylogeny of their RT domain protein sequences. Gene 448, 207–213 (2009)
Kohn, A. B., Moroz, T. P., Barnes, J. P., Netherton, M. & Moroz, L. L. Single-cell semiconductor sequencing. Methods Mol. Biol. 1048, 247–284 (2013)
Moroz, L. L. & Kohn, A. B. Single-neuron transcriptome and methylome sequencing for epigenomic analysis of aging. Methods Mol. Biol. 1048, 323–352 (2013)
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnol. 29, 644–652 (2011)
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 17, 10–12 (2011)
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet. 25, 25–29 (2000)
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000)
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012)
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 32, D138–D141 (2004)
Girardo, D. O., Citarella, M. R., Kohn, A. B. & Moroz, L. L. Automatic transcriptome analysis and quest for signaling molecules in basal metazoans. Int. Comp. Biol. Abstact meeting. P1.136 (2012)
Girardo, D. O., Citarella, M., Kohn, A. B. & Moroz, L. L. Zero-click, automatic assembly, annotation and visualization workflow for comparative analysis of transcriptomes: The quest for novel signaling pathways. Int. Comp. Biol. 11.6 Abstract. (2013)
Ryan, J. F. et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342, 6164 (2013)
Kocot, K. M. et al. Phylogenomics reveals deep molluscan relationships. Nature 477, 452–456 (2011)
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006)
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007)
Chenna, R. et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 (2003)
Jeanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G. & Gibson, T. J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 23, 403–405 (1998)
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 113 (2004)
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000)
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011)
Ptitsyn, A. & Moroz, L. L. Computational workflow for analysis of gain and loss of genes in distantly related genomes. BMC Bioinform. 13, (suppl. 15)(2012)
Moroz, L. L. et al. Neuronal transcriptome of Aplysia: Neuronal compartments and circuitry. Cell 127, 1453–1467 (2006)
Fuller, R. R., Moroz, L. L., Gillette, R. & Sweedler, J. V. Single neuron analysis by capillary electrophoresis with fluorescence spectroscopy. Neuron 20, 173–181 (1998)
Lapainis, T., Rubakhin, S. S. & Sweedler, J. V. Capillary electrophoresis with electrospray ionization mass spectrometric detection for single-cell metabolomics. Anal. Chem. 81, 5858–5864 (2009)
Nemes, P., Knolhoff, A. M., Rubakhin, S. S. & Sweedler, J. V. Metabolic differentiation of neuronal phenotypes by single-cell capillary electrophoresis-electrospray ionization-mass spectrometry. Anal. Chem. 83, 6810–6817 (2011)
Acknowledgements
We thank Friday Harbor Laboratories for facilities during animal collection and Marine Genomics apprenticeships (L.L.M., B.J.S.); E. Dabe, G. Winters, J. Netherton, N. Churches and C. Bostwick for help with animal, tissue, in situ, RNA and DNA assays; and X.-X. Tan, F. Lu and T. Tyazelova for sequencing. We thank F. Nivens for videos and P. L. Williams for database support. This work was supported by NSF (NSF-0744649 and NSF CNS-0821622 to L.L.M.; NSF CHE-1111705 to J.V.S.), NIH (1R01GM097502, R01MH097062, R21RR025699 and 5R21DA030118 to L.L.M.; P30 DA018310 to J.V.S.; R01 AG029360 and 1S10RR027052 to E.I.R.), NASA NNX13AJ31G (to K.M.H., L.L.M. and K.M.K.), NSERC 458115 and 211598 (J.P.R.), University of Florida Opportunity Funds/McKnight Brain Research and Florida Biodiversity Institute (L.L.M.), Rostock Inc./A.V. Chikunov (E.I.R.), grant from Russian Federation Government 14.B25.31.0033 (Resolution No.220) (E.I.R.). F.A.K., I.S.P. and R.D. were supported by HHMI (55007424), EMBO and MINECO (BFU2012-31329 and Sev-2012-0208). Contributions of AU Marine Biology Program 117 and Molette laboratory 22.
