
Abstract
Oil palm is the most productive oil-bearing crop. Although it is planted on only 5% of the total world vegetable oil acreage, palm oil accounts for 33% of vegetable oil and 45% of edible oil worldwide, but increased cultivation competes with dwindling rainforest reserves. We report the 1.8-gigabase (Gb) genome sequence of the African oil palm Elaeis guineensis, the predominant source of worldwide oil production. A total of 1.535 Gb of assembled sequence and transcriptome data from 30 tissue types were used to predict at least 34,802 genes, including oil biosynthesis genes and homologues of WRINKLED1 (WRI1), and other transcriptional regulators1, which are highly expressed in the kernel. We also report the draft sequence of the South American oil palm Elaeis oleifera, which has the same number of chromosomes (2n = 32) and produces fertile interspecific hybrids with E. guineensis2 but seems to have diverged in the New World. Segmental duplications of chromosome arms define the palaeotetraploid origin of palm trees. The oil palm sequence enables the discovery of genes for important traits as well as somaclonal epigenetic alterations that restrict the use of clones in commercial plantings3, and should therefore help to achieve sustainability for biofuels and edible oils, reducing the rainforest footprint of this tropical plantation crop.
Similar content being viewed by others
Main
The genus Elaeis (tribe Cocoseae) is in the family Arecaceae4, one of the oldest families of flowering plants, with fossils dating from the Cretaceous period5. The genus consists of two species, E. guineensis from West Africa6 and E. oleifera from Central and South America. E. guineensis has a higher yield, but E. oleifera has higher unsaturated fatty acid content, lower height, and resistance to disease7. Commercial cultivation of oil palm commenced on the West African coast in the early twentieth century8. In southeast Asia, where it is one of the most important commercial crops, the first recorded oil palm was brought from Africa through Mauritius and Amsterdam in 1848 (ref. 9), when four seedlings were planted as ornamentals in the Bogor Botanical Gardens in Java. Commercial cultivation began in the early twentieth century and despite the long breeding cycle (10 to 12 years) and large land requirement for field trials10, high yield breeding materials (up to 12 tonnes per hectare per year (t ha−1 yr−1) (ref. 9)) have been developed in less than 100 years. As such, the largely undomesticated oil palm is an ideal candidate for genomic-based tools including expressed sequence tags (ESTs)11,12,13 and transcriptome sequencing of the oil palm fruit during development, maturation and ripening1,14 to harness the potential of this remarkably productive crop.
We sequenced the approximately 1.8-Gb E. guineensis genome (AVROS (Algemene Vereniging van Rubberplanter ter Oostkust van Sumatra) pisifera fruit form) to high coverage with a combination of Roche/454 GS FLX Titanium (Roche/454) and Sanger bacterial artificial chromosome (BAC) end sequencing (Supplementary Figs 1–3, Supplementary Tables 1–5 and Methods). The combined total length of the assembly (a fifth genome build or P5-build) is 1.535 Gb. Comparison of the P5-build to genetic linkage maps (Supplementary Fig. 4 and Methods) resulted in 16 genetic scaffolds (one per chromosome; 43% of the P5 scaffold assembly) for a final total of 40,072 scaffolds with an N50 (the scaffold size above which 50% of the total length of the sequence assembly can be found) of 1.27 megabases (Mb) (Supplementary Table 4) and 74% of RefSeq supported genes. For comparison, the genome of E. oleifera was also sequenced with a combination of fragment and linker libraries (Supplementary Table 2 and Methods). Thirty transcriptome libraries were sequenced and assembled, producing 4,528–18,936 isotigs (unique transcript assemblies) per library (Supplementary Fig. 5 and Supplementary Table 6). We sequenced 298,039 reads from methylation-filtered genomic libraries from Deli dura and pisifera genotypes of E. guineensis and E. oleifera (Methods). Methylation-filtered libraries included 90% of the gene models and were enriched 5.6 times for genes with a ‘gene space’ of between 300 and 400 Mb, comparable to that of rice and maize15.
