
Abstract
Eucalypts are the world’s most widely planted hardwood trees. Their outstanding diversity, adaptability and growth have made them a global renewable resource of fibre and energy. We sequenced and assembled >94% of the 640-megabase genome of Eucalyptus grandis. Of 36,376 predicted protein-coding genes, 34% occur in tandem duplications, the largest proportion thus far in plant genomes. Eucalyptus also shows the highest diversity of genes for specialized metabolites such as terpenes that act as chemical defence and provide unique pharmaceutical oils. Genome sequencing of the E. grandis sister species E. globulus and a set of inbred E. grandis tree genomes reveals dynamic genome evolution and hotspots of inbreeding depression. The E. grandis genome is the first reference for the eudicot order Myrtales and is placed here sister to the eurosids. This resource expands our understanding of the unique biology of large woody perennials and provides a powerful tool to accelerate comparative biology, breeding and biotechnology.
Similar content being viewed by others


Main
A major opportunity for a sustainable energy and biomaterials economy in many parts of the world lies in a better understanding of the molecular basis of superior growth and adaptation in woody plants. Part of this opportunity involves species of Eucalyptus L’Hér, a genus of woody perennials native to Australia1. The remarkable adaptability of eucalypts coupled with their fast growth and superior wood properties has driven their rapid adoption for plantation forestry in more than 100 countries across six continents (>20 million ha)2, making eucalypts the most widely planted hardwood forest trees in the world. The subtropical E. grandis and the temperate E. globulus stand out as targets of breeding programmes worldwide. Planted eucalypts provide key renewable resources for the production of pulp, paper, biomaterials and bioenergy, while mitigating human pressures on native forests3. Eucalypts also have a large diversity and high concentration of essential oils (mixtures of mono- and sesquiterpenes), many of which have ecological functions as well as medicinal and industrial uses. Predominantly outcrossers1 with hermaphroditic animal-pollinated flowers, eucalypts are highly heterozygous and display pre- and postzygotic barriers to selfing to reduce inbreeding depression for fitness and survival4.
To mitigate the challenge of assembling a highly heterozygous genome, we sequenced the genome of ‘BRASUZ1’, a 17-year-old E. grandis genotype derived from one generation of selfing. The availability of annotated forest tree genomes from two separately evolving rosid lineages, Eucalyptus (order Myrtales) and Populus (order Malpighiales5), in combination with genomes from domesticated woody plants (for example, Vitis, Prunus, Citrus), provides a comparative foundation for addressing fundamental evolutionary questions related to the biology of woody perennials. Moreover, the unique palaeogeographic evolution of Eucalyptus, that is, isolation from other members of the rosid clade, enables disentangling of the events that led to the modern members of the rosids by characterizing shared and unique whole-genome duplication events and syntenic gene space with other sequenced genomes. The draft genome of E. grandis suggests that the Eucalyptus genome has been shaped by an early lineage-specific genome duplication event and a subsequent high rate of tandem gene duplication.
Sequencing, assembly and annotation
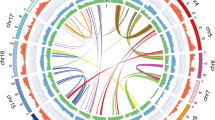
We assembled a non-redundant chromosome-scale reference (V1.0) sequence for BRASUZ1 based on 6.7× whole-genome Sanger shotgun coverage, paired bacterial artificial chromosome (BAC)-end sequencing and a high-density genetic linkage map6 (see Methods and Supplementary Information section 1). An estimated 94% of the genome is organized into 11 pseudomolecules (605 megabases (Mb), Fig. 1). Anchoring the genome assembly to an independent linkage map7 revealed that the remaining 4,941 smaller unanchored scaffolds (totalling 85 Mb) correspond largely to repeat-rich sequences and segments of alternative haplotypes of the assembled chromosomes derived from regions of residual heterozygosity in the otherwise inbred BRASUZ1 genome.

Genome features in 1-Mb intervals across the 11 chromosomes. Units on the circumference show megabase values and chromosomes. a, Gene density (number per Mb, range 6–131). b, Repeat coverage (22–88% per Mb). c, Average expression state (fragments per kilobase of exon per million sequences mapped, FPKM, per gene per Mb, 6–41 per Mb). d, Heterozygosity in inbred siblings (proportion of 28 S1 offspring heterozygous at position, 0.39–0.93). e, Telomeric repeats. f, Tandem duplication density (2–50). g, h, Single nucleotide polymorphisms (SNPs) identified by resequencing BRASUZ1 in 1-Mb bins (g) and per gene (h, 11,656 genes); homozygous regions (∼24%) and genes in green and heterozygous regions and genes in purple. Central blue lines connect gene pairs from the most recent whole-genome duplication event (Supplementary Data 1).
The E. grandis genome encodes a large number of predicted protein-coding loci (36,376) of which 89% are expressed in vegetative and reproductive tissues (Extended Data Fig. 1) plus various classes of non-coding genes (Supplementary Information section 2). Of the 36,376 predicted proteins, 30,341 (84%) are included in gene clusters shared with other rosid lineages (Extended Data Fig. 2). Retrotransposons account for the major portion of the genome (44.5%), with long terminal repeat retrotransposons being the most pervasive class (21.9%). DNA transposons encompass only 5.6% of the genome. For this class, Helitron elements are the most abundant with an estimated 15,000 copies or 3.8% of the genome (Supplementary Information section 2).
Genome evolution and phylogeny
To address the phylogenetic position of Eucalyptus, we performed genome-wide analysis of 17 sequenced plant genomes, generating a matrix of 697,423 aligned amino acid positions from 3,268 orthologue gene clusters (Methods and Supplementary Information section 3). Studies employing broad taxon sampling but a modest number of genes8 have consistently recovered two very well-supported clades of eurosids—the fabids and malvids—and grouped Eucalyptus and other Myrtales with the malvids. Our analysis alternatively places Eucalyptus as a sister taxon to the eurosids (Extended Data Fig. 3) and supports the grouping of Populus and Jatropha (order Malpighiales) with malvids rather than fabids, in agreement with other recent whole-genome studies9,10. The discrepancy between our genome-wide analyses and the angiosperm phylogeny group (APG) consensus highlights important methodological trade-offs between sampling more characters (as in our genome-wide study) versus more taxa (as per APG)11,12.
The evolutionary history of the Eucalyptus genome is marked by a lineage-specific palaeotetraploidy event newly revealed by our genomic analysis, superimposed on the earlier palaeohexaploidy event shared by all eudicots (Fig. 2). The whole-genome duplication (WGD) is estimated to have occurred ∼109.9 (105.9–113.9) million years (Myr) ago (Supplementary Information section 3 and Extended Data Fig. 4) in a Gondwanan ancestor around the time when Australia and Antarctica began to separate from East Gondwana. This WGD event is considerably older than those typically detected in other rosids13 and could have played a pivotal part in the evolution of the Myrtales lineage and its subsequent diversification from other rosid ancestors. The coincidence of the estimated WGD timing and the origin of the Myrtales14 leads to speculation that the WGD event could be directly related to the origin of the clade. More precise timing will require genomic analysis of other families and genera from the Myrtales.

