
Abstract
The discovery of a living coelacanth specimen in 1938 was remarkable, as this lineage of lobe-finned fish was thought to have become extinct 70 million years ago. The modern coelacanth looks remarkably similar to many of its ancient relatives, and its evolutionary proximity to our own fish ancestors provides a glimpse of the fish that first walked on land. Here we report the genome sequence of the African coelacanth, Latimeria chalumnae. Through a phylogenomic analysis, we conclude that the lungfish, and not the coelacanth, is the closest living relative of tetrapods. Coelacanth protein-coding genes are significantly more slowly evolving than those of tetrapods, unlike other genomic features. Analyses of changes in genes and regulatory elements during the vertebrate adaptation to land highlight genes involved in immunity, nitrogen excretion and the development of fins, tail, ear, eye, brain and olfaction. Functional assays of enhancers involved in the fin-to-limb transition and in the emergence of extra-embryonic tissues show the importance of the coelacanth genome as a blueprint for understanding tetrapod evolution.
Similar content being viewed by others
Main
In 1938 Marjorie Courtenay-Latimer, the curator of a small natural history museum in East London, South Africa, discovered a large, unusual-looking fish among the many specimens delivered to her by a local fish trawler. Latimeria chalumnae, named after its discoverer1, was over 1 m long, bluish in colour and had conspicuously fleshy fins that resembled the limbs of terrestrial vertebrates. This discovery is considered to be one of the most notable zoological finds of the twentieth century. Latimeria is the only living member of an ancient group of lobe-finned fishes that was known previously only from fossils and believed to have been extinct since the Late Cretaceous period, approximately 70 million years ago (Myr ago)1. It was almost 15 years before a second specimen of this elusive species was discovered in the Comoros Islands in the Indian Ocean, and only 309 individuals have been recorded in the past 75 years (R. Nulens, personal communication)2. The discovery in 1997 of a second coelacanth species in Indonesia, Latimeria menadoensis, was equally surprising, as it had been assumed that living coelacanths were confined to small populations off the East African coast3,4. Fascination with these fish is partly due to their prehistoric appearance—remarkably, their morphology is similar to that of fossils that date back at least 300 Myr, leading to the supposition that, among vertebrates, this lineage is markedly slow to evolve1,5. Latimeria has also been of particular interest to evolutionary biologists, owing to its hotly debated relationship to our last fish ancestor, the fish that first crawled onto land6. In the past 15 years, targeted sequencing efforts have produced the sequences of the coelacanth mitochondrial genomes7, HOX clusters8 and a few gene families9,10. Nevertheless, coelacanth research has felt the lack of large-scale sequencing data. Here we describe the sequencing and comparative analysis of the genome of L. chalumnae, the African coelacanth.
Genome assembly and annotation
The African coelacanth genome was sequenced and assembled using DNA from a Comoros Islands Latimeria chalumnae specimen (Supplementary Fig. 1). It was sequenced by Illumina sequencing technology and assembled using the short read genome assembler ALLPATHS-LG11. The L. chalumnae genome has been reported previously to have a karyotype of 48 chromosomes12. The draft assembly is 2.86 gigabases (Gb) in size and is composed of 2.18 Gb of sequence plus gaps between contigs. The coelacanth genome assembly has a contig N50 size (the contig size above which 50% of the total length of the sequence assembly can be found) of 12.7 kilobases (kb) and a scaffold N50 size of 924 kb, and quality metrics comparable to other Illumina genomes (Supplementary Note 1, and Supplementary Tables 1 and 2).
The genome assembly was annotated separately by both the Ensembl gene annotation pipeline (Ensembl release 66, February 2012) and by MAKER13. The Ensembl gene annotation pipeline created gene models using protein alignments from the Universal Protein Resource (Uniprot) database, limited coelacanth complementary DNA data, RNA-seq data generated from L. chalumnae muscle (18 Gb of paired-end reads were assembled using Trinity software14, Supplementary Fig. 2) as well as orthology with other vertebrates. This pipeline produced 19,033 protein-coding genes containing 21,817 transcripts. The MAKER pipeline used the L. chalumnae Ensembl gene set, Uniprot protein alignments, and L. chalumnae (muscle) and L. menadoensis (liver and testis)15 RNA-seq data to create gene models, and this produced 29,237 protein-coding gene annotations. In addition, 2,894 short non-coding RNAs, 1,214 long non-coding RNAs, and more than 24,000 conserved RNA secondary structures were identified (Supplementary Note 2, Supplementary Tables 3 and 4, Supplementary Data 1–3 and Supplementary Fig. 3). It was inferred that 336 genes underwent specific duplications in the coelacanth lineage (Supplementary Note 3, Supplementary Tables 5 and 6, and Supplementary Data 4).