The guanine–cytosine content of the E. guineensis genome (37%) is similar to that of other plant genomes, including the date palm16, but genes were conspicuous for having a much higher guanine–cytosine content (50%). Gene-finding algorithms (Methods) predicted 158,946 gene candidates covering 92 Mb of exonic gene space (5% of the 1.8-Gb genome sequence) (Supplementary Tables 4 and 5). Of these candidates, 34,802 were similar to known proteins at the peptide sequence level with 96% observed in transcriptome data (Methods). Of the remaining 124,144 candidates, 15,311 were identified in transcriptome data (Supplementary Table 4). Known retroelements (67,169) and other transposons (41,664) made up the remaining 108,833 candidates. Comparison to all repetitive element classes resulted in the identification of 775,703 independent genomic regions matching repetitive sequence elements, corresponding to 282 Mb of sequence (or 18% of the P5-build), with 39% guanine–cytosine content (Supplementary Table 4). Repeat content of the unmapped and unassembled contigs was far higher, as expected, and estimated to be approximately half of the 1.53-Gb P5-build, or 57% of the 1.8-Gb E. guineensis genome.
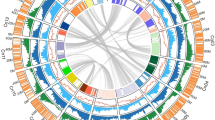
The 16 EG5 chromosomes (Fig. 1) were numbered according to size and compared with previous mapping (Supplementary Table 7) and fluorescence in situ hybridization studies (FISH)17. Gene density (Fig. 1a) was distributed unevenly: five of the smallest six chromosomes had one gene-rich arm, and one repeat-rich arm, as shown previously with FISH17. Known repeat classes matching the TIGR grass repeat database and REPBASE were distributed in gene-poor, methylated regions (Fig. 1b, c), whereas simple di- and trinucleotide repeats17 were mostly within genic regions (Fig. 1d). Potential centromeric regions were identified using an internally repetitive pericentromeric repeat17 (Fig. 1e, purple), whereas highly conserved TTTAGGG telomeric repeat arrays were identified at the extreme ends of 7 of the 32 chromosome arms (Fig. 1e, green). The most prominent 5S ribosomal RNA cluster (Fig. 1e, orange) was found on the largest chromosome, whereas the only telocentric chromosome was one of the two smallest, as described previously17. Two interstitial telomere repeat arrays on chromosomes 2 and 14 were embedded within putative centromeric regions. Robertsonian fusions of telocentric chromosome ends may have given rise to these two chromosomes18, and date palm has 18 chromosomes16, consistent with this hypothesis. Typical of monocot genomes19, the most abundant repetitive elements were copia (33%) and gypsy (8%) retroelements, as well as other long terminal repeat (LTR) retrotransposons (6%) (Supplementary Fig. 6 and Supplementary Methods). Interestingly, 47% of all repeats observed were uncharacterized previously, with 73% absent from E. oleifera and 99% absent from Musa acuminata (banana). The distribution of repeats in methylation-filtered reads indicated that RIRE1 and other copia elements are especially heavily methylated.

E. guineensis has 16 chromosome pairs, ordered by size, which correspond to 16 linkage groups identified by genetic mapping (Supplementary Table 7). Tracks displayed are: a, gene density; b, methyl-filtered read density; c, retroelement density; d, simple sequence repeats; e, low copy number repetitive elements, including telomere repeat TTTAGGG (green), 5S rRNA (orange) and pericentromeric repeats (purple); f, regional G-C content (range 0.3–0.45); g, genetically mapped scaffolds from the P5-build; and h, segmental duplications. Densities for telomere repeats are exaggerated for visual clarity.
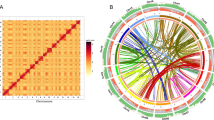
Comparison of E. guineensis chromosomes to each other revealed that oriented homologous duplicated sequences (segmental duplications) are notably abundant (Fig. 1 and Supplementary Fig. 7). Analysis of conserved gene order revealed that the duplications were retained in E. oleifera, so that segmental duplications pre-dated the divergence of the African and South American oil palm (Supplementary Fig. 8a). Given that most of the genome is represented by segmental duplications, and not triplications, we conclude that oil palm is a palaeotetraploid, in line with speculation based on cytogenetics and restriction fragment length polymorphism (RFLP) mapping17,20. These duplications do not span the putative pericentromeric regions (Fig. 1), indicating that most centromeres arose after polyploidization, consistent with extensive chromosome restructuring. Homologues of 94.4%, 83.5% and 80.2% of the genes from Phoenix dactylifera, M. acuminata and Arabidopsis respectively, were found in E. guineensis (Fig. 2a, Supplementary Table 8 and Methods). Each E. guineensis duplication matched unique scaffolds in the date palm genome (Supplementary Fig. 8b), indicating that date palms have most of the segmental duplications found in oil palm. Polyploidization has been inferred by chromosome counts in only a limited number of Arecaceae21, and a likely scenario is that the progenitor of both palms arose as a polyploid. We performed a similar analysis of the banana genome22 and found extensive synteny between each oil palm chromosome and several chromosomes from banana (Supplementary Fig. 9), confirming that duplication events in the M. acuminata genome occurred after the Musa–Elaeis split, as proposed previously22.

