
Abstract
The discovery of rare genetic variants is accelerating, and clear guidelines for distinguishing disease-causing sequence variants from the many potentially functional variants present in any human genome are urgently needed. Without rigorous standards we risk an acceleration of false-positive reports of causality, which would impede the translation of genomic research findings into the clinical diagnostic setting and hinder biological understanding of disease. Here we discuss the key challenges of assessing sequence variants in human disease, integrating both gene-level and variant-level support for causality. We propose guidelines for summarizing confidence in variant pathogenicity and highlight several areas that require further resource development.
Similar content being viewed by others


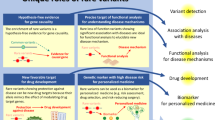
Main
High-throughput sequencing approaches can generate detailed catalogues of genetic variation in both disease patients and the general population. However, for these technologies to have the greatest medical impact we must be able to separate genuine disease-causing or disease-associated genetic variants reliably from the broader background of variants present in all human genomes that are rare, potentially functional, but not actually pathogenic (Box 1) for the disease or phenotype under investigation.
Many, but unfortunately not all, variants that have been causally associated with rare and common genetic disorders represent robust and correct conclusions. False assignments of pathogenicity can have severe consequences for patients, resulting in incorrect prognostic, therapeutic or reproductive advice, and for the research enterprise, resulting in misallocation of resources for basic and therapeutic research. Unfortunately, although the vast majority of genes reported as causally linked to monogenic diseases are true positives, false assignments of causality at the variant level are a substantial issue. One recent analysis of 406 published severe disease mutations observed in 104 newly sequenced individuals reported that 122 (27%) of these were either common polymorphisms or lacked direct evidence for pathogenicity1. Other studies have identified numerous alleged severe-disease-causing variants in the genomes of population controls2,3. In other cases, well-powered follow-up studies of high-profile reported mutations have cast serious doubts on initial reports assigning disease causality to sequence variants4,5, but the vast majority of false-positive findings probably remain undetected. As the volume of patient sequencing data increases it is critical that candidate variants are subjected to rigorous evaluation to prevent further misannotation of the pathogenicity of variants in public databases.
This paper describes the challenges in reliably investigating the role of sequence variants in human disease, and approaches to evaluate the evidence supporting variant causality. It represents the conclusions of a working group of experts in genomic research, analysis and clinical diagnostic sequencing convened by the US National Human Genome Research Institute.
We focus on the application of genome-scale approaches to investigating rare germline variants, defined here as variants with a minor allele frequency of <1%. Our recommendations are most relevant for variants with relatively large effects on disease risk. Our intended scope encompasses the vast majority of variants implicated in severe monogenic diseases as well as rare, large-effect risk variants in complex disease6, but excludes the common, small-effect variants typically identified by genome-wide association studies of complex traits7.
Unambiguous assignment of disease causality for sequence variants is often impossible, particularly for the very low-frequency variants underlying many cases of rare, severe diseases. Consequently, we refer in this manuscript to the concept of implicating a gene or sequence variant: that is, the process of integrating and assessing the evidence supporting a role for that gene or variant in pathogenesis. We emphasize the primacy of strong genetic support for causation for any new gene, which may then be supplemented and extended with ancillary support from functional and informatic studies.
Our recommendations centre on five key areas: study design; gene-level implication; variant-level implication; publication and databases; and implications for clinical diagnosis. Core guidelines for researchers are summarized in Box 2. We also provide a list of factors to consider in the analyses of candidate variants in presumed monogenic diseases (Supplementary Information) and a list of resources for assessing pathogenicity (Supplementary Table 1).
Study design
Investigators seeking to identify pathogenic variants should select technological and analytical approaches based on the most likely genetic architecture of the disease of interest. Rare, high-penetrance protein-coding variants can be cost-effectively captured by exome sequencing, which is rapidly becoming the first-line approach for presumed monogenic disorders8. Cytogenomic arrays and genotyping of linkage panels remain useful approaches for the identification of copy number variation and for identifying co-segregating haplotypes within large Mendelian (especially dominant) disease families, respectively. Optimal approaches to discovering rare pathogenic variants in complex diseases remain unclear: exome sequencing9, deep and low-coverage whole-genome sequencing10 and/or next-generation genotyping arrays with enhanced coverage of protein-coding variants are all being applied in research settings. As the cost of sequencing declines, we expect that deep whole-genome sequencing will soon become the technology of choice for investigating all genetic architectures.