The closest living fish relative of tetrapods
The question of which living fish is the closest relative to ‘the fish that first crawled on to land’ has long captured our imagination: among scientists the odds have been placed on either the lungfish or the coelacanth16. Analyses of small to moderate amounts of sequence data for this important phylogenetic question (ranging from 1 to 43 genes) has tended to favour the lungfishes as the extant sister group to the land vertebrates17. However, the alternative hypothesis that the lungfish and the coelacanth are equally closely related to the tetrapods could not be rejected with previous data sets18.
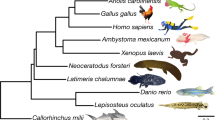
To seek a comprehensive answer we generated RNA-seq data from three samples (brain, gonad and kidney, and gut and liver) from the West African lungfish, Protopterus annectens, and compared it to gene sets from 21 strategically chosen jawed vertebrate species. To perform a reliable analysis we selected 251 genes in which a 1:1 orthology ratio was clear and used CAT-GTR, a complex site-heterogeneous model of sequence evolution that is known to reduce tree-reconstruction artefacts19 (see Supplementary Methods). The resulting phylogeny, based on 100,583 concatenated amino acid positions (Fig. 1, posterior probability = 1.0 for the lungfish–tetrapod node) is maximally supported except for the relative positions of the armadillo and the elephant. It corroborates known vertebrate phylogenetic relationships and strongly supports the conclusion that tetrapods are more closely related to lungfish than to the coelacanth (Supplementary Note 4 and Supplementary Fig. 4).

Multiple sequence alignments of 251 genes with a 1:1 ratio of orthologues in 22 vertebrates and with a full sequence coverage for both lungfish and coelacanth were used to generate a concatenated matrix of 100,583 unambiguously aligned amino acid positions. The Bayesian tree was inferred using PhyloBayes under the CAT + GTR + Γ4 model with confidence estimates derived from 100 gene jack-knife replicates (support is 100% for all clades but armadillo + elephant with 45%)48. The tree was rooted on cartilaginous fish, and shows that the lungfish is more closely related to tetrapods than the coelacanth, and that the protein sequence of coelacanth is evolving slowly. Pink lines (tetrapods) are slightly offset from purple lines (lobe-finned fish), to indicate that these species are both tetrapods and lobe-finned fish.
The slowly evolving coelacanth
The morphological resemblance of the modern coelacanth to its fossil ancestors has resulted in it being nicknamed ‘the living fossil’1. This invites the question of whether the genome of the coelacanth is as slowly evolving as its outward appearance suggests. Earlier work showed that a few gene families, such as Hox and protocadherins, have comparatively slower protein-coding evolution in coelacanth than in other vertebrate lineages8,10. To address the question, we compared several features of the coelacanth genome to those of other vertebrate genomes.
Protein-coding gene evolution was examined using the phylogenomics data set described above (251 concatenated proteins) (Fig. 1). Pair-wise distances between taxa were calculated from the branch lengths of the tree using the two-cluster test proposed previously20 to test for equality of average substitution rates. Then, for each of the following species and species clusters (coelacanth, lungfish, chicken and mammals), we ascertained their respective mean distance to an outgroup consisting of three cartilaginous fishes (elephant shark, little skate and spotted catshark). Finally, we tested whether there was any significant difference in the distance to the outgroup of cartilaginous fish for every pair of species and species clusters, using a Z statistic. When these distances to the outgroup of cartilaginous fish were compared, we found that the coelacanth proteins that were tested were significantly more slowly evolving (0.890 substitutions per site) than the lungfish (1.05 substitutions per site), chicken (1.09 substitutions per site) and mammalian (1.21 substitutions per site) orthologues (P < 10−6 in all cases) (Supplementary Data 5). In addition, as can be seen in Fig. 1, the substitution rate in coelacanth is approximately half that in tetrapods since the two lineages diverged. A Tajima’s relative rate test21 confirmed the coelacanth’s significantly slower rate of protein evolution (P < 10−20) (Supplementary Data 6).