a, Venn Diagram illustrating the proportion of shared gene family clusters in E. guineensis, M. acuminata, A. thaliana and P. dactylifera. Number of genes (clusters) compared were: A. thaliana, 27,416 (15,399); M. acuminata, 36,529 (16,874); P. dactylifera, 28,882 (15,979) and E. guineensis, 34,802 (16,374). b, Gene ontology classifications of oil palm and date palm. c, Ratio of gene number (oil palm:date palm) in each gene ontology classification. Deg, degradation; FA, fatty acid; hexoses-P, hexose phosphate pathway; mal and pyr met, malate and pyruvate metabolism; mt, mitochondrial; OPP, oxidative pentose phosphate; pt PLP syn, plastidial phospholipid synthesis; TAG, triacylglycerol; TPs, transporters.
The 34,802 sequence similarity gene predictions (Supplementary Table 4) were annotated for gene ontology terms (Fig. 2b, c), with a focus on oil biosynthesis (Methods). Oil synthesis in the kernel commences at 11 to 12 weeks after anthesis (WAA) and is complete by 15 to 16 WAA at which stage mesocarp oil synthesis starts, reaching a peak at 20 WAA23. In plants, de novo fatty acid synthesis (FAS) is compartmentalized in plastids, whereas triacylglycerol (TAG) synthesis occurs in the cytoplasm. Although the oil palm accumulates markedly higher TAG than the date palm1, the number of genes involved in TAG biosynthesis is notably similar in both palms. In contrast, FAS genes have higher representation in the oil palm genome (Fig. 2b). Apart from the transcriptomes of 30 tissues (Fig. 3a, b, Supplementary Fig. 5 and Supplementary Table 6), in-depth sequencing of kernel and mesocarp (Fig. 3c and Supplementary Table 9) indicated that the transcriptome signatures are similar in both tissues with plastidial FAS genes upregulated compared to TAG. Thus, the enzymes of the Kennedy pathway for TAG assembly must cope with the high flux of de novo fatty acids in oil palm. FAS genes were upregulated markedly just before the onset, and then declined during the peak of lipid accumulation. This contrasts with previous reports that FAS transcripts continue to increase in the mesocarp1. This may reflect the tenera fruit form used in this study, as dura has a longer maturation period (22 WAA)24 than tenera (20 WAA)23,25. WRI1 regulates oil accumulation in the oil palm mesocarp1 and we found highest mesocarp expression of WRI1 just before lipid accumulation onset. However, kernel showed 75-fold higher WRI1 expression compared to mesocarp (Fig. 3c and Supplementary Table 9), implying its pivotal role in kernel oil synthesis. LEAFY COTYLEDON1 (LEC1), LEC2, ABSCISIC ACID INSENSITIVE3 (ABI3) and FUSCA3, which activate WRI1 in oilseeds, were not found in mesocarp transcriptomes, but were well-represented in the kernel, with LEC1 and ABI3 showing higher expression at the start and completion of oil accumulation, respectively. Interestingly, the transcriptional regulator PICKLE (PKL) was expressed in both the mesocarp and kernel (Supplementary Table 9).

a, b, Number of lipid-synthesis-related (a) and carbohydrate-synthesis-related (b) gene transcripts in different tissues. c, Comparison of gene-expression levels between kernel and mesocarp tissue before and at peak of oil accumulation.
Genes involved in sucrose degradation and the oxidative pentose phosphate pathway were more highly represented in oil palm than date palm (Fig. 2b). Pentose phosphates are recycled into glucose 6-phosphate to fuel glycolytic pathways, and import of these cytosolic metabolites requires specific transporters on the plastid envelope. Although these transporter genes are upregulated strongly in oil palm1, they are similarly represented in date and oil palm genomes (Fig. 2b). Thus, channelling of sugars destined for oil synthesis is regulated at the transcriptome level in oil palm. Additional insights important to TAG biosynthesis, fruit ripening and abscission are provided in Supplementary Notes.
