
Abstract
About 8,000 years ago in the Fertile Crescent, a spontaneous hybridization of the wild diploid grass Aegilops tauschii (2n = 14; DD) with the cultivated tetraploid wheat Triticum turgidum (2n = 4x = 28; AABB) resulted in hexaploid wheat (T. aestivum; 2n = 6x = 42; AABBDD)1,2. Wheat has since become a primary staple crop worldwide as a result of its enhanced adaptability to a wide range of climates and improved grain quality for the production of baker’s flour2. Here we describe sequencing the Ae. tauschii genome and obtaining a roughly 90-fold depth of short reads from libraries with various insert sizes, to gain a better understanding of this genetically complex plant. The assembled scaffolds represented 83.4% of the genome, of which 65.9% comprised transposable elements. We generated comprehensive RNA-Seq data and used it to identify 43,150 protein-coding genes, of which 30,697 (71.1%) were uniquely anchored to chromosomes with an integrated high-density genetic map. Whole-genome analysis revealed gene family expansion in Ae. tauschii of agronomically relevant gene families that were associated with disease resistance, abiotic stress tolerance and grain quality. This draft genome sequence provides insight into the environmental adaptation of bread wheat and can aid in defining the large and complicated genomes of wheat species.
Similar content being viewed by others

Main
We selected Ae. tauschii accession AL8/78 for genome sequencing because it has been extensively characterized genetically (Supplementary Information). Using a whole genome shotgun strategy, we generated 398 Gb of high-quality reads from 45 libraries with insert sizes ranging from 200 bp to 20 kb (Supplementary Information). A hierarchical, iterative assembly of short reads employing the parallelized sequence assembler SOAPdenovo3 achieved contigs with an N50 length (minimum length of contigs representing 50% of the assembly) of 4,512 bp (Table 1). Paired-end information combined with an additional 18.4 Gb of Roche/454 long-read sequences was used sequentially to generate 4.23-Gb scaffolds (83.4% were non-gapped contiguous sequences) with an N50 length of 57.6 kb (Supplementary Information). The assembly represented 97% of the 4.36-Gb genome as estimated by K-mer analysis (Supplementary Information). We also obtained 13,185 Ae. tauschii expressed sequence tag (EST) sequences using Sanger sequencing, of which 11,998 (91%) could be mapped to the scaffolds with more than 90% coverage (Supplementary Information).
To aid in gene identification, we performed RNA-Seq (53.2 Gb for a 117-Mb transcriptome assembly) on 23 libraries representing eight tissues including pistil, root, seed, spike, stamen, stem, leaf and sheath (Supplementary Information). Using both evidence-based and de novo gene predictions, we identified 34,498 high-confidence protein-coding loci. FGENESH4 and GeneID models were supported by a 60% overlap with either our ESTs and RNA-Seq reads, or with homologous proteins. More than 76% of the gene models had a significant match (E value ≤ 10−5; alignment length ≥ 60%) in the GenBank non-redundant database. An additional 8,652 loci were predicted as low-confidence genes as a result of incomplete gene structure or limited expression data support (Supplementary Information). We also predicted a total of 2,505 transfer RNA, 358 ribosomal RNA, 35 small nuclear RNA and 78 small nucleolar RNA genes (Supplementary Information).
We found that more than 65.9% of the Ae. tauschii genome was composed of different transposable element (TE) families (Supplementary Information). About 5 × 106 Illumina reads of Ae. tauschii were mapped to hexaploid wheat repetitive sequences and we found that a comparable percentage of reads (more than 62.3%) could be classified as part of a TE sequence (Supplementary Fig. 6). This estimate is similar to that derived from a previous survey of Roche/454 sequences5. There were 410 different TE families, of which the 20 most abundant contributed more than 50% of the Ae. tauschii genome (Supplementary Table 9). A single peak of increased insertion activity was estimated to occur about 3–4 Myr ago by measuring the similarity of the assembled LTR retrotransposons (Supplementary Information), suggesting that the expansion of the Ae. tauschii genome was relatively recent and coincided with the abrupt climate change during the Pliocene Epoch6.
