
Abstract
Most great ape genetic variation remains uncharacterized1,2; however, its study is critical for understanding population history3,4,5,6, recombination7, selection8 and susceptibility to disease9,10. Here we sequence to high coverage a total of 79 wild- and captive-born individuals representing all six great ape species and seven subspecies and report 88.8 million single nucleotide polymorphisms. Our analysis provides support for genetically distinct populations within each species, signals of gene flow, and the split of common chimpanzees into two distinct groups: Nigeria–Cameroon/western and central/eastern populations. We find extensive inbreeding in almost all wild populations, with eastern gorillas being the most extreme. Inferred effective population sizes have varied radically over time in different lineages and this appears to have a profound effect on the genetic diversity at, or close to, genes in almost all species. We discover and assign 1,982 loss-of-function variants throughout the human and great ape lineages, determining that the rate of gene loss has not been different in the human branch compared to other internal branches in the great ape phylogeny. This comprehensive catalogue of great ape genome diversity provides a framework for understanding evolution and a resource for more effective management of wild and captive great ape populations.
Similar content being viewed by others

Main
We sequenced great ape genomes to a mean of 25-fold coverage per individual (Table 1, Supplementary Information and Supplementary Table 1) sampling natural diversity by selecting captive individuals of known wild-born origin as well as individuals from protected areas in Africa (Fig. 1a). We also included nine human genomes—three African and six non-African individuals11 . Variants were called using the software package GATK (ref. 12) (Methods), applying several quality filters, including conservative allele balance filters, and requiring that genomes showed <2% contamination between samples (Methods and Supplementary Information). In order to assess the quality of single nucleotide variant (SNV) calls, we performed three sets of independent validation experiments with concordance rates ranging from 86% to 99% depending on allele frequency, the great ape population analysed and the species reference genome used (Supplementary Information and Supplementary Table 2). In total, we discovered 84.0 million fixed substitutions and 88.8 million segregating sites of high quality (Table 1 and Supplementary Table 3), providing the most comprehensive catalogue of great ape genetic diversity to date. From these variants we also constructed a list of potentially ancestry-informative markers (AIMs) for each of the surveyed populations, although a larger sampling of some subspecies is still required (Supplementary Information).

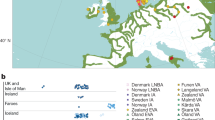
a, Geographical distribution of great ape populations across Indonesia and Africa sequenced in this study. The formation of the islands of Borneo and Sumatra resulted in the speciation of the two corresponding orangutan populations. The Sanaga River forms a natural boundary between Nigeria–Cameroon and central chimpanzee populations whereas the Congo River separates the bonobo population from the central and eastern chimpanzees. Eastern lowland and western lowland gorillas are both separated by a large geographical distance. b, Heterozygosity estimates of each of the individual species and subspecies are superimposed onto a neighbour-joining tree from genome-wide genetic distance estimates (branch lengths in units of substitutions per bp). Arrows indicate heterozygosities previously reported30 for western and central chimpanzee populations. c, Runs of homozygosity among great apes. The relationship between the coefficient of inbreeding (FROH) and the number of autozygous >1 megabase segments is shown. Bonobos and eastern lowland gorillas show an excess of inbreeding compared to the other great apes, suggesting small population sizes or a fragmented population. d, Genetic structure based on clustering of great apes. All individuals (columns) are grouped into different clusters (K = 2 to K = 6, rows) coloured by species and according to their common genetic structure. Most captive individuals, labelled on top, show a complex admixture from different wild populations. A signature of admixture, for example, is clearly observed in the known hybrid Donald, a second-generation captive predicted to be a 15% admixture of central chimpanzee on a western background consistent with its pedigree. A grey line at the bottom denotes new groups at K = 6 in agreement with the location of origin or ancestral admixture.
We initially explored the genetic relationships between individuals by constructing neighbour-joining phylogenetic trees from both autosomal and mitochondrial genomes (Supplementary Information). The autosomal tree identified separate monophyletic groupings for each species or subspecies designation (Supplementary Fig. 8.5.1) and supports a split of extant chimpanzees into two groups. Nigeria–Cameroon and western chimpanzees form a monophyletic clade (>97% of all autosomal trees); central and eastern chimpanzees form a second group (72% of all autosomal trees).