To place palms on the evolutionary tree, evidence-based gene models from each species were combined with a previous seed plant data set26 to form a matrix of 1,685 gene partitions (858,954 patterns) and 107 taxa. P. dactylifera, E. guineensis and E. oleifera are present in 1,206, 1,229 and 1,190 partitions, respectively. All three were well separated from other monocots (Fig. 4), including nearest neighbours Musa (banana), Curcuma (turmeric) and Zingiber (ginger). Phylogenetic dating using conservative constraints (Methods) predicted a divergence 65 million years (Myr) ago between date and oil palm, and 51 Myr between E. oleifera and E. guineensis. This is comparable with divergence between Old and New world relatives such as African Sorghum bicolor (sorghum) and American Zea mays (maize) panicoid grasses (26 Myr). Unlike maize and sorghum, however, E. guineensis and E. oleifera give rise to fertile hybrids2, consistent with the vicariant hypothesis for phylogeographical divergence, in which geographically isolated species are under no selective pressure to evolve reproductive isolation27.

A carefully annotated subset of proteins from E. guineensis, E. oleifera and P. dactylifera were included in a matrix of 1,685 gene partitions (858,954 patterns) and 107 taxa. This matrix is extracted from partitions with at least 30 taxa present in a much larger matrix. A maximum likelihood tree of monocotyledonous taxa is shown, along with bootstrap values when less than 100 (Methods). Scale bar indicates the mean number of substitutions per site.
The genome sequence of oil palm will be a rich resource for oil palm breeders, geneticists and evolutionary biologists alike. It has revealed that palms are ancient tetraploids, and that the African and South American species probably diverged in the Old and New Worlds. Over-represented genes in lipid and carbohydrate metabolism are expressed differentially in mesocarp and kernel, accounting for the different properties of palm fruit and palm kernel oils. The genome sequence will also enable mapping of somaclonal epigenetic alterations that restrict the use of clones in commercial plantings. The dense representation of sequenced scaffolds on the genetic map will facilitate identification of genes responsible for important yield and quality traits. The genome sequence of this tropical plantation crop is an important step in achieving the goals that are critical to the sustainability challenges associated with growing demands for biofuels and edible oils.
Methods Summary
Genome sequencing and assembly, genetic mapping and gene annotation
Sequencing reads (Roche/454) were generated from genomic and BAC pool DNA fragment and paired-end linker libraries (Supplementary Figs 1–3 and Supplementary Tables 1–3). BAC end sequencing was performed using the Sanger method. Sequence data were assembled as described (Methods). The genetic map for the selfed Nigerian tenera palm T128 was constructed as described (Methods). Gene predictions, genome and protein comparisons and gene ontology annotation were performed as described (Methods).
Transcriptome sequencing
Thirty transcriptome libraries were constructed and transcriptome sequences were generated (Roche/454) (Methods). Transcriptome libraries from kernel and mesocarp were constructed and deep sequenced by Illumina HiSeq 2000 (Methods).
Methylation filtration
Methylation filtration libraries were constructed as described15 and resulting clones were sequenced using the Sanger method (Methods).
Phylogenetic analysis
Open reading frames were assembled from evidence-based gene models and combined with a large seed plant data set26(Methods).
Online Methods
Genome assembly
The assembly of E. guineensis (AVROS, pisifera fruit form) genome P5-build was constructed from sequences from a total of 148 linker libraries and 81 fragment libraries (Roche/454). Reads were generated from genomic DNA fragment libraries (53.5 million reads), BAC pool DNA fragment libraries (3.6 million reads), and from a series of genomic (89.1 million reads) and BAC (8.6 million reads) paired-end linker libraries. In total, 46.8 billion bases of raw sequence were generated, representing approximately 26-fold raw sequence coverage of the 1.8-Gb oil palm genome. Sequence data were assembled using the Newbler algorithm28 (Supplementary Table 4). The seventh assembly and genome build of E. oleifera (O7-build) was constructed from a total of 127 linker libraries and 68 fragment libraries. In total, 130 million E. oleifera reads, representing approximately 25-fold raw sequence coverage, were generated (Supplementary Table 2).
High information content restriction fragment fingerprints were generated for 124,286 BAC clones from the E. guineensis genome with an average size of 150 kb. BAC fingerprints were used to construct a physical map of the reference genome (Supplementary Figs 2 and 3). In addition, BAC ends from this library were sequenced using Sanger sequencing to generate 235,613 paired reads with an average read length of approximately 600 bp. BAC end reads were used to create the shotgun assembly, as well as to locate individual BACs within the assembled genome.