Author information
Authors and Affiliations
Contributions
L.L.M. conceived the project, designed the experiments and wrote the manuscript. A.B.K., A.P.G., D.R., E.K., F.G., R.S.,T.P.M., E.I.R. and L.L.M. prepared gDNA, RNA samples and performed sequencing; I.S.P., F.A.K., V.V.S., F.Y., M.R.C., A.B.K. and L.L.M. did assemblies, gene model prediction and annotations; K.M.K. and K.M.H. performed phylogenomic analysis; A.P., A.B.K. and L.L.M. worked on gene family gain/loss analysis; F.A.K. and R.D. characterized protein divergence; A.B.K., L.L.M. and K.M. characterized homeodomain complement; S.D., C.D., J.V.S. and L.L.M. performed capillary electrophoresis/microchemical metabolomic assays; A.P.G., A.B.K., E.B. and E.I.R. did small RNA sequencing and analysis; K.M.B. and J.P.R. characterized immune gene complement; V.V.K. and J.J. characterized transposons; T.P.N. and L.L.M. performed immunolabelling, electron microscopy and pharmacological assays; Y.V.B. and L.L.M. performed pharmacological, electrophysiological and imaging assays on muscles; D.O.G., M.R.C., A.B.K. and L.L.M. performed secretory peptide prediction; A.B.K. and L.L.M. analysed RNA-seq data; A.B.K. performed methylation analysis; B.J.S., A.B.K. and L.L.M. analysed developmental data; J.J.S., D.O.G., Y.B., R.B., A.F., A.B.K. and L.L.M. performed in situ hybridization experiments; C.E.M. identified species and wrote their description and biology; all authors contributed to preparation of the manuscript and the text.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
The genome and transcriptome data are available at DDBJ/EMBL/GenBank AVPN00000000; the BioProject PRJNA213480 and SRA SRP001155; and (http://neurobase.rc.ufl.edu/Pleurobrachia). Principal Investigator: Leonid L. Moroz.
Extended data figures and tables
Extended Data Figure 1 Anatomy and neuromuscular systems of the ctenophore Pleurobrachia bachei.
a–e, Anatomy of the ctenophore, Pleurobrachia bachei A. Agassiz, 1860. Natural colouration of the major organs in live animal is shown. a, Details of the transparent Pleurobrachia body are shown including, b, the pharynx and tentacle sheaths (pockets). Eight rows of comb plates, called ctenes, are made of giant compound cilia that diffract light, creating iridescence. c, Comb rows in Pleurobrachia are constantly beating. The mouth and the aboral organ (AO) are located at the opposite poles of the animal (a, c). The AO controls complex coordinated behaviours of the animal. d, Ciliated furrows connect the AO and the ctenes to mediate behaviour. e, Tentacles have numerous contractile tentillae used to capture food with specialized glue cells or colloblasts (see also Fig. 1e). f–h, Pleurobrachia neural nets and muscles. f, Comb plate muscles (red) were revealed using in situ hybridization for β-tubulin and subepithelial neural net (green) revealed by tyrosinated α-tubulin immunostaining. g, In this image comb cilia (green) were stained using tyrosinated α-tubulin antibodies (green) whereas underlying comb plate muscles were visualized by phalloidin (a muscle marker) that did not stain neurons. h, Organization of the subepithelial neural net around the mouth as revealed by tyrosinated α-tubulin antibodies (whole-mount preparation). Scale bars: 120 μm (f); 100 μm (g); 200 μm (h). See Supplementary Methods 10 and 11.
Extended Data Figure 2 DNA methylation and active DNA demethylation in Pleurobrachia bachei.
CpG DNA methylation facilitates the elimination of CpG dinucleotides over evolutionary time66. a, Histogram shows relative occurrences of different dinucleotides in genomes of P. bachei (red bars), Drosophila melanogaster (green bars, no DNA methylation) and Homo sapiens (blue bars). The P. bachei genome contains 2.3% CpG dinucleotides, which is much lower than the expected random frequency and, therefore, indicative of a genome that undergoes methylation compared to humans66. b, DNMT genealogy tree. The enzyme DNA methyltransferase (DNMT), which catalyses transfer of a methyl group to DNA to form 5- methyl cytosine (5-mC), is present in Pleurobrachia. c, TET family of enzymes catalyses active DNA demethylation via formation of 5-hydroxymethyl cytosine (5-hmC, the 6th DNA base). RNA-seq profiling reveals differential expression for DNMT and TET-like genes during development and in adult P. bachei. Both DNMT and TET-like genes are predominantly expressed during cleavage starting from the 1st division. However, the TET-like gene is also highly expressed in adult combs (asterisk). The y axis shows a normalized expression level for each transcript. d, ELIZA-based colorimetric assays validate the presence of both 5-mC and 5-hmC in the P. bachei genome (the rat brain is used as a positive control; n = 6 for Pleurobrachia and n = 3 for rat; data shown as mean ± s.e.m., see Supplementary Methods 8 and Supplementary Data section 3 for details).