Genome-wide patterns of heterozygosity (Fig. 1b) reveal a threefold range in single nucleotide polymorphism (SNP) diversity. Non-African humans, eastern lowland gorillas, bonobos and western chimpanzees show the lowest genetic diversity (∼0.8 × 10−3 heterozygotes per base pair (bp)). In contrast, central chimpanzees, western lowland gorillas and both orangutan species show the greatest genetic diversity (1.6 × 10−3 – 2.4 × 10−3 heterozygotes per bp). These differences are also reflected by measures of inbreeding from runs of homozygosity13 (Fig. 1c and Supplementary Information). Bonobos and western lowland gorillas, for example, have similar distributions of tracts of homozygosity as human populations that have experienced strong genetic bottlenecks (Karitiana and Papuan). Eastern lowland gorillas appear to represent the most inbred population, with evidence that they have been subjected to both recent and ancient inbreeding.
To examine the level of genetic differentiation between individuals we performed a principal component analysis (PCA) of SNP genotypes (Supplementary Information). Chimpanzees were stratified between subspecies with PC1 separating western and Nigeria–Cameroon chimpanzees from the eastern and central chimpanzees and PC2 separating western and Nigeria–Cameroon chimpanzees. In gorillas, PC1 clearly separates eastern and western gorillas, whereas the western lowland gorillas are distributed along a gradient of PC2, with individuals from the Congo and western Cameroon positioning in opposite directions along the axis. The isolated Cross River gorilla is genetically more similar to Cameroon western lowland gorillas and can be clearly differentiated with PC3 (Supplementary Fig. 8.2.9).
We explored the level of shared ancestry among individuals within each group14 using an admixture model (FRAPPE). In chimpanzees, the four known subspecies are clearly distinguished when fitting the model using four ancestry components (K = 4) (Fig. 1d). Additional substructure is identified among the eastern chimpanzees Vincent and Andromeda (K = 6), who hail from the most eastern sample site (Gombe National Park, Tanzania). As in Gonder et al.2, we have identified three Nigeria–Cameroon samples (Julie, Tobi and Banyo, K = 3–5) with components of central chimpanzee ancestry. However, taking central chimpanzees and the remaining Nigeria–Cameroon chimpanzees as ancestral populations shows no evidence of gene flow by either the F3 statistic or HapMix. This indicates that these three samples are not the result of a recent admixture and may represent a genetically distinct population (Supplementary Information).
In gorillas, following the separation of eastern and western lowland species (K = 2), an increasing number of components further subdivide western lowland populations distinguishing Congolese and Cameroonian gorillas—a pattern consistent with the structure observed in the PCA analysis (Supplementary Fig. 8.2.9). One striking observation is the extent of admixed ancestry predicted for captive individuals when compared to wild-born. Our analysis suggests that most captive individuals included in this study are admixed from two or more genetically distinct wild-born populations leading to an erosion of phylogeographic signal. This finding is consistent with microsatellite analyses of captive gorillas15 and the fact that great ape breeding programs have not been managed at the subspecies level.
As great apes have been evolving on separate lineages since the middle Miocene, we attempted to reconstruct the history of these various species and subspecies by applying methods sensitive to branching processes, changes in effective population size (Ne), and gene flow occurring at different time scales. Using a combination of speciation times inferred from a haploid pairwise sequential Markovian coalescent (PSMC) analysis16, a coalescent hidden Markov model (CoalHMM)3 and incomplete lineage sorting approaches, we were able to estimate the most ancient split times and effective population sizes among the great ape species. By combining these estimates with an approximate Bayesian computation (ABC)17 analysis applied to the more complex chimpanzee phylogeny, we constructed a composite model of great ape population history over the last ∼15 million years (Fig. 2). This model presents a complete overview of great ape divergence and speciation events in the context of historical effective population sizes.

Population splits and effective population sizes (Ne) during great ape evolution. Split times (dark brown) and divergence times (light brown) are plotted as a function of divergence (d) on the bottom and time on top. Time is estimated using a single mutation rate (μ) of 1 × 10−9 mut bp−1 year−1. The ancestral and current effective population sizes are also estimated using this mutation rate. The results from several methods used to estimate Ne (COALHMM, ILS COALHMM, PSMC and ABC) are coloured in orange, purple, blue and green, respectively. The chimpanzee split times are estimated using the ABC method. The x axis is rescaled for divergences larger than 2 × 10−3 to provide more resolution in recent splits. All the values used in this figure can be found in Supplementary Table 5. The terminal Ne correspond to the effective population size after the last split event.