Before assembly, all Roche/454 sequence runs were screened for quality based on average read length, linker library efficiency and library redundancy using a custom pipeline based on the SSAHA program29. For linker positive reads, library insert sizes were estimated by aligning both ends to a draft assembly of oil palm, and measuring the intervening sequence in cases in which both ends matched a single draft scaffold. In order to minimize the negative impact of chimaeras in the linker libraries, we removed all identical reads that were due to library redundancy, not independent observations. In addition to the Roche/454 data, a set of 235,613 BAC end reads from the Origen_1 BAC library were included in the P5 assembly, with an average BAC size of 150 kb. These data were assembled using the Newbler algorithm28 on a Dell PowerEdge R910 server with 512 Gb of RAM, and 32 cores running Ubuntu Linux 10.04. The P5 assembly took 15 days and 5 h to complete.
Genetic map construction
Two genetic maps were constructed. The first mapping population was derived from the self-pollination of the Nigerian tenera palm T128. The mapping population and map-construction methodology are described elsewhere30. The second mapping population comprised 87 palms obtained from a cross between Ulu Remis Deli dura (ENL48) and Yangambi pisifera (ML161) grown at the FELDA Research Station at Jerantut, Malaysia. Linkage analysis for the dura × pisifera (P2) cross was performed using both JoinMap 4.0 and GenStat 14th edition. JoinMap was used to examine markers and identify loci showing distorted segregation (chi-squared test). JoinMap was initially used to construct the two parental framework maps at recombination frequency ≤ 0.2 and a nearest neighbour stress value of ≤ 4 (centimorgan) using the maximum likelihood mapping algorithm as described31. The density of the linkage maps was later increased by mapping additional co-dominant markers into the parental framework maps using the maximum likelihood mapping algorithm in GenStat 14. The integrated map was built using the maximum likelihood mapping method in GenStat14 by combining data from markers on both the two parental maps. Comparison between the integrated map and the parental maps was visualized using MapChart 2.2 (ref. 32).
Genetic-map integration and chromosome-sequence construction
The 1,511 markers used in the generation of the T128_codominant and P2_DxP maps were compared to scaffolds from the P5-build using the exonerate33 program with an ungapped alignment and a minimum identity match of 97%. Markers that did not uniquely map to P5 scaffolds were discarded, and one scaffold marker ordering was created for each of the two maps. After reviewing shared scaffolds between the two maps, a final order was determined for ordering 169 scaffolds, and ordering and orienting 124 scaffolds based on multiple markers. The scaffold sequences were then concatenated in order and reverse complemented as required to create 16 linkage groups based chromosome sequences. During map integration, LG15 (T128_codominant naming convention) appeared significantly shorter than a previous integration based on the P4 assembly with the P2_regression map. This was owing to map instability introduced into the P2_DxP map generation in which the P2_regression was more stable. After review, the LG15 chromosome sequence was extended based on mapping of P5 scaffolds onto the P4/P2_regression version.
Gene identification and annotation
Based on the longest set of scaffolds representing 10% of the original P1-build, we used the SNAP34 gene finder to identify initial candidate gene predictions for the E. guineensis genome. Initial SNAP runs were performed using the rice (Os.hmm) gene model. Initial genes discovered were compared with the RefSeq35 database as well as the TIGR Gramineae repeat database in order to remove retroelements and pseudogenes. The remaining transcripts were then screened for missing start and stop codons, as well as other warnings from SNAP. The remaining set was then used as input to the SNAP programs FATHOM and FORGE according to the SNAP documentation to create a new hidden Markov model (HMM) with greater specificity to E. guineensis. The same screening process was applied again and four iterations of the training were used resulting in the final ‘pisif_2_22_11.hmm’ gene model.
For the Glimmer35 predictions, assembled E. guineensis transcriptome sequences (groups A, C, D and G in Supplementary Table 6) were translated from start to stop with a size selection ranging from 500 to 5,000 nucleotides. These were then compared to complete coding sequences for Magnoliophyta from GenBank using BLASTX (E-value < 10−10). Transcripts with homology starting at position one of the Magnoliophyta targets were selected for further analysis. The transcripts were then screened using BLASTClust (NCBI) and CD-HIT36 to reduce the number of genes to meet Glimmer’s training requirements. Exon boundaries were determined by mapping to the previous P4-build, and were used to create an E. guineensis HMM.