In selecting technological and analytical approaches for a new study, investigators should consider formal power calculations11 incorporating predicted distributions of allele frequencies and effect sizes for pathogenic variants, genetic and phenotypic heterogeneity of available cohorts, population frequency of the disease, and available sample sizes. Although parameter values may be uncertain, current knowledge of the genetics of the disease and similar traits can be used to constrain likely ranges. In particular, for many diseases there is overwhelming evidence that both locus and allelic heterogeneity is high, such as in autism, epilepsy and schizophrenia. A study design that assumes low locus and allelic heterogeneity would be certain to fail for these conditions, and this fact would be revealed by even casual evaluations of power for reasonable genetic models. Gene discovery for conditions with low locus heterogeneity and sufficiently high-penetrance mutations is occasionally possible by sequencing a single family12; however, most gene-discovery applications will require substantially larger sample sizes: multiple unrelated families for rare monogenic conditions, and thousands to tens of thousands of patients and controls for complex disorders9,13.
To assemble large sample sizes will typically require pooling of patient cohorts by multiple investigators. Although such consortium approaches are desirable, investigators should be mindful of systematic differences among cohorts stemming from technical biases, population stratification, and genetic and phenotype heterogeneity. For studies of complex traits, many quality-control methods developed for genome-wide association studies of common variants will also apply to rare variant studies14, but DNA sequencing data face a different and typically more challenging set of quality considerations, particularly when data sets are combined for meta-analysis. In addition, new methods may need to be developed to address population stratification of rare variants15, which show stronger geographic clustering than common variants16; to minimize the impact of stratification, controls should be matched closely to the ancestry of patient samples.
For presumed monogenic diseases, the availability of multiple families with very similar clinical phenotypes substantially increases power for gene discovery. For cases in which there is a single affected proband and no family history, investigators should consider sequencing the unaffected parents of the probands, permitting efficient discovery of de novo mutations and compound heterozygous genotypes. Investigators should begin by examining sequence variation in genes known to be associated with that phenotype, and assessing sequence coverage of the coding sequences and splice junctions for these genes before exploring the possibility of new candidate genes in the affected individuals.
Gene-level implication
To implicate a variant as pathogenic requires that the DNA sequence affected by that variant has a role in the disease process. For genes not previously reported as causal, investigators must simultaneously demonstrate evidence for a role of a candidate gene and one or more variants disrupting it. Even if the candidate gene has been previously implicated in the same or a similar disease phenotype, the overall support from published sources should be carefully assessed and reported. Multiple classes of evidence may potentially contribute to pathogenic inferences at the level of both gene and variant, and include genetic, informatic and experimental data (Table 1 and Supplementary Information). However, in keeping with the history of the field of human genetics, we emphasize the critical primacy of robust statistical genetic support for the implication of new genes, which may then be supplemented with ancillary experimental or informatic evidence supporting a mechanistic role for the gene in the disease in question.
Historically, gene-level implication in monogenic diseases has relied first on identifying a narrow set of candidate genes through genetic data such as linkage analyses or experimental data on biochemical function, and then identifying rare, probably damaging variants (altering the normal levels or biochemical function of a gene or gene product) in one of the candidate genes in multiple affected patients. The increasing availability of large-scale sequencing data now allows genome-scale approaches to gene discovery, in which the distribution of rare, predicted gene-disrupting variants in patients is systematically compared to population controls or well-validated null models to identify genes with an excess of potentially pathogenic variants for clinical and functional follow-up.
It is worth emphasizing that the whole-genome sequence data sets are in some ways more prone to misinterpretation than earlier analyses because of the sheer wealth of candidate causal mutations in any human genome, many of which may provide a compelling story about how the variant may influence the trait; a problem that has been referred to as the ‘narrative potential’ of human genomes17. To avoid such biases the evidence supporting any candidate gene should be contrasted wherever possible with the evidence observed at other presumably non-disease-related genes (for example, by ranking the gene among all others and reporting the probability of a similar or greater contrast being observed by chance). Formal genome-wide statistical approaches to monogenic-disease gene discovery will require considerable methods development, but general guidelines for establishing the significance of variation can be considered here. As we discuss below, these considerations apply equally to assessing the significance of rare variation in common disease studies.