PSMC analyses of historical Ne (Fig. 3) suggests that the ancestral Pan lineage had the largest effective population size of all lineages >3 million years ago (Myr), after which time the population of the common ancestor of both bonobos and chimpanzees experienced a dramatic decline. Both PSMC and ABC analyses support a model of subsequent increase in chimpanzee Ne starting ∼1 Myr, before their divergence into separate subspecies. Following an eastern chimpanzee increase in Ne (∼500 thousand years ago, kyr), the central chimpanzees reached their zenith ∼200–300 kyr followed by the western chimpanzee ∼150 kyr. Although the PSMC profiles of the two subspecies within each of the major chimpanzee clades (eastern/central and Nigeria–Cameroon/western) closely shadow each other between 100 kyr and 1 Myr, the western chimpanzee PSMC profile is notable for its initial separation from that of the other chimpanzees, followed by its sudden rise and decline (Fig. 3 and Supplementary Information). The different gorilla species also show variable demographic histories over the past ∼200 kyr. Eastern lowland gorillas have the smallest historical Ne, consistent with smaller present-day populations and a history of inbreeding (Fig. 1c). A comparison of effective population sizes with the ratio of non-synonymous to synonymous substitutions finds that selection has acted more efficiently in populations with higher Ne, consistent with neutral theory (Supplementary Information).

Inferred historical population sizes by pairwise sequential Markovian coalescent analysis. The lower x axis gives time measured by pairwise sequence divergence and the y axis gives the effective population size measured by the scaled mutation rate. The upper x axis indicates scaling in years, assuming a mutation rate ranging from 10−9 to 5 × 10−10 per site per year. The top left panel shows the inference for modern human populations. In the rest of the three panels, thin light lines of the same colour correspond to PSMC inferences on 100 rounds of bootstrapped sequences.
Although the phylogeny of bonobos and western, central and eastern common chimpanzees has been well established based on genetic data18, there is still uncertainty regarding their relationship to Nigeria–Cameroon chimpanzees2,19. Regional neighbour-joining trees and a maximum-likelihood tree estimated from allele frequencies both show that Nigeria–Cameroon and western chimpanzees form a clade. A complex demographic history has been previously reported for chimpanzees with evidence of asymmetrical gene flow among different subspecies. For instance, migration has been identified from western into eastern chimpanzees4, two subspecies that are currently geographically isolated. We find support for this using the D-statistic, a model-free approach that tests whether unequal levels of allele sharing between an outgroup and two populations that have more recently diverged (D(H,W;E,C)>16 s.d.). However, no previous genome-wide analysis that has examined gene flow included chimpanzees from the Nigeria–Cameroon subspecies and a comparison of them with eastern chimpanzees results in a highly significant D-statistic (D(H,E;W,N)>25 s.d.). Furthermore, TreeMix, a model-based approach that identifies gene flow events to explain allele frequency patterns not captured by a simple branching phylogeny, infers a signal of gene flow between Nigeria–Cameroon and eastern chimpanzees (P = 2 × 10300). A more detailed treatment of gene flow applying different models and methods may be found in the Supplementary Information.
Genetic diversity is depressed at or close to genes in almost all species (Supplementary Fig. 11.1) with the effect less pronounced in subspecies with lower estimated Ne, consistent with population genetic theory. When we compare the relative level of X chromosome and autosomal (X/A) diversity across great apes as a function of genetic distance from genes, the eastern lowland gorillas and Bornean orangutans are outliers, with substantially reduced X/A diversity compared to the neutral expectation of 0.75, regardless of the distance to genes. This pattern is consistent with a recent reduction in effective population size20, clearly visible in the PSMC analysis for both species (Fig. 3). However, bonobos also demonstrate a relatively constant level of X/A diversity regardless of distance from genes, with values very much in line with neutral expectations. All other subspecies demonstrate a pattern consistent with previous studies in humans21 where X/A diversity is lower than 0.75 close to genes and higher farther away from genes.