a, Paralogous gene pairs in Eucalyptus for the identified palaeohexaploidization (bottom) and palaeotetraploidization (top) events. Each line represents a duplicated gene, and colours reflect origin from the seven ancestral chromosomes (A1, A4, A7, A10, A13, A16, A19). b, Number of synonymous substitutions per synonymous site (Ks) distributions of Eucalyptus paralogues (top) and Eucalyptus–Vitis orthologues (bottom). Blue bars (top) indicate Ks values for 378 gene pairs from the palaeotetraploidization WGD event (red dot), and red bars show Ks values for 274 gene pairs of the palaeohexaploidization event (red star). c, Evolutionary scenario of genome rearrangements from the Eudicot ancestor to Eucalyptus and other sequenced plant genomes; palaeohistory modified from ref. 49.
The Eucalyptus genome exhibits substantial conservation of synteny with other rosids as has been demonstrated for the basal rosid lineage represented by Vitis vinifera15. Extending the method previously described5 we identified 480 pairwise segments of conserved synteny between Eucalyptus and Vitis (Supplementary Information section 3). These segments include 68% of Eucalyptus genes and 76% of Vitis genes used in the analysis. The WGD in the Eucalyptus lineage relative to Vitis is clearly revealed by the 2:1 pattern in which two different Eucalyptus regions are typically collinear with a single region in Vitis. However, the gene content of these segments varies, as more than 95% of the paralogues in Eucalyptus have been lost subsequent to the WGD (a total of 5,896 Vitis genes have 6,158 synteny-confirmed orthologues in Eucalyptus). Half of the total length of the orthologous segments is contributed by segments longer than 1.83 Mb in Eucalyptus and 2.35 Mb in Vitis, suggesting that the loss of redundant genes after the WGD in Eucalyptus was accompanied by a compaction of those parts of the genome.
Eucalyptus chromosome 3, the largest single chromosome in the Eucalyptus genome, is the only chromosome that does not contain inter-chromosomal segmental duplications (Fig. 1), having fused with its WGD homologue. A similar situation occurs in Populus chromosome XVIII. Interestingly, Eucalyptus chromosome 3 and Populus chromosome XVIII nearly exclusively contain the ancestral eudicot chromosome 2 (Fig. 2c), despite their independent WGDs. There are no other examples among the currently sequenced dicotyledon genomes that contain a sole single copy of an ancestral chromosome. Moreover, in Eucalyptus and Populus, all other ancestral chromosomes appear to be dispersed and rearranged among the extant chromosomes (Fig. 2c). The conserved gene content and order (Supplementary Information section 3) on these chromosomes in two distantly related species could be due to: (1) convergent selection and positional stoichiometry of genes related to long-lived perennial woody habit that favours preservation of certain genes in syntenic order; and/or (2) merged ancestral chromosome structure (that is, multiple telomeres and centromeres on one chromosome) that suppresses gene expression, recombination and/or successive rearrangement. Eucalyptus chromosome 3 has the lowest average gene expression metrics of any of the Eucalyptus chromosomes (Fig. 1c), favouring the second hypothesis. Alternatively, there are several clusters of shared syntenic genes that appear to be related to perennial habit, including homologues of NAM (no apical meristem, PF02365) and senescence-associated protein (PF02365), several syntenic sets of disease-resistance genes, as well as genes related to cell-wall formation (Supplementary Data 2).
We also find that E. grandis has the largest number of genes in tandem repeats (12,570, 34% of the total) reported among sequenced plant genomes (Table 1 and Supplementary Information section 3). The low frequency of contig breaks separating tandem gene pairs (Extended Data Fig. 5) and conserved gene order on independent BAC clones spanning two large tandem gene arrays (Supplementary Data 3 and Supplementary Information 1) support the accuracy of the assembly across highly similar tandem copies. Tandem duplication often involves stress-response genes that are retained in a lineage-specific fashion, suggesting that tandem duplication is important for adaptive evolution in dynamically changing environments16. For example, more than 80% of the S-domain receptor-like kinase (SDRLK) subfamily occurs in tandem arrays (Supplementary Data 4). There also seems to be a bias in gene retention following tandem duplication in comparison to segmental and whole-genome duplication17. Even within the genus Eucalyptus, tandem duplication appears to be dynamic, for example, a cluster of MYB transcription factor genes in E. globulus lacks four of the nine tandem duplicates found in E. grandis (Extended Data Fig. 6).
Despite having the same number of chromosomes (n = 11) and highly co-linear genomes18, eucalypts vary considerably in genome size. E. grandis (640 Mb19) and E. globulus (530 Mb19) represent different sections (Latoangulatae and Maidenaria) within the subgenus Symphyomyrtus20, estimated to have diverged in the past 36 million years21. Resequencing of the subtropical E. grandis (BRASUZ1) and a representative of the temperate E. globulus (‘X46’, Supplementary Information section 3) revealed that many small, non-transposable element (TE)-derived changes distributed throughout the genome (164,813 regions; mean length 538 bp, median 230 bp, maximum 30,610 bp, total 88.7 Mb) account for nearly all of the genome size difference between the two species. Recent TE activity accounts for only 2 Mb of the size difference. This is in contrast to other studies in closely related plant species that report a predominant role for TEs in genome size evolution22. Using sequence data from other Eucalyptus species taxonomically positioned around the E. grandis–E. globulus split (J. Tibbits, unpublished data), we estimate that since divergence, E. grandis has gained 58 Mb and lost 12 Mb, while E. globulus has gained 15 Mb and lost 24 Mb, suggesting more active genome size evolution than was apparent from previous estimates.
Genetic load and heterozygosity
Eucalypts are preferentially outcrossing with late-acting post-zygotic self-incompatibility resulting in outcrossing rates that can exceed 90%1, high levels of nucleotide variation23,24 and accumulation of genetic load and expression of inbreeding depression4. A microsatellite survey of BRASUZ1 and its inbred siblings indicated putative hotspots of genetic load (Supplementary Information section 4). To investigate the distribution of preserved heterozygosity further, we resequenced an unrelated (outbred) E. grandis parental genotype M35D2 and 28 of its S1 offspring. The offspring were genotyped using 308,784 high-confidence heterozygous sites (within 22,619 genes) identified in M35D2 (Methods and Supplementary Information section 4). Contrary to Mendelian expectation of 50% retained heterozygosity after selfing, we observed 52% to 79% heterozygosity in the 28 S1 offspring (average of 66%). In all chromosomes except 5 and 11, heterozygosity was high (>80%) in long chromosome segments with peaks at >90% on chromosomes 6, 7 and 9 (Fig. 1d). Despite the strong bias towards heterozygosity in these regions, a small proportion of either homozygous haplotype was always present, suggesting that there are genetic backgrounds in which homozygosity of any particular gene is not lethal. One exception is on chromosome 4, where a 25-Mb region is completely devoid of one homozygous class across all surveyed genotypes (Extended Data Fig. 7 and Supplementary Information section 4).
The genetic architecture of genetic load and contribution of individual loci to inbreeding depression are largely unknown for woody perennials and present a barrier to rapid domestication via recurrent inbred mating. Our results suggest that a model of genome-wide cumulative effects of many small recessive alleles affecting overall fitness and survival best explains the architecture of inbreeding depression in Eucalyptus. This result is consistent with recent genome-wide selection experiments in Eucalyptus showing that a multifactorial model of a few hundred small effects throughout the genome contribute additively to height growth25, in contrast to earlier suggestions of the existence of a relatively small number of loci of larger effect as reported in several biparental QTL mapping studies26.