Our paramount recommendation is that for genome-wide analyses of rare variants for both Mendelian and complex disorders, formal calculation of statistical significance should be used to evaluate the strength of evidence of a set of findings, following the well-established standard of maintaining overall type I (false discovery) error rates below 5%. For example, investigators should not simply assume that the presence of two or more independently occurring de novo mutations in the same gene within a sequenced cohort is definitive evidence of a causal role for that gene18,19; such a threshold results in ever increasing numbers of false positives as the number of sequenced cases increases. To illustrate this, consider the recent situation of four exome sequencing studies, involving a total of 945 families with a child affected by autism20,21,22,23, which together observed four independent de novo missense mutations in TTN. Nevertheless, the investigators did not consider TTN to have a causal role in autism, and appropriately so: using a statistical model similar to previously published approaches6,22,24 that accounts for gene size (TTN has the largest coding sequence of any gene in the genome), mutation rate, number of trios and distribution of exome coverage, 1.96 de novo TTN missense or loss-of-function mutations are predicted by chance, which is not significantly different (P = 0.14) from the four observed.
We consider a single gene as the fundamental unit for monogenic disease gene testing, for all disease models; a disease caused by de novo mutations or a disease caused by inherited dominant or recessive variants. An appropriate framework for detecting pathogenic variants will evaluate all of the variation in a gene compared to a well-calibrated null model specific for the hypothesis being considered (for example, de novo, dominant, recessive).
Although the field has well-established guidelines for declaring significance using linkage data25, it is now important to consider a conservative baseline threshold for declaring significance purely from sequencing data of cases, in the absence of other genealogical information. In this scenario, as the gene is the fundamental unit of analysis, and there is no additional data to constrain the search space for genes, a typical study might perform tests on 21,000 protein-coding genes and 9,000 long non-coding RNA genes26,27. A conservative genome-wide significance threshold corresponding to this testing strategy is a Bonferroni-corrected P value of 1.7 × 10−6 (that is, 0.05 out of 30,000). Importantly, if several different schemes are used to define ‘qualifying mutations’ in such analyses, it is necessary to make further statistical adjustments for each of the different sets of rules that are used.
Formal null models can be specified based on the disease model of interest. As mentioned above, the null model for the case of the de novo mutation analysis should consider confounding variables such as sample size, gene size and mutation rate (which may vary by orders of magnitude among genes). We note that such null models have power even for extremely rare conditions and small sample sizes: the first exome sequencing study of Kabuki syndrome28 initially identified 7 de novo loss-of-function variants in the MLL2 gene in just 10 sequenced patients, a finding that is extremely unlikely by chance under the background mutation model described above (P = 1.9 × 10−28) and that provided compelling evidence implicating this gene as causal.
Formal methods for assessing the significance of observations in rare disease cohorts can also be used to assess, for example, the aggregate evidence for segregation of rare variants in a particular gene when considering inherited variation, building on previously published examples29. In this case, the null model should be a population genetic model, for instance, the site frequency spectrum (SFS) of variation constructed from a well-matched control cohort. The null model of the SFS for a given gene should consider both the mutation rate and selective constraint acting on that gene. When evaluating data from a single case, the probability that the variation in a gene is from the null model can by estimated by first identifying the most pathogenic class of variant present in that gene in that case, and then by calculating the probability of sampling a variant of the same class of pathogenicity from the null SFS. Similarly, when the recessive disease model applies, the most pathogenic class of variant on the paternal and maternal haplotypes is identified, and then the probability of sampling both variants from the null SFS is calculated. This testing framework for inherited variants is easily scaled to include multiple disease cases. Ideally, to avoid false positives, the control cohort upon which the SFS is based would be sequenced and analysed in a manner identical to the disease cases.
Such methods may not yet be applicable to every rare disease scenario, and will require work to extend to more exotic inheritance modes such as parental imprinting or obligate compound heterozygosity30. Although formal methods are established to perform these tests rigorously, researchers should at the very least evaluate and report the level of background variation in an implicated gene in population cohorts, taking advantage of public resources such as the Exome Variant Server (http://evs.gs.washington.edu/EVS/) when implicating a new gene in pathogenesis. Furthermore, the analysis of at least some number of controls, sequenced and analysed in a manner identical to cases, can be critical for avoiding the systematic false positives that remain commonplace in exome and genome sequencing.