We next examined the abundance of transposable elements in the coelacanth genome. Theoretically, transposable elements may make their greatest contribution to the evolution of a species by generating templates for exaptation to form novel regulatory elements and exons, and by acting as substrates for genomic rearrangement22. We found that the coelacanth genome contains a wide variety of transposable-element superfamilies and has a relatively high transposable-element content (25%); this number is probably an underestimate as this is a draft assembly (Supplementary Note 5 and Supplementary Tables 7–10). Analysis of RNA-seq data and of the divergence of individual transposable-element copies from consensus sequences show that 14 coelacanth transposable-element superfamilies are currently active (Supplementary Note 6, Supplementary Table 10 and Supplementary Fig. 5). We conclude that the current coelacanth genome shows both an abundance and activity of transposable elements similar to many other genomes. This contrasts with the slow protein evolution observed.
Analyses of chromosomal breakpoints in the coelacanth genome and tetrapod genomes reveal extensive conservation of synteny and indicate that large-scale rearrangements have occurred at a generally low rate in the coelacanth lineage. Analyses of these rearrangement classes detected several fission events published previously23 that are known to have occurred in tetrapod lineages, and at least 31 interchromosomal rearrangements that occurred in the coelacanth lineage or the early tetrapod lineage (0.063 fusions per 1 Myr), compared to 20 events (0.054 fusions per 1 Myr) in the salamander lineage and 21 events (0.057 fusions per 1 Myr) in the Xenopus lineage23 (Supplementary Note 7 and Supplementary Fig. 6). Overall, these analyses indicate that karyotypic evolution in the coelacanth lineage has occurred at a relatively slow rate, similar to that of non-mammalian tetrapods24.
In a separate analysis we also examined the evolutionary divergence between the two species of coelacanth, L. chalumnae and L. menadoensis, found in African and Indonesian waters, respectively. Previous analysis of mitochondrial DNA showed a sequence identity of 96%, but estimated divergence times range widely from 6 to 40 Myr25,26. When we compared the liver and testis transcriptomes of L. menadoensis27 to the L. chalumnae genome, we found an identity of 99.73% (Supplementary Note 8 and Supplementary Fig. 7), whereas alignments between 20 sequenced L. menadoensis bacterial artificial chromosomes (BACs) and the L. chalumnae genome showed an identity of 98.7% (Supplementary Table 11 and Supplementary Fig. 8). Both the genic and genomic divergence rates are similar to those seen between the human and chimpanzee genomes (99.5% and 98.8%, respectively; divergence time of 6 to 8 Myr ago)28, whereas the rates of molecular evolution in Latimeria are probably affected by several factors, including the slower substitution rate seen in coelacanth. This suggests a slightly longer divergence time for the two coelacanth species.
The adaptation of vertebrates to land
As the species with a sequenced genome closest to our most recent aquatic ancestor, the coelacanth provides a unique opportunity to identify genomic changes that were associated with the successful adaptation of vertebrates to the land environment.
Over the 400 Myr that vertebrates have lived on land, some genes that are unnecessary for existence in their new environment have been eliminated. To understand this aspect of the water-to-land transition, we surveyed the Latimeria genome annotations to identify genes that were present in the last common ancestor of all bony fish (including the coelacanth) but that are missing from tetrapod genomes. More than 50 such genes, including components of fibroblast growth factor (FGF) signalling, TGF-β and bone morphogenic protein (BMP) signalling, and WNT signalling pathways, as well as many transcription factor genes, were inferred to be lost based on the coelacanth data (Supplementary Data 7 and Supplementary Fig. 9). Previous studies of genes that were lost in this transition could only compare teleost fish to tetrapods, meaning that differences in gene content could have been due to loss in the tetrapod or in the lobe-finned fish lineages. We were able to confirm that four genes that were shown previously to be absent in tetrapods (And1 and And2 (ref. 29), Fgf24 (ref. 30) and Asip2 (ref. 31)), were indeed present and intact in Latimeria, supporting the idea that they were lost in the tetrapod lineage.