Extended Data Figure 3 Animal phylogeny.
a, Phylogeny of Metazoa based on 586 genes. Topology inferred using RAxML 7.2.7 and maximum likelihood with the CAT +WAG + F model with all taxa from Supplementary Table 12. Bootstrap support values are listed at each node. Colour coding: purple, ctenophore; yellow, Porifera; pink, Cnidaria; light blue, Bilateria. b, Removal of fast-evolving taxa Trichoplax and Caenorhabditis improves topological robustness. Topology inferred using RAxML 7.2.7 and maximum likelihood with the CAT +WAG + F model with all taxa from Supplementary Table 12 except Trichoplax and Caenorhabditis. Bootstrap support values are listed at each node. c, Removal of distant out-groups such as Fungi and Filasterea further improves topological robustness. Topology inferred using RAxML 7.2.7 using maximum likelihood with the CAT +WAG + F model with all taxa from Supplementary Table 12 except Trichoplax, Caenorhabditis and non-choanoflagellate out-groups. Bootstrap support values are listed at each node. d, Analysis with improved ctenophore taxon sampling based on 114 genes. Topology inferred using RAxML 7.2.7 using maximum likelihood with the CAT +WAG + F model with all taxa from Supplementary Table 13. Bootstrap support values are listed at each node.
Extended Data Figure 4 Identification of tentacle- and comb-specific transcripts.
a, Identification of tentacle-specific transcripts. The left photo shows SEM of a Pleurobrachia tentacle with two branching tentillae densely covered with hundreds of colloblasts or glue cells. Comparative transcriptome (RNA-seq) profiling among major organs allowed us to identify several dozen genes differentially or uniquely expressed in tentacles. The histogram shows illustrative examples of some of these genes with a normalized expression level (y axis) for each represented transcript. One of these Pleurobrachia-specific genes we named tentillin (green arrow). In situ hybridization experiments (n = 9) revealed a remarkable cell-specificity expression pattern for tentillin in all main tentacle branches and tentillae, possible labelling colloblasts or associated secretory cells. Scale bar: 50 μm. b, Identification of comb-specific transcripts. The left photo shows a microscopic image of one comb row from an intact animal. The natural colouration is a reflection of the iridescence patterns produced from large cilia forming combs. Comparative transcriptome (RNA-seq) profiling among major organs allowed us to identify several hundreds of genes differentially or uniquely expressed in combs. The histogram shows illustrative examples of some of these genes with a normalized expression level (y axis) for each represented transcript (see Supplementary Methods 4.2.3.6, 4.2.3.7 and 10, all sequences used in the analysis can be found in Supplementary Tables 29, 30 and 32).
Extended Data Figure 5 Illustrative examples of tissue-specific gene expression in Pleurobrachia bachei.
a, b, Dicer and Argonaut are predominantly expressed in structures associated to sensory and integrative functions. These include the aboral organ, polar fields and combs. Note a relatively weak staining of other cell types in the skin and following ciliated furrows in Dicer and Argonaut preparations. c, d, Pleurobrachia ELAV is expressed in combs and not in neurons. ELAVs are RNA-binding proteins and they are considered as pan-neuronal markers (see Supplementary Data 5.6.1). However, in Pleurobrachia ELAV expression has not been detected in neural tissues or cells with recognizable neuronal-like appearances. In situ hybridization for Pleurobrachia ELAV3 (c, d) shows the highest levels of expression in the adult comb plate but not in any of the neural tissues or organs enriched with neurons such as the aboral organ and polar fields. e, WntX is selectively expressed in the aboral organ (AO) and major conductive pathways of Pleurobrachia, indicating its involvement in integrative and neural-like functions (in situ hybridization on a whole-mount preparation). One of the highest WntX expressions is found in AO and ciliated furrows whereas the polar fields showed a moderate expression level associated to their central regions. In situ hybridization was performed on whole mounts using DIG-labelled probes (see details in the Supplementary Methods, all in situ hybridization were performed at least on 4–5 different animals and these are representative photos for these experiments). Scale bars: 500 μm (a-d); 800 μm (e).