Just as for genome-wide association studies of common variants14, replication of newly implicated disease genes in independent families or population cohorts is critical supporting evidence, and in most cases essential for a novel gene to be regarded as convincingly implicated in disease. For the rarest disorders additional cases for independent replication may be unavailable and it may be impossible to make a compelling statistical case for implication from human genetic data alone. In these cases, gene implication must be based on an integrated analysis of genetic, informatic and experimental evidence.
Provided that it is carried out in a statistically rigorous fashion, ancillary information can be used to boost power for gene discovery. For example, many genome-wide sequencing-based studies treat all protein-altering variants equally while ignoring all other classes of variants. More elegant schemes aimed at prioritizing based on predicted pathogenicity may boost power for such studies. Another approach is to stratify gene candidates by their expression in a tissue appropriate to the disease under analysis. For example, a recent study combined variant- and gene-level stratification to show that the de novo mutation rate in congenital heart disease was similar in cases versus controls, but the odds ratio rose to 7.5 when focusing on de novo mutations predicted to be damaging and to occur in genes expressed in the developing heart31.
Experimental evidence that can contribute to support for gene implication falls into three broad categories, listed here in order of increasing strength. First, experimental data can be used to demonstrate that the normal function of the gene is consistent with the known biology of the disease process, for example by showing that the gene is expressed in tissues relevant to the disease32, or that its protein product co-localizes with, or physically interacts with, the products of other genes previously implicated in the disease33. Second, investigators can demonstrate that a gene product is functionally disrupted by mutations in patients with the disease of interest, as discussed in the variant-level evidence section below. Lastly, disruption of the candidate gene in a model organism can be shown to result in a phenotype that recapitulates the relevant pathology in humans and is unlikely to occur with disruption of genes selected at random34,35.
A complete description of the experimental methods relevant to gene implication falls outside the scope of this manuscript. However, we note that the value of experimental approaches depends critically on the appropriateness of the model system to the human disorder that is being investigated. Whether cell line or animal models will be most appropriate will depend on context: simple cultured cell models may be inappropriate for developmental disorders affecting complex organ systems. For similar reasons, animal models are not well suited for analysis of human-specific aspects of biology.
As noted above, it is also important to consider the specificity of gene-level support; that is, the probability of observing a similar result if the experiment or analysis was performed with a randomly selected gene. For example, if a new candidate gene is implicated in non-syndromic short stature in humans, observing that its orthologue is associated with small body size in knockout mice is relatively uninformative given that a similar phenotype occurs in over 30% of all knockout mouse strains36. Similarly, reports that the product of a gene potentially implicated in a metabolic disorder is localized to mitochondria should also consider that these are complex organelles with many highly expressed genes. Wherever possible, investigators should use informatics approaches to assess such metrics in publicly available high-throughput data sets of functional genomic and model organism phenotype data37. Although it remains challenging to quantify the statistical confidence of functional observations, those that can be convincingly demonstrated to represent very low-probability events under an appropriate null hypothesis provide more compelling support for implicating a given variant. Even in situations in which a formal statistical framework is not possible we emphasize that researchers must assess functional data rigorously and clearly report their limitations.
Variant-level implication
Genetic evidence implicating a variant must be assessed within the context of the considerable background of rare genetic variants in humans. Even healthy individuals carry many rare protein-disrupting variants38, and about half carry at least one de novo protein-altering mutation39. Such variants are therefore not typically sufficient proof of causality when observed in a disease case, even if present in well-established disease genes: genes differ markedly in their tolerance to variation40 and rare variants predicted to be damaging in disease-associated genes are often observed even in population controls41.
In both established and newly implicated disease genes, investigators should formally assess and report the statistical support for association. Family-based studies should also assess co-segregation of candidate variants with disease status. Given that a separate, unobserved pathogenic mutation may lie on the same haplotype as the candidate variant, segregation analysis alone cannot definitively implicate a specific variant as pathogenic, but (at least under an assumption of complete penetrance) lack of segregation can exclude non-pathogenic variants from consideration.