Lignocellulosic biomass production
Whereas woody growth habit (the ability to produce radial secondary tissues from a vascular cambium) is polyphyletic, having appeared and disappeared multiple times across more than 30 diverse taxa27, secondary cell wall formation itself is highly conserved across vascular plants. Large woody plants produce secondary cell walls on a vastly different scale from that of herbaceous plants. Approximately 80% of woody biomass comprises cellulose and hemicellulose, with the remaining biomass primarily composed of lignin28,29,30. A major determinant of industrial processing efficiency lies in secondary cell wall ultrastructure, which is dependent on interactions among these biopolymers. We identified putative functional homologues of genes encoding 18 enzymatic steps of cellulose and heteroxylan biosynthesis (Supplementary Information section 5). Despite the lineage-specific WGD event and the high number of genes in tandem duplications, relative and absolute expression levels (See Methods) suggest that most of the key enzymatic steps involve only one or two functional homologues (Fig. 3), which are highly and specifically expressed in xylem tissue. The xylem expression pattern of genes involved in sucrose catabolism suggests that Eucalyptus uses both direct (SUSY) and indirect (INV) pathways for the production of UDP-glucose (Fig. 3). Notably, the two sucrose synthase 4 homologues (Eucgr.C03199 and Eucgr.C00769) are expressed at high levels in xylem tissue and account for 70% and 18% of total sucrose synthase expression, respectively. These genes, found on chromosome 3 in Eucalyptus, are part of a syntenic set of genes found on Populus chromosome XVIII, indicating that these genes pre-date the speciation events that separate these genera. There are 10 multigene families encoding phenylpropanoid biosynthesis genes that have expanded, mostly through tandem duplication, to include 174 genes in E. grandis (Supplementary Information section 5). Phylogenetic analysis and expression profiling have allowed us to define a core set of 24 genes, as well as five novel lignification candidates, preferentially and highly expressed in developing xylem (Extended Data Fig. 8 and Supplementary Information section 5). These results highlight the central role of tandem gene duplication in shaping functional diversity in Eucalyptus and suggest that subfunctionalization within these expanded gene families has prioritized specific genes for wood formation.

Relative (yellow–blue scale) and absolute (white–red scale) expression profiles of secondary cell-wall-related genes implicated in cellulose and xylan biosynthesis29. Sugar and polymer intermediates are shown in green, while the proteins (enzymes) involved in each step are shown in blue. Detailed protein names, annotation and mRNA-seq expression data are provided in Supplementary Data 5. ST, shoot tips; YL, young leaves; ML, mature leaves; FL, floral buds; RT, roots; PH, phloem, IX, immature xylem. Absolute expression level (FPKM50) is only shown for immature xylem, the target secondary cell-wall-producing tissue. DUF, domain of unknown function; GATL, galacturonosyl transferase-like; GUX, glucuronic acid substitution of xylan; HEX, hexokinase; INV, invertase; IRX, irregular xylem; PGM, phosphoglucomutase; SUSY, sucrose synthase; RWA, reduced wall acetylation; UGD, UDP-glucose dehydrogenase; UGP, UDP-glucose pyrophosphorylase; UXS, UDP-xylose synthase.
Secondary metabolites and oils
It is generally thought that the extremely diverse array of secondary metabolites observed within Eucalyptus defends against a comparably diverse array of biotic pests, pathogens and herbivores encountered across its natural range. Many of the defence compounds are terpenoid based, including the commercially valuable eucalyptus oil, which is composed largely of 1,8-cineole. The conjugation of terpenes with phloroglucinol derivatives31, as well as the formation of monoterpene glucose esters32, leads to the myriad of defence compounds that vary across the genus. E. grandis has the largest observed number of terpene synthase genes among all sequenced plant genomes (n = 113 compared to a range of n = 2 in Physcomitrella to 83 in Vitis, Fig. 4), as well as a marked expansion of several phenylpropanoid gene families (Supplementary Information section 5). Furthermore, a subgroup of R2R3-MYB transcription factor genes known to be involved in the regulation of the phenylpropanoid pathway in Arabidopsis is expanded by tandem duplication in Eucalyptus to yield 16 genes with diverse expression profiles (Extended Data Fig. 9) possibly associated with the wide range of phenylpropanoid-derived compounds found in Eucalyptus.

The phylogenetic tree shows all TPS genes found in eight plant genomes (Supplementary Data 6). TPS subfamilies are indicated on the circumference of the circle. The tree has been rooted between the two major groups of type I and type III TPS. The table shows the number of TPS genes from several species obtained from a Pfam search for the two Pfam motifs (PF01397 and PF03936) found in the TPS genes. Colour coding in the table corresponds to that in the tree. The scale bar (0.3) shows the number of amino acid substitutions per site.
Reproductive biology
The genus Eucalyptus is named for its unusual floral structure derived from the Greek eu-, well, and kaluptos, covered, which refers to the operculum that covers the floral buds before anthesis. The ability to produce large amounts of pollen and seed over long generation times increases the reproductive success of woody perennials33 and impacts on adaptation and population genetics. Interestingly, the evolution and genetic control of the unique floral structure in Eucalyptus may be reflected in the expansion and deletion of genes typically associated with floral structure (for example, the APETALA1/FRUITFUL-like clade, Supplementary Information section 7). SOC1, a type II MADS-box gene that integrates multiple signals related to initiation of flowering, including long days, vernalization and pathways related to gibberellin signalling34, has been markedly expanded in E. grandis compared to other angiosperms (Extended Data Fig. 10). Eucalyptus is a diverse genus of over 700 species distributed in a wide range of environments ranging from tropical, subtropical and temperate forests1. This environmental heterogeneity encompasses extensive variation in the onset, season and intensity of flowering35. Because of SOC1’s diverse roles in environmental control of flowering, the expansion and subfunctionalization of the SOC1 subfamily may have contributed to the evolutionary diversification of Eucalyptus by integrating multiple signals into flowering responses relevant to different geographical zones. Eucalyptus may thus provide a model for the evolution of responses to divergent sets of flowering cues required for wide colonization and speciation.
Conclusions and future directions
The availability of a high-quality reference represents a timely step forward in fundamental studies of adaptation across the diversity of habitats occupied by eucalypt species. The unique biology and evolutionary history of Eucalyptus are reflected in its genome, for example, the expansion of terpene synthesis genes and the large number of tandem repeats, respectively. The coincidence of a lineage-specific WGD with the origin of the Myrtales reinforces the proposed role of genome duplication in angiosperm evolution and underscores the value of additional genome sequencing of families and genera in this important rosid lineage. Future studies of variation in functional genes will provide insights into the relative influences of drift and selection on Eucalyptus evolution and identify mechanisms of speciation and adaptive divergence. Such insight will lead to improved understanding of the response of eucalypts to environmental change. Comparative analysis of the E. grandis genome with those of other large perennials will add crucial insights into the evolutionary innovations that have made eucalypts keystone species that shape biodiversity in diverse ecosystems. The prospect of accelerating breeding cycles for productivity and wood quality via genomic prediction of complex traits25 and association genetics is enhanced by the release of the Eucalyptus genome. Genome-enabled derivation of an integrative data framework based on large-scale genotypic and phenotypic data sets will offer increasingly valuable insights into the complex connections between individual genomic elements and the extraordinary phenotypic variation in Eucalyptus.