Extended Data Figure 6 Absence of serotonin in ctenophores.
Here we used nanolitre volume sampling, capillary electrophoresis separation and wavelength-resolved native fluorescence detection as described for ultra-sensitive assay of 5-hydroxytryptamine (serotonin or 5-HT) and related metabolites (a, the top electropherogram and the table with standards used). Limits of detection (LODs) range from the low attomole to the femtomole range, with 5-HT LODs being approximately 20–50 attomoles. b, c, Using this assay we failed to detect 5-HT in Pleurobrachia (b, n = 6) but 5-HT was reliably detected in the hemichordate Saccoglossus (c) and molluscs62. See details in Supplementary Methods 17 and Supplementary Table 22 for quantification.
Extended Data Figure 7 l-glutamate as a transmitter candidate in Pleurobrachia bachei.
a, The ionotropic glutamate receptors (iGluRs) are diverse and underwent substantial adaptive radiation within the Ctenophora lineage. Phylogenetic analysis shows Pleurobrachia iGluRs share highest identity to each other forming a distinct branch on the tree topology (Supplementary Data 5.9). b, Differentially expressed iGluR subtypes in Pleurobrachia bachei (red and green labelling with fluorescent in situ hybridization protocols). Dark blue fluorescence is DAPI nuclear staining. AO, aboral organ. Scale bars: 100 μm (top left and top middle); 60 μm (top right); 50 μm (bottom left); 30 μm (bottom middle); 200 μm (bottom right). c–f, Glutamate-induced action potentials and currents in muscle cells. c, Typical responses of ctenophore muscle cells to glutamate pulses recorded extracellularly (as individual action potentials/contractions from a single muscle cell in response to local application of glutamate, 1 mM), and d, from the same cell in whole-cell current clamp mode with clear action potentials. e, Isolated muscle cell. Scale bar, 25 μm. f, Glutamate-activated whole-cell currents recorded from the same cell (as in c). Time course of application is depicted by the diagram below the voltage signal. Two responses (inward current) are shown. The holding potential was −70 mV (Supplementary Methods 13–16). g, Representative electropherograms show capillary electrophoresis separation with laser-induced fluorescence detection from different organs in Pleurobrachia bachei (n = 5) for transmitter candidate identification. The bottom electropherograms are standards (Supplementary Methods 17 and Supplementary Tables 23–25 for quantification).
Extended Data Figure 8 Quest for putative secretory molecules in ctenophores.
a, Computational pipeline for prediction of secretory products in Pleurobrachia and the overview of secretory products predicted from the Pleurobrachia gene models (Supplementary Method and Data sections 4.2.3.7 and 5.11, respectively). b, Expression of novel secretory molecules in ctenophores (DIG-labelled in situ hybridization, Supplementary Data 5.11 and Supplementary Methods 10). Each of the predicted secretory prohormone was selected based on its unique and/or highly differential expression pattern as revealed by RNA-seq profiling. Ctenophorin is uniquely expressed in polarized cells around the mouth of Pleurobrachia and we found its homologues in all ctenophore species we sequenced. Tentillin is a Plerobrachia-specific gene, which is uniquely expressed in polarized secretory-like cells in tentillae and tentacles. Jansonin’s expression is primarily restricted to polarized cells located in the aboral organ and polar fields. For comparison, we showed a different but also cell-specific expression pattern of BarX transcription factor in cells of unknown identify localized in polar fields, comb plates and tentacles (see b4). c, d, Most predicted secretory products are expressed later in development and in adult organs of Pleurobrachia (RNA-seq). c, Expression patterns of 72 predicted prohormones in P. bachei indicates that 20 of them are present and differentially expressed in development (Supplementary Table 32 for all Pleurobrachia precursor sequences). Surprisingly 5 of these precursor mRNAs were found starting from the second cleavage stage whereas the rest are predominantly expressed on day 3 of development. This correlates to the first appearance of neurons in Pleurobrachia cydippid larva (see Supplementary Data 5.11 and Supplementary Method section 4.2.3.6 for the RNA-seq analysis).