It has been proposed that loss of gene function may represent a common evolutionary mechanism to facilitate adaptation to changes in an environment22. There has been speculation that the success of humans may have, in part, been catalysed by an excess of beneficial loss-of-function mutations23. We thus characterized the distribution of fixed loss-of-function mutations among different species of great apes identifying nonsense and frameshift mutations resulting from SNVs (n = 806) and indels (n = 1080) in addition to gene deletion events (n = 96) (Supplementary Table 4). We assigned these events to the phylogeny and determined that the number of fixed loss-of-function mutations scales proportionally to the estimated branch lengths (R2 = 0.987 SNVs, R2 = 0.998 indels). In addition, we found no evidence of distortion on the terminal branches of the tree compared to point mutations based on a maximum likelihood analysis (Supplementary Information). Thus, the human branch in particular showed no excess of fixed loss-of-function mutations even after accounting for human-specific pseudogenes24 (Supplementary Information).
Our analysis provides one of the first genome-wide views of the major patterns of evolutionary diversification among great apes. We have generated the most comprehensive catalogue of SNPs for chimpanzees (27.2 million), bonobos (9.0 million), gorillas (19.2 million) and orangutans (24.3 million) (Table 1) to date and identified several thousand AIMs, which provides a useful resource for future analyses of ape populations. Humans, western chimpanzees and eastern gorillas all show a remarkable dearth of genetic diversity when compared to other great apes. It is striking, for example, that sequencing of 79 great ape genomes identifies more than double the number of SNPs obtained from the recent sequencing of more than a thousand diverse humans25—a reflection of the unique out-of-Africa origin and nested phylogeny of our species.
We provide strong genetic support for distinct populations and subpopulations of great apes with evidence of additional substructure. The common chimpanzee shows the greatest population stratification when compared to all other lineages with multiple lines of evidence supporting two major groups: the western and Nigeria–Cameroon and the central and eastern chimpanzees. The PSMC analysis indicates a temporal order to changes in ancestral effective population sizes over the last two million years, previous to which the Pan genus suffered a dramatic population collapse. Eastern chimpanzee populations reached their maximum size first, followed by the central and western chimpanzee. The Nigeria–Cameroon chimpanzee population size appears much more constant.
Despite their rich evolutionary history, great apes have experienced drastic declines in suitable habitat in recent years26, along with declines in local population sizes of up to 75% (ref. 27). These observations highlight the urgency to sample from wild ape populations to more fully understand reservoirs of genetic diversity across the range of each species and to illuminate how basic demographic processes have affected it. The >80 million SNPs we identified in this study may now be used to characterize patterns of genetic differentiation among great apes in sanctuaries and zoos and, thus, are of great importance for the conservation of these endangered species with regard to their original range. These efforts will greatly enhance conservation planning and management of apes by providing important information on how to maintain genetic diversity in wild populations for future generations.
Methods Summary
We sequenced to a mean coverage of 25× (Illumina HiSeq 2000) a total of 79 great ape individuals, representing 10 subspecies and four genera of great apes from a variety of populations across the African continent and Southeast Asia. SNPs were called using GATK12 after BWA28 mapping to the human genome (NCBI Build 36) using relaxed mapping parameters. Samples combined by species were realigned around putative indels. SNP calling was then performed on the combined individuals for each species. For indels, we used the GATK Unified Genotyper to produce an initial set of indel candidates applying several quality filters and removing variants overlapping segmental duplications and tandem repeats. We also removed groups of indels clustering within 10 bp to eliminate possible artefacts in problematic regions. Conservative allelic imbalance filters were used to eliminate false heterozygotes that may affect demographic analyses, some of which are sensitive to low levels of contamination. We estimate that the application of this filter resulted in a 14% false negative rate for heterozygotes. Our multispecies study design facilitated this assessment of contamination, which may remain undetected in studies focused on assessing diversity within a single species. The amount of cross-species contamination was estimated from the amount of non-endogenous mitochondrial sequence present in an individual. Because we wished to compare patterns of variation between and within species, we report all variants with respect to coordinates of the human genome reference. For FRAPPE analyses, we used MAF0.06 (human, orangutan and bonobo) and 0.05 (chimpanzee and gorilla) to remove singletons. For most of the analyses, we only used autosomal markers, except in the X/A analysis. To determine the amount of inbreeding, we calculated the heterozygosity genome-wide in windows of 1 megabase with 200-kilobase sliding windows. We then clustered together the neighbouring regions to account for runs of homozygosity. For the PSMC analyses, we called the consensus bases using SAMtools29. Underlying raw sequence data are available through the Sequence Read Archive (SRA) (PRJNA189439 and SRP018689). Data generated in this work are available from (http://biologiaevolutiva.org/greatape/). A complete description of the materials and Methods is provided in the Supplementary Information.