Methods Summary
We used whole-genome shotgun sequencing (6.73× final sequence coverage from 7.7 million Sanger reads) followed by assembly in Arachne v.20071016 (ref. 36) and high-density genetic linkage mapping to produce chromosome-scale pseudomolecule sequences of the 11 nuclear chromosomes of BRASUZ1. Protein-coding loci were identified using homology-based FgenesH and GenomeScan predictions and ∼260,000 PASA37 EST assemblies from E. grandis and sister species. Gene family clustering was performed with the Inparanoid algorithm38,39 and peptide sequences analysed with Interproscan40, SignalP41, Predotar42, TMHMM43 and orthology-based projections from Arabidopsis. We performed maximum-likelihood-based phylogenetic reconstruction44 of the green plant phylogeny based on 174,020 peptides encoded by single copy orthologous genes from 17 plant genomes. Protein domains and domain arrangements were analysed to identify a core set of domains and arrangements present in rosid lineages represented by Eucalyptus, Vitis, Populus and Arabidopsis. Genome-wide gene expression profiling was performed using Illumina RNA-seq analysis of seven developing tissues from E. grandis. We identified whole-genome duplications using an approach45,46 based on paralogue- and orthologue-specific comparisons of the 36,376 predicted protein-coding genes and further refined the estimated age of the lineage-specific WGD event using a phylogenetic dating approach13. Genome synteny between E. grandis and P. trichocarpa was evaluated using the VISTA pipeline infrastructure47,48. Genome resequencing of E. grandis (BRASUZ1) and sister species E. globulus (X46) genomes was performed with Illumina PE100 DNA sequencing. Lignin, cellulose, xylan, terpene and flowering-related gene families were analysed using a combination of gene annotation, phylogenetic analysis and mRNA-seq expression profiling.
Online Methods
Whole-genome shotgun sequencing and assembly
All sequencing reads were collected with standard Sanger sequencing protocols on ABI 3730XL automated sequencers at the Joint Genome Institute, Walnut Creek, CA. Three different sized libraries were used for the plasmid subclone sequencing process and paired-end sequencing. A total of 3,446,208 reads from the 2.6-kb sized libraries, 3,479,232 reads from the 6.0-kb sized libraries and 518,016 reads from a 36.2–40.6-kb library were sequenced. Two BAC libraries (EG_Ba, 127.5-kb insert and EG_Bb, 155.0-kb insert) were end sequenced to add an additional 294,912 reads for long-range linking.
The sequence reads were assembled using a modified version of Arachne v.20071016 (ref. 36) with parameters maxcliq1 = 100, correct1_passes = 0, n_haplotypes = 2 and BINGE_AND_PURGE = True. The resulting output was then passed through Rebuilder and SquashOverlaps with parameters to merge adjacent assembled alternative haplotypes and subsequently run through another complete Arachne assembly process to finalize the assembly. This produced 6,043 scaffold sequences, with a scaffold L50 of 4.9 Mb and total scaffold size of 692.7 Mb. Scaffolds were screened against bacterial proteins, organelle sequences, GenBank nr and were removed if found to be a contaminant. Additional scaffolds were removed if they (1) consisted of >95% of base pairs that occurred as 24mers four other times in the scaffolds larger than 50 kb; (2) contained a majority of unanchored RNA sequences; or (3) were less than 1 kb in length.
For chromosome-scale pseudomolecule construction, markers from the genetic map were placed using two methods. SSR-based markers were placed using three successive rounds of e-PCR with N = 0, N = 1 and N = 3. Markers that had sequence associated with them, including SNP markers, were placed with BLAT51 and blastn52. A total of 19 breaks (16 in high coverage (>6x), 3 in low coverage (≤6x)) were made in scaffolds based on linkage group discontiguity; a subset of the broken scaffolds were combined using 257 joins to form the 11 pseudomolecule chromosomes. Map joins were denoted with 10,000 repeats of the letter N (Ns). The pseudomolecules contained 605.9 Mb out of 691.3 Mb (88%) of the assembled sequence. The final assembly contains 4,952 scaffolds with a contig L50 of 67.2 kb and a scaffold L50 of 53.9 Mb. The completeness of the resulting assembly was estimated using 1,007,962 ESTs from BRASUZ1. The goal of this analysis was to obtain an estimate of the completeness of the assembly, rather than to do a comprehensive examination of gene space. Briefly, ESTs <300 bp were removed, along with chloroplast, mitochondrial or rDNA ESTs. All duplicate ESTs were placed against the genome using BLAT51. The remaining ESTs were screened for alignments that had ≥90% identity and ≥85% EST coverage. The screened alignments indicated that 98.98% of available expressed gene loci were included in the 11 chromosome assemblies.
Gene prediction
To produce the current gene set, we used the homology-based FgenesH and GenomeScan predictions. The best gene prediction at each locus was selected and integrated with EST assemblies using the PASA program37. The gene set shown in the browser was generated from the input gene models at JGI. The gene prediction pipeline was structured as follows: peptides from diverse angiosperms and ∼260,000 EST assemblies (from ∼2.9 M filtered E. grandis ESTs and ∼2.4 M EST sequences from other closely related (‘sister’) Eucalyptus species, assembled with PASA) were aligned to the genome and their overlaps used to define putative protein-coding gene loci. The corresponding genomic regions were extended by 1 kb in each direction and submitted to FgenesH and GenomeScan, along with related angiosperm peptides and/or ORFs from the overlapping EST assemblies. These two sets of predictions were integrated with expressed sequence information using PASA37 against ∼260,000 Eucalyptus EST assemblies. The results were filtered to remove genes identified as transposon-related.
Gene family cluster and gene ontology analysis
The Inparanoid algorithm38,39 was used to identify orthologous and paralogous genes that arose through duplication events. Clusters were determined using a reciprocal best pair match and then an algorithm for adding in-paralogues was applied. The peptide sequences used were from Arabidopsis lyrata, Arabidopsis thaliana, Brachypodium distachyon, Caenorhabditis elegans, Chlamydomonas reinhardtii, Danio rerio, Ectocarpus siliculosus, Escherichia coli, Eucalyptus grandis, Fragaria vesca, Glycine max, Homo sapiens, Jatropha curcas, Mus musculus, Neurospora crassa, Nostoc punctiforme, Oryza sativa, Phoenix dactylifera, Physcomitrella patens, Populus trichocarpa, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Selaginella moellendorffii, Solanum tuberosum, Sorghum bicolor, Synechocystis pcc6803, Theobroma cacao, Vitis vinifera and Zea mays. The sequences were downloaded from Gramene53,54, Phytozome (http://www.phytozome.net) and Ensembl (http://www.ensembl.org). A functional annotation pipeline similar to the one used for strawberry genome annotation10 was used to infer Gene Ontology55 assignments to 29,841 protein coding genes (∼82%). Peptide sequences were analysed through an integrated approach involving Interproscan40, SignalP41, Predotar42, TMHMM43 and orthology-based projections from Arabidopsis.
Green plant phylogeny
We used an integrated approach of gene orthology clustering10 and an automated workflow for phylogenomic analyses56 to reconstruct land plant phylogeny of peptide sequences. A total of 174,020 peptides encoded by single-copy protein coding orthologous nuclear genes from 17 plant genomes (Supplementary Data 7) were identified, aligned and assembled into a supermatrix resulting from conservative and liberal superalignments that retained 42.26% (697,423 amino acids) and 46.35% (764,978 amino acids) of the original 1,650,340 amino acid concatenated alignment for maximum likelihood phylogenetic reconstruction44, respectively (Supplementary Data 7). These alignments come from 3268 orthologous gene clusters with each cluster carrying single copy genes from at least 9 (50%) species.