Extended Data Figure 9 Diversity of ion channels in Pleurobrachia bachei.
a, Metazoan ion channel complement. The 112 ion channels identified in the Pleurobrachia genome are classified as voltage gated (v) or other gating such as second messengers. Receptor channels (R) are ligand-gated or ionotropic (iGluR, ChRN, HTR3, GABA and CLR) and indicated in grey. Metazoan novelties indicate type of ion channels absent in the choanoflagellates, the sister group to all animals. Coloured squares show channels: (1) primarily absent in ctenophores (pink); (2) secondarily lost in sponges or placozoans (dark yellow); (3) eumetazoan (Cnidaria+Bilareria) innovations (blue); or (4) examples of expansion of certain class of channels in some animal lineages (red). All Pleurobrachia sequences used in the analysis can be found in Supplementary Table 31. b, Ion channels are predominantly expressed in tentacles, combs and aboral organ. Hierarchical clustering of 112 identified ion channels in developmental stages and adult tissues of Pleurobrachia. Adult organs involved in food capture and ciliated locomotion and integrative functions show significantly higher diversity and overall higher level of expression levels for most of ion channel types. Mobile tentacles had the highest expression of voltage-gated channels, in particular Cav and Nav. The legend shows relative expression levels based on RNA-seq data (see Supplementary Methods 4.2.3.6).
Extended Data Figure 10 Two alternative scenarios of neuronal evolution.
a, Single origin of the neural system (monophyly) with possible loss of some neural molecular components in ctenophores as well as the possible secondary loss of the entire nervous systems in sponges and placozoans. b, Multiple origins of neurons in animals as introduced and supported in this study (see main text discussion section for details).
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Tables 1-28 and 34-35, Legends for Supplementary Tables 29-33, Supplementary Data and Supplementary References. (PDF 2064 kb)
Supplementary Table 29
Pleurobrachia bachei Unfiltered Gene Models – see Supplementary Information p.66 for full legend. (XLSX 12754 kb)
Supplementary Table 30
Pleurobrachia bachei Filtered Gene Models - see Supplementary Information p.66 for full legend. (XLSX 8417 kb)
Supplementary Table 31
Analyzed Transcripts - see Supplementary Information p.66 for full legend. (XLSX 3759 kb)
Supplementary Table 32
Predicted Secretory Peptides - see Supplementary Information p.67 for full legend. (XLSX 55 kb)
Supplementary Table 33
This table shows divergence of ctenophore proteins. (XLS 49 kb)
Pleurobrachia Behavior
Free swimming Pleurobrachia bachei or sea gooseberry from North-Western Pacific. Pleurobrachia has different types of locomotion (controlled by comb plates and the aboral organ). The Pleurobrachia genome was sequenced by Moroz et al. Their integrative analysis suggests that neurons evolved more than once. Credit: Frazier Nivens and Leonid L. Moroz (WMV 21235 kb)
Introduction to Ctenophores or Comb Jellies
Comb jellies form the least-investigated group of pre-bilaterian animals with the most elusive genealogy. Ctenophores can be called 'aliens of the sea' due to their unique molecular and tissue organization. These beautiful holoplanktonic predators have highly sophisticated ciliated locomotion mediated by specialized comb plates, giving their name to the phylum Cteno-phora, meaning comb-bearing. The ciliated locomotion mode itself can be viewed as primordial; being preserved in extant ctenophores; it reaches the most extreme specialization with cilia in ctenes approaching several millimeters –200 times the length of cilia in other animals. This video shows representatives of reference ctenophore lineages sequenced by Moroz et al. Credit: Frazier Nivens and Leonid Moroz (WMV 25017 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Moroz, L., Kocot, K., Citarella, M. et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 510, 109–114 (2014). https://doi.org/10.1038/nature13400
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13400
This article is cited by
-
Neuromuscular organization of the benthic ctenophore, Vallicula multiformis
Zoological Letters (2024)
-
Highly conserved and extremely evolvable: BMP signalling in secondary axis patterning of Cnidaria and Bilateria
Development Genes and Evolution (2024)
-
Functional analysis in a model sea anemone reveals phylogenetic complexity and a role in cnidocyte discharge of DEG/ENaC ion channels
Communications Biology (2023)
-
Comb jellies’ unique fused neurons challenge evolution ideas
Nature (2023)
-
Analysis of cnidarian Gcm suggests a neuronal origin of glial EAAT1 function
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