Informatic and/or experimental evidence for variant implication can be used to assess whether a variant is likely to be deleterious in an evolutionary sense (Box 1), which primarily comes from in silico annotation and comparative genomics42, and predict that a variant is damaging in terms of biological function, arising both from computational predictions and experimental assays. Both categories of evidence can support implication, but they do not necessarily demonstrate a causal role for the variant with respect to the trait under study. Again, we stress that hundreds to thousands of coding variants in an individual will typically be labelled as potentially deleterious or damaging, or both; the strength of the resulting evidence for pathogenicity must be considered in the context of this background level of variation.
Measures of evolutionary sequence conservation are widely used indicators of deleteriousness for both protein-coding and non-coding variation42. Such approaches have demonstrated value in prioritizing candidate variants43,44; however, their predictive power is limited by both statistical and biological factors. Many deleterious variants do not show a strong conservation signature, particularly if the gene has been subject to rapid evolution in the human or primate lineage, or if there have been compensatory substitutions in other regions of the protein in ancestral species45. Conversely, strong conservation can be maintained at sites subject to even relatively weak selective pressure, at which variants may have only small effects on disease risk. The power of these methods also depends on the accuracy and phylogenetic scope of the underlying sequence alignments. These limitations should be taken into account when using predictions of deleteriousness as evidence for implication. Even though it is worthwhile to use multiple prediction algorithms, investigators should avoid treating these as though they represent strong or independent lines of evidence for pathogenicity.
Although some classes of variation, such as truncating or splice-site-disrupting variants in the middle of a protein-coding gene, are more likely to be damaging than others, such variants are also enriched for sequencing and annotation errors and may be rescued by alternative RNA splicing, other variants, or local sequence context41. These possibilities should be assessed, and if possible the predicted damaging effect should be confirmed experimentally.
Experimental approaches to investigating the impact of a sequence variant on gene function, or cell or organism phenotype, can also have a role in demonstrating that a variant is damaging to gene function and in identifying the molecular mechanisms underlying a variant’s effect on disease risk. However, great care must be taken to select appropriate experimental methods, which will depend on the class of variant, biological context (for example, tissue type), access to samples and reagents, desired throughput, time and cost. When a gene has already been confidently implicated in disease, and it is known what class of variant is causal (for instance, loss or gain of function as represented by a specific assay), then an experiment that places a variant of unknown significance into such a functional class can be particularly informative.
Evidence derived directly from patient tissue or cells can often be stronger than that from model systems, particularly (for loss-of-function variants) if the molecular defect can be rescued by complementation in a cellular assay. Replicating disease-relevant phenotypes in a heterologous cell line engineered to carry the proposed causal variant can help to rule out effects of a patient’s genetic background on disease outcome. Weaker but still valuable support can be provided by assays performed in model organisms, more artificial cell culture systems, and non-cellular models such as construct-based assays of altered protein–protein interactions or transcript splicing. Models are most valuable if they directly mimic the predicted functional impact of the candidate variants: for example, knockout mice are better models of recessive loss of function than of dominant missense mutations in a candidate gene. In the case of compound heterozygous recessive inheritance—particularly if the proposed mode of action depends on an interaction between allelic variants, such as in TAR (thrombocytopenia with absent radius) syndrome30—it will be necessary to develop cellular assays that incorporate and assess multiple variants simultaneously.
The impact of variation in non-protein-coding regions of the genome—such as splicing and transcriptional enhancers—remains particularly challenging to interpret, but we note that systematic experimental approaches have begun to both highlight the regions of the human genome most likely to have a role in gene regulation46, and to dissect the potential impact of variation within them47. However, given the challenges of predicting impact for non-coding variants, it remains critical to determine whether the purported pathogenic variant does in fact produce the expected effect on expression or splicing of the affected gene, either by demonstrating an unusual expression level in the patient or by in vitro experimentation (such as minigene constructs).
We caution against the assumption that convincingly implicated variants, even in presumed monogenic disorders, are necessarily fully penetrant (that is, sufficient in isolation to cause disease). In fact the penetrance of most reported disease-associated mutations has not been accurately assessed with current data owing to the biases associated with sample ascertainment. Indeed, the prevalence of reported severe-disease-causing mutations in population controls2,3 suggests that incomplete penetrance, false assignment of pathogenicity, or wider-than-appreciated ranges of expressivity are a substantially more common feature of reported Mendelian disease mutations than generally appreciated. Accurate estimates of penetrance require characterization of reported mutations in large, well-phenotyped population cohorts48,49,50. Further large-scale studies of this kind should be a priority for the field.