Accession codes
Primary accessions
Sequence Read Archive
Referenced accessions
Sequence Read Archive
References
Gonder, M. K. et al. A new west African chimpanzee subspecies? Nature 388, 337 (1997)
Gonder, M. K. et al. Evidence from Cameroon reveals differences in the genetic structure and histories of chimpanzee populations. Proc. Natl Acad. Sci. USA 108, 4766–4771 (2011)
Hobolth, A., Christensen, O. F., Mailund, T. & Schierup, M. H. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 3, e7 (2007)
Hey, J. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 27, 921–933 (2010)
Becquet, C. & Przeworski, M. A new approach to estimate parameters of speciation models with application to apes. Genome Research 17, 1505–1519 (2007)
Mailund, T. et al. A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species. PLoS Genet. 8, e1003125 (2012)
Coop, G. & Przeworski, M. An evolutionary view of human recombination. Nature Rev. Genet. 8, 23–34 (2007)
Bustamante, C. D. et al. Natural selection on protein-coding genes in the human genome. Nature 437, 1153–1157 (2005)
Hahn, B. H. AIDS as a zoonosis: scientific and public health implications. Science 287, 607–614 (2000)
Keele, B. F. et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313, 523–526 (2006)
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012)
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 43, 491–498 (2011)
Pemberton, T. J. et al. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 91, 275–292 (2012)
Tang, H., Peng, J., Wang, P. & Risch, N. J. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 28, 289–301 (2005)
Nsubuga, A. M., Holzman, J., Chemnick, L. G. & Ryder, O. A. The cryptic genetic structure of the North American captive gorilla population. Conserv. Genet. 11, 161–172 (2010)
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011)
Beaumont, M. A., Zhang, W. & Balding, D. J. Approximate Bayesian computation in population genetics. Genetics 162, 2025–2035 (2002)
Morin, P. A. et al. Kin selection, social structure, gene flow, and the evolution of chimpanzees. Science 265, 1193–1201 (1994)
Bjork, A., Liu, W., Wertheim, J. O., Hahn, B. H. & Worobey, M. Evolutionary history of chimpanzees inferred from complete mitochondrial genomes. Mol. Biol. 28, 615–623 (2011)
Pool, J. E. & Nielsen, R. Population size changes reshape genomic patterns of diversity. Evolution 61, 3001–3006 (2007)
Hammer, M. F. et al. The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nature Genet. 42, 830–831 (2010)
Olson, M. V. & Varki, A. Sequencing the chimpanzee genome: insights into human evolution and disease. Nature Rev. Genet. 4, 20–28 (2003)
Olson, M. V. When less is more: gene loss as an engine of evolutionary change. Am. J. Hum. Genet. 64, 18–23 (1999)
Wang, X., Grus, W. E. & Zhang, J. Gene losses during human origins. PLoS Biol. 4, e52 (2006)
The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010)
Junker, J. et al. Recent decline in suitable environmental conditions for African great apes. Divers. Distrib. 18, 1077–1091 (2012)
Campbell, G., Kuehl, H., N’Goran Kouamé, P. & Boesch, C. Alarming decline of West African chimpanzees in Côte d’Ivoire. Curr. Biol. 18, R903–R904 (2008)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
The Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005)
Acknowledgements
We thank the following funding agencies: ERC Starting Grant (260372) to T.M.-B.; NIH grants HG002385 to E.E.E., R01_HG005226 to K.R.V., A.E.W., M.F.H., L.S. and J.D.W., GM100233 and NSF HOMINID grant 1032255 to D.R. and He.Li.; MICINN (Spain) BFU2011-28549 to T.M.-B., BFU2010-19443 to Ja.Be., Spanish Government and FEDER for grants BFU2009-13409-C02-02 and BFU2012-38236 to A.N. and J.P.-M., Direcció General de Recerca, Generalitat de Catalunya (Grup de Recerca Consolidat 2009 SGR 1101) to Ja.Be., D.C., A.N. and T.M.-B.; ERC Advanced Grant (233297) and Max Planck Society to S. Paabo; Danish Council for Independent Research Natural Sciences to H.S.; Spanish Grant (CGL-2010-20170) and Zoo de Barcelona (Beca PRIC) to A.R.