Transcriptome analysis
Reads (Roche/454) from each library and from all libraries (Fig. 3a, b and Supplementary Table 6) were assembled into isotigs respectively. The isotigs from each library were blasted on A. thaliana gene models with a threshold of E-value < 10−5. The best hit A. thaliana gene model was assigned to the homologue of the query isotig. Based on Bourgis’ annotation1, copy numbers were given to each gene in each category group. The final copy numbers of each functional group were scaled on the number of genes in each group. To estimate expression level of genes in mesocarp and kernel tissues, Illumina HiSeq 2000 reads from each library were mapped to assembled isotigs from all E. guineensis reads by using the Burrows–Wheeler Aligner. Gene group expression levels were calculated as the number of mapped reads on each isotig divided by the total number of isotigs, multiplied by 100,000, and scaled by the number of genes in each gene group. Both copy number and read coverage were the mean of measures from two biological replicates. Data were analysed as described above for Roche/454 data, except that expression levels were calculated as transcripts per million tags.
Methylation-filtered library analysis
Methylation-filtered (‘GeneThresher’) genomic DNA libraries were constructed as described15,37 to select unmethylated clones (depleted of most repetitive sequences38) by propagation in McrBC+ strains of Escherichia coli. Briefly, nuclear DNA was extracted individually from young leaves of Deli dura and AVROS pisifera of E. guineensis and from E. oleifera. For each of the three DNA populations, genomic shotgun libraries were constructed as described37. Sequences were generated from one end of each cloned insert by ABI 3730 sequencing (Life Technologies), generating 298,039 reads (73,390 from Deli dura, 101,327 from AVROS pisifera and 123,322 from E. oleifera).
Segmental duplication analysis
Chromosomes from the EG5-linked assembly were screened in a self–self comparison test using the MUMmer3 set of tools39. Final alignments were carried out on chromosome pairs using the PROmer program (optional parameters – d 0.5 –c 200), alignments were reviewed visually with the MUMmer plot program, and approximate boundaries for segmental duplications were recorded (Supplementary Fig. 7 and Supplementary Table 7).
To test for the existence of observed segmental duplications in other genomes, we performed comparative genomics of each half of the 16 segmental duplications in E. guineensis with the E. oleifera and P. dactylifera scaffold sets. Comparisons were performed using a custom analysis pipeline based on the MUMmer program (optional parameters –n –l 30 –b –c –L). MUMmer output was summarized as an offset-sorted overlap plot showing where query scaffolds share local alignment with a reference chromosome. Output was reviewed to verify that each half of the proposed segmental duplication was present in the query genome, and that the scaffolds matching were different for each half of the duplication in E. guineensis.
Genome comparison by gene models
NCBI TBLASTN program was used to compare A. thaliana, O. sativa, P. dactylifera and E. guineensis predicted proteins on the genomes of each of the four species, with a threshold of E-value < 10−5. At this level of conservation, matches represent shared gene families between the query and target genomes. By comparing predicted proteins to genome sequence, biases introduced by gene prediction methods are minimized compared to a direct gene-level comparison. Comparisons between the gene models from one species and its own genome are close to, but less than 100%, owing to the limit of sensitivity of TBLASTN at this E-value cutoff. Public database sources for comparative genome sequences and gene models are provided in Supplementary Methods.
Gene clustering and Venn diagram
CD-HIT clustering algorithm36 was used to look for homologous protein sequences among A. thaliana, O. sativa, P. dactylifera and E. guineensis at 40% similarity level. This algorithm avoids all-versus-all BLAST search by using a short word filter.
Gene ontology annotation
E. guineensis gene model protein sequences were queried against A. thaliana annotated gene model protein sequences by using BLASTP40 with a threshold of E-value < 10−5. The blast output file was then loaded into Blast2GO41. Blast2GO performed gene ontology annotation by using an annotation rule on the found ontology terms. The most specific annotations were assigned on each sequence with default parameters in the annotation rule.
Phylogenetic analysis
A carefully annotated set of open reading frames was assembled from evidence-based gene models in each species. The oil palm open reading frames were combined with a large seed plant data set published previously26, with updated gene models for Arabidopsis thaliana, Oryza sativa and Solanum lycopersicum, as well as the addition of P. dactylifera, M. acuminata, Carica papaya and Selaginella molendorffii (outgroup) for a total of 107 taxa. Orthologues were identified using OrthologID42, and a sub-matrix with at least 30 taxa present per partition was extracted for phylogenetic analysis. This sub-matrix contains 1,685 gene partitions with 858,954 patterns. Maximum likelihood analysis was carried out using RAxML43 with the JTT+F+Γ model and 100 bootstrap replicates.
Accession codes
Accessions
BioProject
GenBank/EMBL/DDBJ
Data deposits
The E. guineensis and E. oleifera BioProjects are available for download at http://genomsawit.mpob.gov.my and have been registered at the NCBI under BioProject accessions PRJNA192219 and PRJNA183707. The Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accessions ASJS00000000 (E. guineensis) and ASIR00000000 (E. oleifera).The versions described in this paper are versions ASJS00000000 and ASIR00000000.