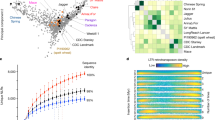
We constructed a high-density genetic map using an F2 population of 490 individuals derived from a cross between the Ae. tauschii accessions Y2280 and AL8/78. The map, whose total length was 1059.8 centimorgans (cM), consisted of 151,083 single nucleotide polymorphism (SNP) markers developed by restriction-site-associated DNA (RAD) tag sequencing technology (Supplementary Fig. 13). Together with bin-mapped wheat ESTs7, SNPs and tags8, the genetic map was used to align 30,303 scaffolds (1.72 Gb; 30,697 genes) to chromosomes (Supplementary Information). The Ae. tauschii genes and scaffolds were also anchored to barley9 and Brachypodium chromosome maps10 (Fig. 1 and Supplementary Fig. 17). Calculation of Ka/Ks ratios (the ratio of non-synonymous substitutions to synonymous substitutions) for pairs of conserved orthologous genes showed that the average values between Ae. tauschii and barley (20,892 genes), Brachypodium (17,231 genes), rice (16,370 genes) and sorghum (18,623 genes) were 0.2214, 0.1888, 0.1736 and 0.1726, respectively, which indicated that most gene lineages evolved under purifying selection in Ae. tauschii. A total of 628 genes exhibited Ka/Ks ratios of more than 0.8 when compared with the other four species, indicating potential positive selection (innermost circle of Fig. 1). These genes were assigned to a wide range of molecular functions by using Gene Ontology (GO) analyses (Supplementary Table 14).

The inner circle represents the seven Ae. tauschii chromosomes scaled according to the genetic map incorporating genome scaffolds. Red points show the Ka/Ks ratios between anchored Ae. tauschii genes and their putative orthologues in Brachypodium. Moving outwards, the second circle compares Ae. tauschii against the seven barley chromosomes9. The heatmaps show the density distribution of barley cDNA loci that are aligned with Ae. tauschii genes. The outer two circles illustrate Brachypodium chromosomes according to conserved synteny with Ae. tauschii. The coloured lines below each chromosome identify putative orthologous gene pairs between Ae. tauschii genes, barley genes and Brachypodium genes.
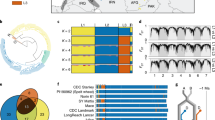
Ae. tauschii proteins were clustered with those of Brachypodium, rice, sorghum and barley (full-length complementary DNAs), and formed 23,202 orthologous groups (at least two members; Supplementary Information). In total, we identified 11,289 (barley/Ae. tauschii) and 14,675 (Brachypodium/Ae. tauschii) orthologous gene pairs. We found that 8,443 gene groups contained sequences from all five grass genomes, and 234 were specific to Pooideae (Ae. tauschii, Brachypodium and barley) and 587 were specific to Triticeae (Ae. tauschii and barley) (Fig. 2a). Enrichment analyses of both Pfam domains and GO terms showed that genes encoding NBS-LRR (nucleotide-binding-site leucine-rich repeat) proteins were over-represented in Ae. tauschii relative to Brachypodium and rice11,12 (Supplementary Information). These observations are consistent with those reported in a recent study13. A total of 1,219 Ae. tauschii genes were similar to NBS-LRR genes (R gene analogues (RGAs))11,14 (Supplementary Information). This number is double that in rice (623) and sixfold that in maize (216)12, indicating that the RGA family has substantially expanded in Ae. tauschii. We mapped 878 RGAs (72%) to specific positions across wheat chromosomes by using molecular marker–genome sequence alignment, which provides a large number of potential disease resistance loci for further investigation.

a, Distribution of orthologous gene families in Ae. tauschii, Brachypodium, sorghum, rice and barley. The number of gene families is represented in each intersection of the Venn diagram. The first number under the species name indicates the total number of genes annotated for a particular species, and the second indicates the number of genes in groups for that organism. The difference between the two accounts for singleton genes that were not present in any cluster. b, The composition of transcription factors (TFs) in Ae. tauschii and Brachypodium composed of more than 30 members.
We found more genes for the cytochrome P450 family in Ae. tauschii (485) than in sorghum (365), rice (333), Brachypodium (262) or maize (261). This family of genes is important for abiotic stress response, especially in biosynthetic and detoxification pathways15. Using 178 manually curated cold-acclimation-related genes such as the CCAAT-binding factor (CBF) transcription factors16, late-embryogenesis-abundant proteins (LEA) and osmoprotectant biosynthesis proteins (Supplementary Information) as queries, we identified 216 cold-related genes in the Ae. tauschii genome, in contrast to 164 genes in Brachypodium, 132 in rice, 159 in sorghum and 148 in maize. Some of these genes were specific to Ae. tauschii or to Pooideae, including those encoding ice-recrystallization inhibition protein 1 precursor, DREB2 transcription factor α isoform and cold-responsive LEA/RAB-related COR protein. Expression analysis of RNA-Seq data showed that most of these Ae. tauschii-specific and Pooideae-specific genes were constitutively expressed in Ae. tauschii (Supplementary Fig. 23). In addition, 1,489 transcription factors (TFs) in 56 families were identified by using Pfam DNA-binding domains (Supplementary Information). Ae. tauschii had an excess of such TFs as MYB-related genes (103, in contrast with 66 in Brachypodium and 95 in maize), and these are also thought to be involved in various stress responses17. The M-type MADS-box genes (58, in contrast with 23 in Brachypodium and 34 in maize) are involved in regulation in plant reproduction18 (Fig. 2b and Supplementary Table 18). ARACNe19 co-expression analysis using RNA-Seq data predicted an expression network of 1,283 interactions (Supplementary Fig. 25), in which 13 TFs were associated with the expression of drought tolerance genes20 (Supplementary Table 20).