We also note the underappreciated importance of calibrating the accuracy of functional assays by large-scale testing of variants confidently established to be non-pathogenic (for example, common missense polymorphisms in the gene of interest). Such experiments establish a baseline estimate for the impact of well-tolerated variants on the assay in question.
Publication and data sharing
As noted above, there are many false positives in disease-mutation databases, stemming largely from erroneous assignment of pathogenicity both in clinical diagnostic laboratories and in the primary literature1,2,51. To reduce this burden will require robust, centralized repositories of mutation data, incorporating explicit, structured evidence for variant pathogenicity and systems for rapid correction of entries. To incentivize both research and clinical laboratories to deposit variation data into open repositories, and to update evidence for or against implication, is a key challenge to be addressed by funding bodies, journals, research consortia, clinical organizations and others52. We are hopeful that such activities can be coordinated around the US National Center for Biotechnology Information (NCBI)’s newly launched ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), which will also interface with existing efforts in this space including the LOVD (Leiden Open (source) Variation Database)53 and other locus-specific databases, OMIM (Online Mendelian Inheritance in Man; http://omim.org/) and DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources)54.
In some cases—such as diseases that are extremely rare or have high degrees of locus heterogeneity—it may be impossible to obtain definitive evidence implicating a specific gene or variant with available sample sizes. In such cases we acknowledge that the suggestive evidence pointing to a gene’s potential implication can nevertheless be valuable in future clinical and research investigations, and should not be excluded from publications or the public domain. However, it is incumbent on investigators, reviewers and journals to be explicit in describing the supporting evidence and the degree of confidence in causality for each proposed gene association and reported variant.
Finally, we emphasize the value of sharing sequence and phenotype data from clinical and research samples to the fullest possible extent. Many investigators and research funders consider responsible data sharing to be a moral and professional imperative55. In many cases, particularly for extremely rare phenotypes, individual laboratories that are not actively recruiting subjects will evaluate only a handful of samples. Sharing of sequence data among testing laboratories has often been restricted, so that many potentially pathogenic mutations and associated phenotypes are known only to individual laboratories. The availability of genome-wide variant calls and detailed clinical phenotype descriptions from such patients in centralized repositories—which will require substantial investment both in informatic infrastructure and new ethical frameworks—would permit more rapid accumulation of evidence for novel genes, and continuous reanalysis to refine the classification of potentially implicated variants and the genotype–phenotype map of human disease. Models for successful data sharing efforts in rare disease already exist in the field of copy number variation with the DECIPHER database54 and the International Standards for Cytogenomic Arrays Consortium (https://www.iscaconsortium.org/), aided by an increasing number of rare-disease resource consortia, and several ambitious efforts to establish clear global standards for genomic data sharing are now underway56.
Added challenges in clinical settings
Although this summary is focused on research, research findings provide the foundation for clinical interpretation. Questionable attributions of causality based on weak research evidence can be readily propagated through research databases and can be misinterpreted clinically as stronger than they truly are. Thus, even researchers who do not explicitly provide diagnosis to patients should be aware that their published findings may be used as support for decisions made in clinical settings.
Clinical laboratories face similar challenges in assessing variant pathogenicity as do researchers, but with the added pressures of diagnostic urgency and the potentially severe consequences of misdiagnosis. Although guidelines are available for variant interpretation in a diagnostic setting57, analytical frameworks for next-generation sequencing data are only beginning to emerge58,59. Responsible application of these technologies will require standards for test validation, variant interpretation and return of results.
The results of genetic and genomic testing are increasingly being used in medical decision-making, including recommendations for prophylactic mastectomy, cardiac defibrillator implantation, tumour therapy and prenatal diagnosis. These actions are neither generally inappropriate nor uniformly incorrect; however, the potential for harm due to misinterpretation of variants is substantial. Although physicians must often make medical decisions using imperfect or ambiguous data, it is critical that healthcare providers be made aware of the varying levels of certainty in the evidence for implicating a variant in disease, both through the consistent use of variant classification terminologies and descriptions of the supporting evidence or lack thereof.
Conclusions
High-throughput DNA sequencing technologies provide unprecedented opportunities to discover new genes and variants underlying human disease, but these discoveries must be rigorously performed and replicated to prevent the proliferation of false-positive findings.