Protein domain analysis
Domains and domain arrangements were compared within the rosids to distinguish a core set of domains and arrangements present in all rosids and those shared by one or more of the four rosid lineages included in the analysis (Supplementary Data 8). Domains occurring at twice the frequency in Eucalyptus compared to the average abundance in the rosids were defined as overrepresented. If several splice variants were present for one protein, we excluded all but the longest transcript. All proteomes were scanned for domains with the Pfam_scan utility and HMMER 3.0 against the Pfam-A and Pfam-B databases57. For the annotation of Pfam-A domains, we used the model-defined gathering threshold and query sequences were required to match at least 30% of the defining model58. Pfam-B domains were annotated using an e-value cutoff of 10−3. When possible, Pfam-A domains were mapped to clans and consecutive stretches of the same domain were collapsed into one large pseudo-domain59,60. We defined domain arrangements as ordered sets of domains for each protein. For the analysis of arrangements, only Pfam-A domains were used.
Genome-wide mRNA expression profiling
To study the expression of predicted protein-coding and ncRNA genes, RNA-seq reads obtained from Illumina sequencing of seven Eucalyptus tissues (that is, shoot tips, young leaf, mature leaf, flower, roots, phloem and immature xylem, http://www.eucgenie.org/, Hefer et al., unpublished data) were mapped to the Eucalyptus genome using TopHat61 with the Bowtie algorithm62 for performing the alignment. The aligned read files were processed by Cufflinks50, with RNA-seq fragment counts (that is, fragments per kilobase of exon per million fragments mapped (FPKM)) to measure the relative abundance of transcripts. Differential ncRNA expression between the seven Eucalyptus tissues was determined using Cuffdiff50.
ncRNA analysis
To predict ncRNAs in Eucalyptus, the genome sequence was scanned using Infernal63 with the covariance models (that is, a combination of sequence consensus and RNA secondary structure consensus) of 1,973 RNA families in the RFam database v10.1 (refs 64, 65). The bit score cutoff of the Infernal search was set as the TC cutoff value that was used by RFam curators as the trusted cutoff. The Infernal search result was further filtered by an e-value cutoff of 0.01. To examine the ncRNA conservation between Eucalyptus and other plant genomes, the Eucalyptus ncRNA candidate sequences obtained from the Infernal search were used as queries to search against the genome sequences listed above using BLAT51 with a minimum coverage (that is, minimum fraction of query that must be aligned) of 80% and a minimum identity of 60%.
5′ UTR empirical curation
Approximately 2.9 million E. grandis ESTs and ∼700 million RNA-seq reads from seven diverse tissues were used to empirically curate 5′ UTR annotations. At each locus, the predicted, EST and RNA-seq derived 5′ UTR lengths were compared. An empirical annotation was prioritized over an in silico prediction and the longest empirical transcript was preferred. Those loci which had a 5′ UTR reported by only FGenesH retained their annotation as the best current annotation.
Genome evolution
We used the E. grandis genome sequence information (http://www.phytozome.net/eucalyptus.php) to unravel the Myrtales evolutionary palaeo-history leading to the modern Eucalyptus genome structure of 11 chromosomes. Independent intraspecific (that is, paralogue) and interspecific (that is, orthologue) comparisons were necessary to infer gene relationships between Eucalyptus and the other rosid genomes. We applied a robust and direct approach45,46 allowing the characterization of genome duplications by aligning the available genes (36,376) on themselves with stringent alignment criteria and statistical validation.
We used the VISTA pipeline infrastructure47,48 for the construction of genome-wide pairwise DNA alignments between E. grandis and Populus trichocarpa. To align genomes we used a combination of global and local alignment methods. First, we obtained an alignment of large blocks of conserved synteny between the two species by applying Shuffle-LAGAN global chaining algorithm66 to local alignments produced by translated BLAT51. After that we used Supermap, the fully symmetric whole-genome extension to the Shuffle-LAGAN. Then, in each syntenic block we applied Shuffle-LAGAN a second time to obtain a more fine-grained map of small-scale rearrangements such as inversions.
Syntenic regions between Eucalyptus chromosome 3 and Populus chromosome XVIII were defined as segments of contiguous sequence. Each contiguous block of DNA was annotated and cross-compared between the two species. Gene models within the syntenic blocks were compared based on a sliding window representing 10 gene models with an allowance of two intercalated gene models. Genes occurring in tandem repeats on either the Eucalyptus or Populus chromosomes were counted as a single locus in either case. The constructed genome-wide pair-wise alignments can be downloaded from http://pipeline.lbl.gov/downloads.shtml and are accessible for browsing and various types of analysis through Phytozome (http://phytozome.org).
For comparative analysis of the E. grandis and E. globulus genomes, enriched nuclei were extracted using a modified BAC library preparation protocol67 and DNA extracted following Tibbits et al.68. DNA was prepared for sequencing using Illumina TruSeq kits and 100-bp paired-end sequencing was performed on a HiSeq2000. NUCLEAR software (Gydle Inc.) was used to filter for high-quality reads that were then mapped to the E. grandis genome scaffold assembly. The VISION software was used to visualize assemblies and assembly metrics were computed using custom Perl, R and Shell scripts.
Genome function analysis
Using homology to Arabidopsis genes and Pfam domain analysis we identified candidate homologues for lignin, cellulose and xylan biosynthetic genes. All possible family members were identified and their gene expression evaluated in seven developing tissues of E. grandis using Illumina RNA-seq analysis. In particular, we analysed each gene’s expression relative to other family members/isoforms in xylem, as well as relative to the median (∼90,000 FPKM) of xylem expression in the entire transcriptome. Considering each gene’s relative and absolute expression levels, all members expressed over median in xylem were noted (Supplementary Data 5 and Supplementary Data 9). Similarly, a search for conserved protein motifs for the terpene synthase gene family was conducted in eight plant genomes, including E. grandis (Supplementary Information section 6 and Supplementary Data 6). The amino acid sequences were aligned and truncated to compare homologous sites. A maximum likelihood tree was created, rooted by the split between two major types of terpene synthase genes, and nodes were coloured by species.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The E. grandis whole-genome sequences are deposited in GenBank under accession number AUSX00000000. A genome browser and further information on the project are available at http://www.phytozome.net/eucalyptus.php.