-H.; EUPRIM-Net to BPRC; DP1ES022577-04 NIH grant to S.A.T.; NSF Grant 0755823 to M.K.G.; P.G. is supported by the G. Harold and Leila Y. Mathers Foundation. A.N. and T.M.-B. are ICREA Research Investigators (Institut Catala d’Estudis i Recerca Avancats de la Generalitat de Catalunya). J.P.-M. is supported by the Zoo de Barcelona and l’Ajuntament de Barcelona. P.H.S. is supported by an HHMI International Student Fellowship. E.E.E. is an investigator with the Howard Hughes Medical Institute. We are especially grateful to all those who generously provided the samples for the project: O. Thalmann and H. Siedel from Limbe Sanctuary; R. Garriga from Tacugama Sanctuary; W. Schempp (University of Freiburg), Burgers' Zoo; Zoo of Antwerp; Wilhelma Zoo; Givskud Zoo; Ngamba Island Chimpanzee Sanctuary and Centre de Primatologie; Centre International de Recherches Médicales de Franceville; North Carolina Zoological Park; Zoo Atlanta; the Lincoln Park Zoo (Chicago); the Antwerp Zoo and the Limbe Wildlife Centre (Cameroon); D. Travis from University of Minnesota and M. Kinsel from University of Illinois Urbana-Champaign and S. Paabo and L. Vigilant, Max Planck Institute for Evolutionary Anthropology. We thank T. Brown for revising the manuscript, L. Capilla and E. Eyras for technical support, and M. Dierssen for comments on genes expressed in the brain.
Author information
Authors and Affiliations
Contributions
E.E.E. and T.M.-B. designed the study. J.P.-M., P.H.S., J.M.K., J.L.K., B.L.-G., M.D., M.F.-C., J.C.M., C.D.B., E.E.E. and T.M.-B. analysed the raw data and performed the variant calling. J.P.-M., P.H.S., Ma.Ma., J.H.-R., I.H.-H., T.C., C.B., L.V., A.R.-H. and C.C. validated the different variants. J.P.-M., P.H.S., B.L.-G., C.A., F.H., E.E.E. and T.M.-B. analysed large deletions. K.R.V., L.J., A.E.W. and M.F.H. analysed the X/Autosome diversity. D.T., G.S., A.C., C.T., F.C., Ha.La., K.P., M.P., M.L., N.P., D.C., Ja.Be., A.N. and A.M.A. performed selection analyses. J.P.-M., P.H.S., J.M.K., J.L.K., T.D.O’C., He.Li., D.R., K.M., A.H., A.E.H., M.H.S., C.H., J.M.A., T.M., C.D.B., E.E.E. and T.M.-B. analysed different aspects of demography. M.L.W., L.S., T.A., I.K., A.P., F.L., J.A.K., E.L., P.G., H.S., M.K.G., S.A.T., R.A.B., R.E.B., O.A.R. and B.H.H. provided critical samples and participated in the discussion of phylogeny. L.F., R.K.W., Ju.Bl., E.E.E., Ma.Ma., L.A.-C., M.G. and I.G.G. generated genome libraries and produced the genome sequence associated with this project. All authors contributed to data interpretation. J.P.-M., P.H.S., E.E.E. and T.M.-B. drafted the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Underlying raw sequence data are available through the Sequence Read Archive (SRA) (PRJNA189439 and SRP018689).
Supplementary information
Supplementary Information
This file contains Supplementary Text, Figures, Tables and References in Supplementary Sections 1-13 – see contents for details. (PDF 9409 kb)
Supplementary Data
This file contains Supplementary Tables 1-5. (XLSX 354 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-Non-Commercial-ShareAlike 3.0 Unported licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Prado-Martinez, J., Sudmant, P., Kidd, J. et al. Great ape genetic diversity and population history. Nature 499, 471–475 (2013). https://doi.org/10.1038/nature12228
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12228
This article is cited by
-
Comparative genomic analyses provide new insights into evolutionary history and conservation genomics of gorillas
BMC Ecology and Evolution (2024)
-
Generation of chimpanzee induced pluripotent stem cell lines for cross-species comparisons
In Vitro Cellular & Developmental Biology - Animal (2024)
-
A common flanking variant is associated with enhanced stability of the FGF14-SCA27B repeat locus
Nature Genetics (2024)
-
CEGA: a method for inferring natural selection by comparative population genomic analysis across species
Genome Biology (2023)
-
Malaria-driven adaptation of MHC class I in wild bonobo populations
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