Assessment of evidence for variant implication is a two-step process. First, the overall evidence for implication of a gene should be considered, focusing primarily on the statistical support for implication from genetic analyses, potentially supplemented by ancillary data from informatic sources and functional studies. Second, a combined assessment of the genetic, experimental and informatic support for individual candidate variants should be performed. Such assessments should be performed even if the genes or variants have been previously reported as confidently implicated; prior evidence should be continuously re-evaluated with newly available information.
We urge that, whenever possible, investigators assess the results of genetic, informatic and functional analyses within a quantitative statistical framework, such as determining the probability of the observed distribution of genetic variants in cases and controls under the null hypothesis, and the a priori power to detect variants of a specified frequency and effect size. The specificity of experimental or informatic results provided in support of implication should also be assessed whenever possible by asking how often a similar result would be obtained by chance among a set of random variants or genes. In such analyses investigators should take advantage of the increasing availability of genome-scale sequencing and functional data, and help to build these resources by contributing their findings to public databases.
The community should also focus on the ongoing development of resources in several key areas (Box 3). In particular, major improvements in databases of reported pathogenic mutations, including details of the evidence supporting pathogenicity, are urgently needed. Large-scale experiments to assay previously reported disease-associated mutations in additional large, well-phenotyped populations will also be required to confirm pathogenicity and provide robust evidence of penetrance and expressivity. Finally, extensive work is needed to develop formal statistical frameworks for quantifying the strength of the evidence for implication.
Objective, systematic and quantitative evaluation of the evidence for pathogenicity and sharing of these evaluations and data amongst research and clinical laboratories will maximize the chances that disease-causing genetic variants are correctly differentiated from the many rare non-pathogenic variants seen in all human genomes.
References
Bell, C. J. et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 3, 65ra4 (2011)
Xue, Y. et al. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 91, 1022–1032 (2012)
Norton, N. et al. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet 5, 167–174 (2012)
Weng, L. et al. Lack of MEF2A mutations in coronary artery disease. J. Clin. Invest. 115, 1016–1020 (2005)
Hunt, K. A. et al. Rare and functional SIAE variants are not associated with autoimmune disease risk in up to 66,924 individuals of European ancestry. Nature Genet. 44, 3–5 (2012)
Allen, A. S. et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013)
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009)
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Rev. Genet. 12, 745–755 (2011)
Kiezun, A. et al. Exome sequencing and the genetic basis of complex traits. Nature Genet. 44, 623–630 (2012)
Pasaniuc, B. et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nature Genet. 44, 631–635 (2012)
Li, B., Wang, G. & Leal, S. M. SimRare: a program to generate and analyze sequence-based data for association studies of quantitative and qualitative traits. Bioinformatics 28, 2703–2704 (2012)
Johnston, J. J. et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am. J. Hum. Genet. 90, 295–300 (2012)
Zuk, O. et al. Searching for missing heritability: designing rare variant association studies. Proc. Natl Acad. Sci USA 111, E445–E464 (2014)
Chanock, S. J. et al. Replicating genotype–phenotype associations. Nature 447, 655–660 (2007)
O’Connor, T. D. et al. Fine-scale patterns of population stratification confound rare variant association tests. PLoS ONE 8, e65834 (2013)
Mathieson, I. & McVean, G. Differential confounding of rare and common variants in spatially structured populations. Nature Genet. 44, 243–246 (2012)
Goldstein, D. B. et al. Sequencing studies in human genetics: design and interpretation. Nature Rev. Genet. 14, 460–470 (2013)
de Ligt, J. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 (2012)
Rauch, A. et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682 (2012)
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012)
O'Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012)
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012)
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012)
O'Roak, B. J. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 1619–1622 (2012)
Lander, E. & Kruglyak, L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature Genet. 11, 241–247 (1995)A foundational paper establishing a statistical framework for linkage analysis.
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012)
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012)
Ng, S. B. et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature Genet. 42, 790–793 (2010)
Lemaire, M. et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nature Genet. 45, 531–536 (2013)This paper provides useful statistical approaches to assess the strength of the genetic evidence for a recessive disease gene.