Change history
14 August 2013
A GenBank accession number was corrected.
References
Bourgis, F. et al. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc. Natl Acad. Sci. USA 108, 12527–12532 (2011)
Hardon, J. J. & Tan, G. Y. Interspecific hybrids in the genus Elaeis I. crossability, cytogenetics and fertility of F1 hybrids of E. guineensis × E. oleifera. Euphytica 18, 372–380 (1969)
Jaligot, E. et al. Epigenetic imbalance and the floral developmental abnormality of the in vitro-regenerated oil palm Elaeis guineensis. Ann. Bot. 108, 1453–1462 (2011)
Dransfield, J. et al. Genera Palmarum: The Evolution and Classification of Palms (Royal Botanic Gardens Kew, 2008)
Purseglove, J. W. In Tropical Crops (Monocotyledons) 416–510 (Longman, 1972)
Zeven, A. C. The origin of the oil palm. J. Niger. Inst.Oil Palm Res. 4, 218–225 (1965)
Cochard, B., Amblard, P. & Durand-Gasselin, T. Oil palm genetic improvement and sustainable development. Oleagineux Corps Gras Lipides 12, 141–147 (2005)
Hartley, C. The Oil Palm 47–94 (Longman, 1988)
Corley, R. H. V. & Tinker, P. B. In The Oil Palm 4th edn, 1–26 (Blackwell Science, 2003)
Mayes, S., Jack, P. L., Corley, R. H. & Marshall, D. F. Construction of a RFLP genetic linkage map for oil palm (Elaeis guineensis Jacq.). Genome 40, 116–122 (1997)
Jouannic, S. et al. Analysis of expressed sequence tags from oil palm (Elaeis guineensis). FEBS Lett. 579, 2709–2714 (2005)
Ho, C. –L. et al. Analysis and functional annotation of expressed sequence tags (ESTs) from multiple tissues of oil palm (Elaeis guineensis Jacq.). BMC Genomics 8, 381 (2007).
Low, E. T. L. et al. Oil palm (Elaeis guineensis Jacq.) tissue culture ESTs: identifying genes associated with callogenesis and embryogenesis. BMC Plant Biol. 8, 62 (2008)
Tranbarger, T. J. et al. Regulatory mechanisms underlying oil palm fruit mesocarp maturation, ripening, and functional specialization in lipid and carotenoid metabolism. Plant Physiol. 156, 564–584 (2011)
Palmer, L. E. et al. Maize genome sequencing by methylation filtration. Science 302, 2115–2117 (2003)
Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nature Biotechnol. 29, 521–527 (2011)
Castilho, A. M., Vershinin, A. V. & Heslop-Harrison, J. S. Repetitive DNA and the chromosomes in the genome of oil palm (Elaeis guineensis). Ann. Bot. 85, 837–844 (2000)
Richards, E. J., Chao, S., Vongs, A. & Yang, J. Characterization of Arabidopsis thaliana telomeres isolated in yeast. Nucleic Acids Res. 20, 4039–4046 (1992)
Du, J. et al. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 63, 584–598 (2010)
Singh, R. et al. Identification of cDNA RFLP markers and their use for molecular mapping in oil palm. Asia Pac. J. Mol. Biol. Biotechnol. 16, 53–63 (2008)
Wood, T. E. et al. The frequency of polyploid speciation in vascular plants. Proc. Natl Acad. Sci. USA 106, 13875–13879 (2009)
D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012)
Sambanthamurthi, R., Sundram, K. & Tan, Y. Chemistry and biochemistry of palm oil. Prog. Lipid Res. 39, 507–558 (2000)
Bafor, M. E. & Osagie, A. U. Changes in lipid class and fatty acid composition during maturation of mesocarp of oil palm (Elaeis guineensis) variety dura. J. Sci. Food Agric. 37, 825–832 (1986)
Shaarani, S. M., Cárdenas-Blanco, A., Amin, M. H. G., Soon, N. G. & Hall, L. D. Monitoring development and ripeness of oil palm fruit (Elaeis guneensis) by MRI and bulk NMR. Int. J. Agric. Biol. 12, 101–105 (2010)
Lee, E. K. et al. A functional phylogenomic view of the seed plants. PLoS Genet. 7, e1002411 (2011)
Riggins, C. W. & Seigler, D. S. The genus Artemisia (Asteraceae: Anthemideae) at a continental crossroads: molecular insights into migrations, disjunctions, and reticulations among Old and New World species from a Beringian perspective. Mol. Phylogenet. Evol. 64, 471–490 (2012)
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005)
Ning, Z., Cox, A. J. & Mullikin, J. C. SSAHA: a fast search method for large DNA databases. Genome Res. 11, 1725–1729 (2001)
Singh, R. et al. The oil palm SHELL gene controls oil yield and encodes a homologue of SEEDSTICK. Nature http://dx.doi.org/10.1038/nature12356 (this issue)