We predicted a total of 159 (133 families) previously undescribed microRNAs (Supplementary Information), and identified segmental and tandem duplications in 42 members of the miR2118 family that were organized into two groups on 15 scaffolds (Supplementary Fig. 26). The miR399 family, which is involved in the regulation of inorganic phosphate homeostasis in rice21, was expanded (20 members in Ae. tauschii, compared with 11 in rice and 10 in maize), and may contribute to the ability of Ae. tauschii to grow in low-nutrient soils. The expansion of the miR2275 family (eight members in Ae. tauschii, compared with two in rice and four in maize) may contribute to the enhanced disease resistance of Ae. tauschii because phased short interfering RNAs initiated by miR2275 have been implicated in these activities22.
The Ae. tauschii genome served as the source for many grain quality genes in hexaploid wheat, creating a step improvement in the formation of the elastic dough essential for bread making2. Grain quality genes include high-molecular-weight glutenin subunits (HMW-GS), low-molecular-weight glutenin subunits (LMW-GS)23, grain texture proteins (GSP; puroindolines)24 and storage protein activator (SPA)25. We identified two HMW-GS genes, five LMW-GS genes, one Pina gene, two Pinb genes, one GSP gene and one SPA gene in the Ae. tauschii genome sequence (Supplementary Information). As has been shown for the Hardness (Ha) locus24, the GSP, Pina and Pinb genes were also organized in a cluster. RNA-Seq analysis showed that these grain quality genes were expressed predominantly in seeds (Supplementary Fig. 29).
The anchoring of more than 40% of the scaffold sequences to four genetic maps and to syntenic regions of other sequenced grass species provided a structural framework for integrating multiple maps by using shared markers (Fig. 1 and Supplementary Information). The co-localization of genes in scaffolds and genetically mapped quantitative trait loci (QTLs) will directly support map-based gene cloning. On chromosome 2D, for example, the locations of 33 QTLs or genes were integrated with scaffold information (http://ccg.murdoch.edu.au/cmap/ccg-live/) (Fig. 3 and Supplementary Information). Alignment of the Ae. tauschii genetic map with the wheat 2D consensus genetic map was unambiguous, with the exception of some single crossovers that were probably due to repetitive elements (dotted lines in Fig. 3). The genome sequence also provided the basis for the identification of more than 860,126 simple sequence repeats (SSRs), with trimers (37.7%) and tetramers (27.5%) as the most abundant SSR types (Supplementary Information). Together with the 711,907 SNPs identified by resequencing a roughly fivefold coverage of a second accession, Y2280 (Supplementary Information), the genomic resources reported here will promote map-based gene cloning and marker-assisted selection in wheat.

The Ae. tauschii genetic map was integrated with markers, scaffolds and mapped QTLs to assist in marker development and map-based cloning. Left: the Ae. tauschii molecular map used for synteny alignment in Fig. 1 was aligned to chromosome 2D (November 2011 consensus map, CMap; http://ccg.murdoch.edu.au/cmap/ccg-live) where sequence information was available. The original marker at a location is retained in CMap as a synonym. Right: within CMap, details for QTL locations are provided at a greater magnification to show all the markers in the regions of interest. The dotted lines indicate an ambiguous relationship that is most probably due to repetitive sequences.
With its high base accuracy and nearly complete set of gene sequences, the Ae. tauschii draft genome sequence provides an essential reference for studying D genome diversity by re-sequencing additional accessions. Over the past half century, the introduction of new D genome diversity into synthetic wheat has been a major effort to expand bread wheat genetic diversity and to create environmentally resilient lines26,27. The Ae. tauschii genome sequence should aid in identifying new elite alleles for agriculturally important traits to alleviate the worsening plight of global climate and environment changes27.