Albers, C. A. et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon–junction complex subunit RBM8A causes TAR syndrome. Nature Genet. 44, 435–439 (2012)
Zaidi, S. et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013)
Lage, K. et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc. Natl Acad. Sci. USA 105, 20870–20875 (2008)
Franke, L. et al. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 78, 1011–1025 (2006)
Boulding, H. & Webber, C. Large-scale objective association of mouse phenotypes with human symptoms through structural variation identified in patients with developmental disorders. Hum. Mutat. 33, 874–883 (2012)
Webber, C. et al. Forging links between human mental retardation-associated CNVs and mouse gene knockout models. PLoS Genet. 5, e1000531 (2009)
Reed, D. R., Lawler, M. P. & Tordoff, M. G. Reduced body weight is a common effect of gene knockout in mice. BMC Genet. 9, 4 (2008)
Giallourakis, C., Henson, C., Reich, M., Xie, X. & Mootha, V. K. Disease gene discovery through integrative genomics. Annu. Rev. Genomics Hum. Genet. 6, 381–406 (2005)
Tennessen, J. A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012)
Veltman, J. A. & Brunner, H. G. De novo mutations in human genetic disease. Nature Rev. Genet. 13, 565–575 (2012)
Bustamante, C. D. et al. Natural selection on protein-coding genes in the human genome. Nature 437, 1153–1157 (2005)
MacArthur, D. G. et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012)
Cooper, G. M. & Shendure, J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nature Rev. Genet. 12, 628–640 (2011)
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nature Methods 7, 248–249 (2010)
Cooper, G. M. et al. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nature Methods 7, 250–251 (2010)
Kondrashov, A. S., Sunyaev, S. & Kondrashov, F. A. Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl Acad. Sci. USA 99, 14878–14883 (2002)
The ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012)
Patwardhan, R. P. et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nature Biotechnol. 30, 265–270 (2012)
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nature Genet. 43, 838–846 (2011)
Bick, A. G. et al. Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am. J. Hum. Genet. 91, 513–519 (2012)
Flannick, J. et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nature Genet. 1380–1385 (2013)One of the first papers to explore systematically the impact of normal human genetic variation in Mendelian disease genes; the paper shows that many previously reported severe disease mutations are not in fact completely penetrant.
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467 1061–1073 (2010) ; corrigendum 473, 544 (2011)
Editorial. Share alike. Nature 490, 143–144 (2012)
Fokkema, I. F. et al. LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat. 32, 557–563 (2011)
Firth, H. V. et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84, 524–533 (2009)
Walport, M. & Brest, P. Sharing research data to improve public health. Lancet 377, 537–539 (2011)
Global Alliance for Genomics and Health Creating a global alliance to enable responsible sharing of genomic and clinical data. http://genomicsandhealth.org/files/public/White%20Paper%20June%203%20final.pdf (2013)
Richards, C. S. et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet. Med. 10, 294–300 (2008)
Gargis, A. S. et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nature Biotechnol. 30, 1033–1036 (2012)
Rehm, H. L. et al. ACMG Clinical Laboratory Standards for Next Generation Sequencing. Genet. Med. (in the press). (2013)
Acknowledgements
This paper was inspired by the deliberations of an expert working group convened by the US National Human Genome Research Institute (NHGRI) on 12 and 13 September 2012 to address the challenges of assigning disease causality to genetic variants. The authors acknowledge B. M. Neale, L. E. Duncan, K. E. Samocha, E. T. Lim and C. G. C. MacArthur for contributions to the manuscript.
Author information
Authors and Affiliations
Contributions
D.G.M., T.A.M. and C.G. planned the project and led the writing group. D.G.M., T.A.M., C.G., D.P.D., H.L.R. and J.S. served as the organizing committee. D.G.M., T.A.M., D.P.D., H.L.R., J.S., G.R.A., D.R.A., R.B.A., S.E.A., E.A.A., J.C.B., L.G.B., D.F.C., G.M.C., N.J.C., M.J.D., M.B.G., D.B.G., J.N.H., S.M.L., L.A.P., J.A.S., S.R.S., D.V., B.F.V., W.W. and C.G. attended a September 2012 workshop, contributed guidelines from their own expertise, and reviewed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Supplementary Table 1. (PDF 155 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-Non-Commercial-ShareAlike 3.0 Unported licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
MacArthur, D., Manolio, T., Dimmock, D. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014). https://doi.org/10.1038/nature13127
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13127
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