Jansen, J. Construction of linkage maps in full-sib families of diploid outbreeding species by minimizing the number of recombinations in hidden inheritance vectors. Genetics 170, 2013–2025 (2005)
Voorrips, R. E. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002)
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005)
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004)
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004)
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006)
Bedell, J. A. et al. Sorghum genome sequencing by methylation filtration. PLoS Biol. 3, e13 (2005)
Ouyang, S. & Buell, C. R. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 32, D360–D363 (2004)
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005)
Chiu, J. C. et al. OrthologID: automation of genome-scale ortholog identification within a parsimony framework. Bioinformatics 22, 699–707 (2006)
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006)
Acknowledgements
We acknowledge the contributions of N. Ahmad, M. Marjuni and N. Abdullah for sampling of oil palm materials. We thank MoGene, GeneWorks, Beijing Genome Institute, Tufts University Core Facility and Macrogen for sequencing services. We thank D. Stevenson for discussions. We appreciate the constant support of Y. M. Choo, and the Ministry of Plantation Industries and Commodities, Malaysia. The project was endorsed by the Cabinet Committee on the Competitiveness of the Palm Oil Industry (CCPO) and funded by the Malaysian Palm Oil Board. E.K.L., R.D. and R.A.M. are supported by a grant from NSF 0421604 (Genomics of Comparative Seed Plant Evolution).
Author information
Authors and Affiliations
Contributions
R.S., M.O.-A., E-T.L.L. and R.S. conceptualized the research programme. R.S., M.O.-A., E.-T.L.L., R.N., N.L., J.M.O., S.W.S., R.K.W., R.S.F., R.A.M. and R.S. designed experiments and coordinated the project. R.S., M.O.A., E.-T.L.L., M.A.A.M., L.C.-L.O., S.-E.O., J.N., B.B., M.A.B., S.W.S., J.M.O. and R.S. conducted laboratory experiments and were involved in data analysis. E.-T.L.L., R.R., K.-L.C., M.A.H., N.A., S.W.S., D.H. and M.H. performed bioinformatics analyses. E.K.L. and R.D. carried out phylogenetic analysis. D.K., J.L.G. and R.A.W. built BAC libraries and carried out BAC end sequencing. R.S., M.O.A., E.-T.L.L., R.N., N.L., S.W.S., J.M.O., R.A.M. and R.S. participated in preparing and revising the manuscript. R.A.M. and R.S. supervised data generation and analysis.
Corresponding authors
Ethics declarations
Competing interests
R.S.F., R.K.W. and R.A.M. are consultants for Orion Genomics LLC. R.K.W. serves on the Board of Directors for Orion Genomics LLC.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-9, Supplementary Tables 1-9, Supplementary Methods, Supplementary Notes and Supplementary references. (PDF 8053 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-Non-Commercial-ShareAlike 3.0 Unported licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Singh, R., Ong-Abdullah, M., Low, ET. et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 500, 335–339 (2013). https://doi.org/10.1038/nature12309
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12309
This article is cited by
-
EgMADS3 directly regulates EgLPAAT to mediate medium-chain fatty acids (MCFA) anabolism in the mesocarp of oil palm
Plant Cell Reports (2024)
-
Interaction between phenylpropane metabolism and oil accumulation in the developing seed of Brassica napus revealed by high temporal-resolution transcriptomes
BMC Biology (2023)
-
Multiplex CRISPR/Cas9 gene-editing platform in oil palm targeting mutations in EgFAD2 and EgPAT genes
Journal of Genetic Engineering and Biotechnology (2023)
-
Molecular insights into the VIRESCENS amino acid sequence and its implication in anthocyanin production in red- and yellow-fruited cultivars of date palm
Scientific Reports (2023)
-
Identification and Analysis of MADS-Box Genes Expressed in the Mesocarp of Oil Palm Fruit (Elaeis guineensis Jacq.)
Biochemical Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