References
Byrne, M. Phylogeny, diversity and evolution of eucalypts. in Plant Genome: Biodiversity and Evolution, Part E: Phanerogams-Angiosperm Vol. 1 (eds Sharma, A. K. & Sharma, A. ) 303–346 (Science Publishers, 2008)
Iglesias, I. & Wiltermann, D. in Eucalyptologics Information Resources on Eucalypt Cultivation Worldwide http://www.git-forestry.com (GIT Forestry Consulting, retrieved, 29 March 2009)
Bauhus, J., van der Meer, P. J. & Kanninen, M. Ecosystem Goods and Services from Plantation Forests 254 (Earthscan, 2010)
Costa e Silva, J., Hardner, C., Tilyard, P. & Potts, B. M. The effects of age and environment on the expression of inbreeding depression in Eucalyptus globulus. Heredity 107, 50–60 (2011)
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006)
Kullan, A. R. K. et al. High-density genetic linkage maps with over 2,400 sequence-anchored DArT markers for genetic dissection in an F2 pseudo-backcross of Eucalyptus grandis × E. urophylla. Tree Genet. Genomes 8, 163–175 (2012)
Petroli, C. D. et al. Genomic characterization of DArT markers based on high-density linkage analysis and physical mapping to the Eucalyptus genome. PLoS ONE 7, e44684 (2012)
Wang, H. et al. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc. Natl Acad. Sci. USA 106, 3853–3858 (2009)
D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012)
Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nature Genet. 43, 109–116 (2011)
Martin, W., Deusch, O., Stawski, N., Grunheit, N. & Goremykin, V. Chloroplast genome phylogenetics: why we need independent approaches to plant molecular evolution. Trends Plant Sci. 10, 203–209 (2005)
Jeffroy, O., Brinkmann, H., Delsuc, F. & Philippe, H. Phylogenomics: the beginning of incongruence? Trends Genet. 22, 225–231 (2006)
Fawcett, J. A., Maere, S. & Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl Acad. Sci. USA 106, 5737–5742 (2009)
Bell, C. D., Soltis, D. E. & Soltis, P. S. The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303 (2010)
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007)
Hanada, K., Zou, C., Lehti-Shiu, M. D., Shinozaki, K. & Shiu, S. H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148, 993–1003 (2008)
Freeling, M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453 (2009)
Hudson, C. J. et al. High synteny and colinearity among Eucalyptus genomes revealed by high-density comparative genetic mapping. Tree Genet. Genomes 8, 339–352 (2012)
Grattapaglia, D. & Bradshaw, H. D. Nuclear DNA content of commercially important Eucalyptus species and hybrids. Can. J. For. Res. 24, 1074–1078 (1994)
Brooker, M. I. H. A new classification of the genus Eucalyptus L'Her. (Myrtaceae). Aust. Syst. Bot. 13, 79–148 (2000)
Crisp, M. D., Burrows, G. E., Cook, L. G., Thornhill, A. H. & Bowman, D. M. Flammable biomes dominated by eucalypts originated at the Cretaceous-Palaeogene boundary. Natuer Commun. 2, 193 (2011)
Ågren, J. A. & Wright, S. I. Co-evolution between transposable elements and their hosts: a major factor in genome size evolution? Chromosome Res. 19, 777–786 (2011)
Külheim, C., Hui Yeoh, S., Maintz, J., Foley, W. & Moran, G. Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genomics 10, 452 (2009)
Novaes, E. et al. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 9, 312 (2008)
Resende, M. D. et al. Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol. 194, 116–128 (2012)
Grattapaglia, D. et al. Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet. Genomes 8, 463–508 (2012)
Groover, A. T. What genes make a tree a tree? Trends Plant Sci. 10, 210–214 (2005)
Boerjan, W., Ralph, J. & Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 54, 519–546 (2003)
Mizrachi, E., Mansfield, S. D. & Myburg, A. A. Cellulose factories: advancing bioenergy production from forest trees. New Phytol. 194, 54–62 (2012)
Scheller, H. V. & Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 61, 263–289 (2010)
Eschler, B. M., Pass, D. M., Willis, R. & Foley, W. J. Distribution of foliar formylated phloroglucinol derivatives amongst Eucalyptus species. Biochem. Syst. Ecol. 28, 813–824 (2000)
Goodger, J. Q. & Woodrow, I. E. α,β-Unsaturated monoterpene acid glucose esters: structural diversity, bioactivities and functional roles. Phytochemistry 72, 2259–2266 (2011)
Petit, R. J. & Hampe, A. Some evolutionary consequences of being a tree. Annu. Rev. Ecol. Evol. Syst. 37, 187–214 (2006)
Lee, J. & Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 61, 2247–2254 (2010)
House, S. M. Reproductive biology of eucalypts. in Eucalypt Ecology: Individuals to Ecosystems (ed. Woinarski, J. ) 30–56 (Cambridge Univ. Press, 1997)
Jaffe, D. B. et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13, 91–96 (2003)
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003)
Remm, M., Storm, C. E. & Sonnhammer, E. L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314, 1041–1052 (2001)
Ostlund, G. et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 38, D196–D203 (2010)
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009)
Bendtsen, J. D., Nielsen, H., von Heijne, G. & Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795 (2004)
Small, I., Peeters, N., Legeai, F. & Lurin, C. Predotar: A tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4, 1581–1590 (2004)
Sonnhammer, E. L., von Heijne, G. & Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182 (1998)
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006)
Salse, J., Abrouk, M., Murat, F., Quraishi, U. M. & Feuillet, C. Improved criteria and comparative genomics tool provide new insights into grass paleogenomics. Brief. Bioinform. 10, 619–630 (2009)
Salse, J. et al. Reconstruction of monocotelydoneous proto-chromosomes reveals faster evolution in plants than in animals. Proc. Natl Acad. Sci. USA 106, 14908–14913 (2009)
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004)
Dubchak, I., Poliakov, A., Kislyuk, A. & Brudno, M. Multiple whole-genome alignments without a reference organism. Genome Res. 19, 682–689 (2009)
Salse, J. In silico archeogenomics unveils modern plant genome organisation, regulation and evolution. Curr. Opin. Plant Biol. 15, 122–130 (2012)
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 28, 511–515 (2010)
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Youens-Clark, K. et al. Gramene database in 2010: updates and extensions. Nucleic Acids Res. 39, D1085–D1094 (2011)
Jaiswal, P. Gramene database: a hub for comparative plant genomics. Methods Mol. Biol. 678, 247–275 (2011)
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet. 25, 25–29 (2000)
Robbertse, B., Yoder, R. J., Boyd, A., Reeves, J. & Spatafora, J. W. Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Curr. 3, RRN1213 (2011)
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011)
Buljan, M., Frankish, A. & Bateman, A. Quantifying the mechanisms of domain gain in animal proteins. Genome Biol. Evol. 11, R74 (2010)
Ekman, D., Bjorklund, A. K., Frey-Skott, J. & Elofsson, A. Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions. J. Mol. Biol. 348, 231–243 (2005)
Forslund, K., Henricson, A., Hollich, V. & Sonnhammer, E. L. Domain tree-based analysis of protein architecture evolution. Mol. Biol. Evol. 25, 254–264 (2008)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009)
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009)
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009)
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003)
Gardner, P. P. et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39, D141–D145 (2011)
Brudno, M. et al. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731 (2003)
Peterson, D. G., Kevin, S. & Stephen, M. Isolation of milligram quantities of nuclear DNA from tomato (Lycopersicon esculentum), a plant containing high levels of polyphenolic compounds. Plant Mol. Biol. Rep. 15, 148–153 (1997)
Tibbits, J. F. G., McManus, L. J., Spokevicius, A. V. & Bossinger, G. A rapid method for tissue collection and high-throughput isolation of genomic DNA from mature trees. Plant Mol. Biol. Rep. 24, 81–91 (2006)
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002)
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599 (2007)
Acknowledgements
The work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. The research and writing of the manuscript was supported, in part, by the Office of Biological and Environmental Research in the US Department of Energy Office of Science under contract DE-AC05-00OR22725 as part of the US DOE Bioenergy Center. Funding for additional components of the study was provided by the Brazilian Ministry of Science, Technology and Innovation (MCTI) through its research funding agencies (CNPq and FINEP), the Brazilian Federal District Research Foundation (FAP-DF), the public-private Genolyptus network of Brazilian forestry companies, the Tree Biosafety and Genomics Research Cooperative (TBGRC, Oregon State University), South African forestry companies Sappi and Mondi, the Technology and Human Resources for Industry Programme (THRIP, UID 80118), the South African Department of Science and Technology (DST) and National Research Foundation (NRF, UID 18312 and 86936), the Laboratoire d’Excellence (LABEX TULIP ANR-10-LABX-41), the Agence Nationale pour la Recherche (Project Tree For Joules ANR-2010-KBBE-007-01; Fundação para a Ciência e Tecnologia (FCT, P-KBBE/AGR_GPL/0001/2010), the Centre National pour la Recherche Scientifique (CNRS), the University Paul Sabatier Toulouse III (UPS). Part of this work was carried out using the Stevin Supercomputer Infrastructure at Ghent University, funded by Ghent University, the Hercules Foundation, and the Flemish Government–department EWI. We also acknowledge S. Oda and E. Gonzalez of Suzano Paper and Pulp for providing genetic material of E. grandis genotypes BRASUZ1, G7J1, M35D2 and their progeny used for genome sequencing and resequencing, Forestal Mininco (Chile) for genetic material of X46, the E. globulus genotype used for genome resequencing, M. Hinchee and W. Rottmann of ArborGen for EST sequences used to support gene annotation, Sappi (South Africa) for genetic material of the population used for genetic linkage mapping, and Sappi and Mondi (South Africa) for E. grandis tissues used for RNA sequencing. We acknowledge M. O’Neill of the University of Pretoria for technical assistance with E. grandis RNA sequencing.