Methods Summary
We selected Ae. tauschii (2n = 14) accession AL8/78 for sequencing. Plants were grown at 25 °C in a darkened chamber for two weeks; DNA was extracted from leaf tissue and purified with a standard phenol/chloroform extraction protocol. Sequencing libraries were constructed and sequenced on Illumina next-generation sequencing platforms (GAII and HiSequation (2000)). High-quality reads were assembled with SOAPdenovo3. Repeat sequences were identified by combining de novo approaches and sequence similarity at the nucleotide and protein levels. Gene models were predicted by combining homology-based, de novo and RNA-Seq-based methods. RNA-Seq reads were assembled with CAP3 (ref. 28) and CD-Hit29 and were mapped to the draft genome with Tophat30. See Supplementary Information for details and additional analyses.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Sequence Read Archive
Data deposits
The genome sequence and the annotation are available from the National Centre for Biotechnology Information (NCBI) as BioProject ID PRJNA182898. This Whole Genome Shotgun project is deposited at DDBJ/EMBL/GenBank under accession number AOCO000000000. The version described in this paper is the first version, AOCO010000000. The Illumina sequencing reads are available in the Sequence Read Archive under accession number SRA030526, RNA-Seq sequences under SRA062662, and resequencing short reads under SRA063175. Genomic data are also available at the Comprehensive Library for Modern Biotechnology (CLiMB) repository under doi:10.5524/100054.
Change history
03 April 2013
The participants and affiliations of the International Wheat Genome Sequencing Consortium were added to the Supplementary Information.
References
Salamini, F., Ozkan, H., Brandolini, A., Schafer-Pregl, R. & Martin, W. Genetics and geography of wild cereal domestication in the near east. Nature Rev. Genet. 3, 429–441 (2002)
Dubcovsky, J. & Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316, 1862–1866 (2007)
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010)
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000)
Wicker, T. et al. A whole-genome snapshot of 454 sequences exposes the composition of the barley genome and provides evidence for parallel evolution of genome size in wheat and barley. Plant J. 59, 712–722 (2009)
Williams, M. et al. Pliocene climate and seasonality in North Atlantic shelf seas. Phil. Trans. R. Soc. A 367, 85–108 (2009)
Qi, L. L. et al. A chromosome bin map of 16,000 expressed sequence tag loci and distribution of genes among the three genomes of polyploid wheat. Genetics 168, 701–712 (2004)
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J. L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7, e32253 (2012)
Mayer, K. F. et al. Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23, 1249–1263 (2011)
The International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010)
McHale, L., Tan, X., Koehl, P. & Michelmore, R. W. Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212 (2006)
Luo, S. et al. Dynamic nucleotide-binding-site and leucine-rich-repeat-encoding genes in the grass family. Plant Physiol. 159, 197–210 (2012)
Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710 (2012)
Yue, J. X., Meyers, B. C., Chen, J. Q., Tian, D. & Yang, S. Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes. New Phytol. 193, 1049–1063 (2012)
Schuler, M. A. & Werck-Reichhart, D. Functional genomics of P450s. Annu. Rev. Plant Biol. 54, 629–667 (2003)
Thomashow, M. F. Molecular basis of plant cold acclimation: insights gained from studying the CBF cold response pathway. Plant Physiol. 154, 571–577 (2010)
Lata, C., Yadav, A. & Prasad, M. in Abiotic Stress Response in Plants—Physiological, Biochemical and Genetic Perspectives ( Shanker, A. & Venkateswarlu, B., eds) 269–296 (InTech, 2011)
Masiero, S., Colombo, L., Grini, P. E., Schnittger, A. & Kater, M. M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23, 865–872 (2011)
Margolin, A. et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7 (Suppl. 1). S7 (2006)
Mao, X. et al. Transgenic expression of TaMYB2A confers enhanced tolerance to multiple abiotic stresses in Arabidopsis. Funct. Integr. Genomics 11, 445–465 (2011)
Hu, B. & Chu, C. Phosphate starvation signaling in rice. Plant Signal. Behav. 6, 927–929 (2011)
Shivaprasad, P. V. et al. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 24, 859–874 (2012)
Gupta, R. B., Singh, N. K. & Shepherd, K. W. The cumulative effect of allelic variation in LMW and HMW glutenin subunits on dough properties in the progeny of two bread wheats. Theor. Appl. Genet. 77, 57–64 (1989)
Chantret, N. et al. Molecular basis of evolutionary events that shaped the hardness locus in diploid and polyploid wheat species (Triticum and Aegilops). Plant Cell 17, 1033–1045 (2005)
Ravel, C. et al. Nucleotide polymorphism in the wheat transcriptional activator Spa influences its pattern of expression and has pleiotropic effects on grain protein composition, dough viscoelasticity, and grain hardness. Plant Physiol. 151, 2133–2144 (2009)
Talbert, L. E., Smith, L. Y. & Blake, N. K. More than one origin of hexaploid wheat is indicated by sequence comparison of low-copy DNA. Genome 41, 402–407 (1998)
Trethowan, R. M. & Mujeeb-Kazi, A. Novel germplasm resources for improving environmental stress tolerance of hexaploid wheat. Crop Sci. 48, 1255–1265 (2008)
Huang, X. & Madan, A. CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877 (1999)
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009)
Acknowledgements
We thank J. M. Wan for support and encouragement; J. Dvorak and M. C. Luo for the AL8/78 line; C. Y. Jin, X. Y. Li, L. C. Zhang, L. Pan and J. C. Zhang for material preparation; Y. H. Lv for providing helpful palaeogeological information; D. M. Appels for producing the CMap database of molecular genetic maps; K. Edwards for providing the details of the SNP-based map for Avalon × Cadenza; L. Goodman for assistance in editing the manuscript; and M. W. Bevan, Y. B. Xu and C. Zou for critical readings of the manuscript. This work was supported by grants from the National 863 Project (2012AA10A308 and 2011AA100104), the International S&T Cooperation Program of China (2008DFB30080), the National Natural Science Foundation of China (31171548 and 31071415), the National Basic Research Program of China (2010CB125900), the Core Research Budget of the Non-profit Governmental Research (201013) and the National Program on R&D of Transgenic Plants (2011ZX08009-001 and 2011ZX08002-002).