Author information
Authors and Affiliations
Contributions
A.A.M., D.G. and G.A.T. are the lead investigators and contributed equally to the work. J.Sc., J.J., J.G., R.D.H., D.M.G., I.D., A.P., U.H., D.S.R., E.L., H.T., D.B. and K.B. contributed to the assembly, annotation and sequence analysis, S.H.B., D.K. and D.G. to BAC library construction, G.P., S.H.B., M.R.P., D.A.F. and D.G. to various parts of biological sample collection, preparation and quality control, U.H., K.V., L.S., Y.V.d.P., F.M. and J.Sa. to genome duplication analyses, D.M.G., P.J. and J.E. to gene family clustering, U.H., P.J., J.E., A.L., A.E.B. and J.W.S. to green plant phylogeny, R.D.H., D.M.G., P.J., S.N. and R.R. to InterPro and Gene Ontology based functional annotation, X.Y., C.-Y.Y., T.L., T.J.T., M.R.P. and G.J.P. to non-coding RNA analyses, I.v.J., E.M., F.J. and A.A.M. to 5′ UTR analysis, M.R., E.M., C.A.H., K.V.d.M, F.J. and A.A.M. to RNA sequencing and expression profiling, A.R.K., E.B.-B., E.M. and A.A.M. to protein domain and arrangement analysis, D.A.S., J.T., P.R. and A.S. to E. globulus genome resequencing and analysis, U.H., M.S., V.C., H.S.C., J.P., J.G.-P. and G.J.P. to tandem duplicate analysis, K.V., V.A., P.D., C.S., C.A.H., E.R., M.R., A.A.M., S.H.S., R.C.J. and M.A.-F. to MADS box analyses, E.M., D.P., P.J., F.J. and A.A.M. to cellulose, xylan and CAZyme analysis, V.C., J.P., C.D., E.M., A.A.M. and J.G.-P. to lignin biosynthesis genes analysis, J.G.-P., S.G.H., C.A.H., M.S., N.S., H.C.-W., H.S.C., J.T. and P.R. to NAC and MYB analysis, C.K., P.J. and W.F. to terpene synthase gene family analysis, G.J.P. and R.C.T. to transposable elements analysis, U.H., A.R.K.K., C.P.S., C.D.P., D.A.F., O.B.S.-J., D.G. and A.A.M. to genetic mapping, U.H., D.A.F., M.R.P., P.S. and D.G. to genetic load and heterozygosity analysis, and S.N. and P.J. to SDRLK gene family analysis. B.M.P., D.A.S., R.E.V. and M.B. contributed to taxonomic and biological background text. G.A.T. headed and K.B. managed the sequencing project, D.S.R. coordinated the bioinformatics activities, A.A.M., D.G. and G.A.T. wrote and edited most of the manuscript. All authors read and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 RNA-seq-based expression evidence for predicted Eucalyptus grandis gene models.
Gene expression was assessed with Illumina RNA-seq analysis (240 million RNA sequences from six tissues, mapped to 36,376 E. grandis genes, V1.1 annotation). Genes were counted as expressed in a tissue if a minimum of FPKM = 1.0 was observed in the tissue. A total of 23,485 gene models (64.6%) were detected in all six tissues compared here and 32,697 (89.9%) in at least one of the six tissues. Expression profiles for individual genes are accessible in the Eucalyptus Genome Integrative Explorer (EucGenIE, http://www.eucgenie.org/).
Extended Data Figure 2 Sharing of protein-coding gene families, protein domains and domain arrangements in Eucalyptus, Arabidopsis, Populus and Vitis.
a, The four rosid lineages have a total of 16,048 protein coding gene clusters (from a total of 35,118 identified in 29 sequenced genomes; see Methods and Supplementary Information section 3) of which a core set of 6,926 clusters are shared among all four lineages. Of the 36,376 high-confidence annotated gene models in E. grandis, 30,341 (84%) are included in 10,049 clusters. E. grandis has 851 unique gene clusters (that is, not shared with any of the three other rosid genomes, but shared with at least one other of the 29 genomes). b, A total of 3,160 Pfam A domains are shared among the four rosid lineages, the majority of which are single-domain arrangements (3,138 shared among the four lineages). Thirteen PfamA domains were only detected in Eucalyptus and 392 domain arrangements are specific to Eucalyptus in this four-way comparison.
Extended Data Figure 3 Green plant phylogeny based on shared gene clusters from 17 sequenced plant genomes.
The phylogenetic tree was generated by RAxML analysis including at least one protein from at least half of the species per protein cluster in a concatenated MUSCLE alignment adjusted by Gblocks with liberal settings (Supplementary Data 7). The corresponding bootstrap partitions are provided at each node. The tree was rooted with Physcomitrella (a moss) as outgroup. The Myrtales lineage represented by Eucalyptus grandis is supported as sister to fabids and malvids (core rosid) clades together with the basal rosid lineage Vitales, whereas Populus trichocarpa (Malpighiales) is grouped with malvids.
Extended Data Figure 4 Dating of the Eucalyptus lineage-specific whole-genome duplication event.
a, Eucalyptus Ks whole-paranome (the set of all duplicate genes in the genome) age distribution. On the x axis the Ks is plotted (bin size of 0.1); on the y axis the number of retained duplicate paralogous gene pairs is plotted. b, Eucalyptus Ks anchor age distribution. On the x axis the Ks is plotted (bin size of 0.1); on the y axis the number of retained duplicate anchors is plotted. Anchors falling within the Ks range of 0.8–1.5 were used for absolute dating. c, Eucalyptus absolute dated anchors from the most recent WGD. The smooth green curve represents the maximum likelihood normal fit of dated anchors derived from the most recent WGD in Eucalyptus, whereas the blue dots represent a histogram of the raw data. The dashed line indicates the ML estimate of the distribution mode, whereas the dotted lines delimit the corresponding 95% confidence intervals. The mode of dated anchors is estimated at 109.93 Myr ago with its lower and upper 95% boundaries at 105.96 and 113.91 Myr ago, respectively. d, Genome duplication pattern in the core eudicot (rosid and asterid) ancestor and lineages leading to Solanum (asterid), Vitis and Eucalyptus (basal rosids) and the core rosids. The three Eucalyptus (E1–E3), Vitis (V1–V3) and Solanum (S1–S3) orthologues were generated by the shared hexaploidy event (purple box, ∼130 to 150 Myr ago) and an additional set of Eucalyptus orthologues (E1'–E3′) were created in the lineage-specific WGD (orange boxes, ∼110 Myr ago).
Extended Data Figure 5 Genome-wide analysis of tandem gene assemblies.
The number and distribution of contig breaks was evaluated for pairs of tandem genes (located within 50 kb of each other). a, Distribution of the number of contig breaks between gene pairs (blue bars) and cumulative proportion of gene pairs separated by contig breaks (black line). b, Distribution of the number of contig breaks per separation distance showing that the number of breaks is positively correlated with separation distance. The red line shows the distribution of distance between gene pairs with three or more contig breaks. c, Distribution of KS divergence of tandem gene pairs in clusters with exactly two tandem genes showing a gradient of similarity (that is, age of duplication) expected for authentic tandem gene pairs. d, Rate of tandem gene duplication (TD) and gene loss in Eucalyptus grandis (Eg), Populus trichocarpa (Pt), 2Vitis vinifera (Vv) and Arabidopsis thaliana (At). All of the rosid genomes (except Arabidopsis) exhibit constant rates of tandem duplication and loss. The rate of tandem gene duplication in Eucalyptus has been stable and consistently higher than in Populus and Vitis. 1 Myr ∼ 0.0026 transversions at fourfold degenerate sites, consistent with Populus and Eucalyptus having diverged ∼100 Myr ago.
Extended Data Figure 6 Illumina PE100 read coverage of the ∼760-kb region containing a R2R3–MYB tandem gene array.
Illumina PE100 reads generated from BRASUZ1 (E. grandis) and X46 (E. globulus) were aligned to the E. grandis (BRASUZ1, V1.0) genome assembly, and insert (green bars) and sequence (blue line) coverage investigated for the ∼760-kb region including a R2R3–MYB tandem array (details in Supplementary Data 3) in the E. grandis genome assembly. a, Read coverage profile of the BRASUZ1 reads mapped to the region showing 1× relative coverage across all nine of the tandem duplicates (red blocks) in the region, and b, X46 (E. globulus) reads mapped to the region showing 1× relative coverage on approximately half of the region with some tandem duplicates apparently absent from the E. globulus genome. Note that insert coverage (green bars) is relatively higher for E. globulus (X46, panel b) due to the larger insert size of the genomic library sequenced for X46 (∼300 bp) than for BRASUZ1 (∼150 bp).
Extended Data Figure 7 Alternative homozygous classes observed in the 28 M35D2 siblings as a function of position on chromosomes 1–11.
Several peaks of conserved heterozygosity (peaks >80%) are seen on all chromosomes except 5 and 11. A region of 25 Mb on chromosome 4 from 11 to 36 Mb is completely devoid of homozygous versions of one of the alleles (red line), but has roughly 25–32% of the siblings homozygous for the other allele (green line) and the rest heterozygous in a roughly 1:4 ratio. The blue line is the total proportion of siblings out of 28 that are heterozygous in the region. One would expect 50% under the null model, but almost the entire chromosome is biased towards heterozygosity. In several other regions (for example, chromosomes 6, 7, 9 and 10) both homozygous classes are depleted, suggesting the presence of genetic load at different loci along the two parental homologues and explaining the strong selection for heterozygosity in such regions.
Extended Data Figure 8 Genes involved in lignin biosynthesis in woody tissues of Eucalyptus.
Relative (yellow–blue scale) and absolute (white–red scale) expression profiles of secondary cell-wall-related genes implicated in lignin biosynthesis. Detailed gene annotation and mRNA-seq expression data are provided in Supplementary Data 9. Five novel Eucalyptus candidates that have not previously been associated with lignification are indicated by asterisks (Carocha et al., unpublished data). ST, shoot tips; YL, young leaves; ML, mature leaves; FL, floral buds; RT, roots; PH, phloem, IX, immature xylem. Absolute expression level (FPKM50) is only shown for immature xylem.
Extended Data Figure 9 Phylogenetic tree of R2R3 MYB sequences from subgroups expanded and/or preferentially found in woody species.
A total of 133 amino acid sequences from Eucalyptus grandis (50), Vitis vinifera (34), Populus trichocarpa (40), Arabidopsis thaliana (6) and Oryza sativa (3) corresponding to three woody-expanded (subgroups 5, 6 and AtMYB5 based on Arabidopsis classification) and five woody-preferential subgroups (I through V). The latter do not contain any Arabidopsis nor Oryza sequences. Sequences were aligned using MAFFT with the FFT-NS-i algorithm69 (Supplementary Data 10). Evolutionary history was inferred constructing a Neighbour-joining tree with 1,000 bootstrap replicates (bootstrap support is shown next to branches) using MEGA5 (ref. 70). The evolutionary distances were computed using the Jones-Taylor-Thornton substitution model and the rate variation among sites was modelled with a gamma distribution of 1. Positions containing gaps and missing data were not considered in the analysis. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. RNA-seq-based relative transcript abundance data for six different tissues, expressed in FPKM values (fragments per kilobase of exon per million fragments mapped), are shown for each Eucalyptus gene next to each subgroup. ST, shoot tips; YL, young leaves; ML, mature leaves; FL, flowers; PH, phloem; and IX, immature xylem.
Extended Data Figure 10 Phylogenetic tree of type II MIKC MADS box proteins.
Neighbour-joining consensus tree of the type II MIKC sub-clade using protein sequences from Eucalyptus grandis, Arabidopsis thaliana, Populus trichocarpa and Vitis vinifera (Supplementary Data 11). Bootstrap values from 1,000 replicates were used to assess the robustness of the tree. Bootstrap values lower than 40% were removed from the tree. Eucalyptus genes are denoted with green dots, Arabidopsis genes with red dots, Populus genes with yellow dots and Vitis genes with blue dots. The gene model numbers from Populus and Vitis were abbreviated to better fit in the figure (P. trichocarpa, Pt; V. vinifera, Vv).
Supplementary information
Supplementary Information
This file contains Supplementary Notes, Supplementary Tables 1-18, a list of the Supplementary Data files and Supplementary References. (PDF 1946 kb)
Supplementary Data
This file contains Supplementary Data files 1-2 – see Supplementary Information p.52 for details. (ZIP 5659 kb)
Supplementary Data
This file contains Supplementary Data files 3-7 - see Supplementary Information p.52 for details. (ZIP 24352 kb)
Supplementary Data
This file contains Supplementary Data files 8-22 - see Supplementary Information p.52 for details. (ZIP 17386 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Myburg, A., Grattapaglia, D., Tuskan, G. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014). https://doi.org/10.1038/nature13308
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13308
This article is cited by
-
Transcriptome analysis of the transition from primary to secondary growth of vertical stem in Eucalyptus grandis
BMC Plant Biology (2024)
-
Genomic and transcriptomic studies on flavonoid biosynthesis in Lagerstroemia indica
BMC Plant Biology (2024)
-
A two-sequence motif-based method for the inventory of gene families in fragmented and poorly annotated genome sequences
BMC Genomics (2024)
-
Exploring the versatility of sesquiterpene biosynthesis in guava plants: a comparative genome-wide analysis of two cultivars
Scientific Reports (2024)
-
Reference genes for Eucalyptus spp. under Beauveria bassiana inoculation and subsequently infestation by the galling wasp Leptocybe invasa
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