Author information
Authors and Affiliations
Consortia
Contributions
J.Z.J., Ji.W., Xu L., H.M.Y., Z.H.H., Lo.M., Ju.W., X.Y.K., X.Y.Z., R.L.J. and Y.Z.M. initiated the project and designed the study. X.Y.K., G.Y.Z., R.A., L.F.G., Qu.H., H.H.K., B.K., X.C.X., R.Z.Z. G.M.L. and C.M. performed the research. S.C.Z., Y.R.L., W.M.H., M.P., C.Z., D.L., C.G., M.S., K.M., Y.T., F.Y.Z., S.K.P., J.W.L., Q.S.L., Ji.C., C.Y.G., G.Y.H., Y.D.L., P.L., J.Y.W., J.Y.X. and J.L.G. generated and analysed the data. Q.J.X., H.F.Z. and Z.W.Q. developed the high-density genetic map. J.Z.J., S.C.Z., Lo.M., R.A., G.Y.Z., X.Y.K. and T.W. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
A list of participants and their affiliations appears in the Supplementary Information.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data 1-5 (see Contents List for details), Supplementary Tables 1-10, 12, 15-16, 18, 20-24 (see separate files for Supplementary Tables 11, 13, 14, 17, 19), Supplementary Figures 1-12, 16-29 (see separate files for Supplementary Figures 13, 14 and 15), additional references and affiliations for the International Wheat Genome Sequencing Consortium. This file was replaced on 3 April 2013 to add the affiliations. (PDF 6613 kb)
Supplementary Figure 13
This file contains the genetic map, which was constructed using RAD tag sequencing technology. (PDF 2509 kb)
Supplementary Figure 14
This file shows the anchoring of Ae. tauschii scaffolds to three SSR-based genetic maps. (PDF 773 kb)
Supplementary Figure 15
This file contains the CMap, which is based on comparisons of three SNP-based maps. (PDF 1193 kb)
Supplementary Table 11
This table contains Scaffold information anchored on the constructed genetic map. (XLSX 658 kb)
Supplementary Table 13
This file contains Scaffolds and genes anchored on seven chromosomes. (XLSX 961 kb)
Supplementary Table 14
This file contains Gene Ontology analysis of those 628 genes potentially under selection. (XLS 89 kb)
Supplementary Table 17
This file contains Ae. tauschii cold-related genes summary. (XLSX 129 kb)
Supplementary Table 19
This file contains Ae. tauschii TF gene summary. (XLSX 987 kb)
Supplementary Information
This file contains the gene ID in the TF co-expression figure for Supplementary Figure 25. (XLSX 17 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Jia, J., Zhao, S., Kong, X. et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95 (2013). https://doi.org/10.1038/nature12028
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12028
This article is cited by
-
A high-resolution genotype–phenotype map identifies the TaSPL17 controlling grain number and size in wheat
Genome Biology (2023)
-
Weed genomics: yielding insights into the genetics of weedy traits for crop improvement
aBIOTECH (2023)
-
Fine mapping of two recessive powdery mildew resistance genes from Aegilops tauschii accession CIae8
Theoretical and Applied Genetics (2023)
-
Low-affinity SPL binding sites contribute to subgenome expression divergence in allohexaploid wheat
Science China Life Sciences (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