We functionally annotated more than 50 genes lost in tetrapods using zebrafish data (gene expression, knock-downs and knockouts). Many genes were classified in important developmental categories (Supplementary Data 7): fin development (13 genes); otolith and ear development (8 genes); kidney development (7 genes); trunk, somite and tail development (11 genes); eye (13 genes); and brain development (23 genes). This implies that critical characters in the morphological transition from water to land (for example, fin-to-limb transition and remodelling of the ear) are reflected in the loss of specific genes along the phylogenetic branch leading to tetrapods. However, homeobox genes, which are responsible for the development of an organism’s basic body plan, show only slight differences between Latimeria, ray-finned fish and tetrapods; it would seem that the protein-coding portion of this gene family, along with several others (Supplementary Note 9, Supplementary Tables 12–16 and Supplementary Fig. 10), have remained largely conserved during the vertebrate land transition (Supplementary Fig. 11).
As vertebrates transitioned to a new land environment, changes occurred not only in gene content but also in the regulation of existing genes. Conserved non-coding elements (CNEs) are strong candidates for gene regulatory elements. They can act as promoters, enhancers, repressors and insulators32,33, and have been implicated as major facilitators of evolutionary change34. To identify CNEs that originated in the most recent common ancestor of tetrapods, we predicted CNEs that evolved in various bony vertebrate (that is, ray-finned fish, coelacanth and tetrapod) lineages and assigned them to their likely branch points of origin. To detect CNEs, conserved sequences in the human genome were identified using MULTIZ alignments of bony vertebrate genomes, and then known protein-coding sequences, untranslated regions (UTRs) and known RNA genes were excluded. Our analysis identified 44,200 ancestral tetrapod CNEs that originated after the divergence of the coelacanth lineage. They represent 6% of the 739,597 CNEs that are under constraint in the bony vertebrate lineage. We compared the ancestral tetrapod CNEs to mouse embryo ChIP-seq (chromatin immunoprecipitation followed by sequencing) data obtained using antibodies against p300, a transcriptional coactivator. This resulted in a sevenfold enrichment in the p300 binding sites for our candidate CNEs and confirmed that these CNEs are indeed enriched for gene regulatory elements.
Each tetrapod CNE was assigned to the gene whose transcription start site was closest, and gene-ontology category enrichment was calculated for those genes. The most enriched categories were involved with smell perception (for example, sensory perception of smell, detection of chemical stimulus and olfactory receptor activity). This is consistent with the notable expansion of olfactory receptor family genes in tetrapods compared with teleosts, and may reflect the necessity of a more tightly regulated, larger and more diverse repertoire of olfactory receptors for detecting airborne odorants as part of the terrestrial lifestyle. Other significant categories include morphogenesis (radial pattern formation, hind limb morphogenesis, kidney morphogenesis) and cell differentiation (endothelial cell fate commitment, epithelial cell fate commitment), which is consistent with the body-plan changes required for land transition, as well as immunoglobulin VDJ recombination, which reflects the presumed response differences required to address the novel pathogens that vertebrates would encounter on land (Supplementary Note 10 and Supplementary Tables 17–24).
A major innovation of tetrapods is the evolution of limbs characterized by digits. The limb skeleton consists of a stylopod (humerus or femur), the zeugopod (radius and ulna, or tibia and fibula), and an autopod (wrist or ankle, and digits). There are two major hypotheses about the origins of the autopod; that it was a novel feature of tetrapods, and that it has antecedents in the fins of fish35 (Supplementary Note 11 and Supplementary Fig. 12). We examine here the Hox regulation of limb development in ray-finned fish, coelacanth and tetrapods to address these hypotheses.
In mouse, late-phase digit enhancers are located in a gene desert that is proximal to the HOX-D cluster36. Here we provide an alignment of the HOX-D centromeric gene desert of coelacanth with those of tetrapods and ray-finned fishes (Fig. 2a). Among the six cis-regulatory sequences previously identified in this gene desert36, three sequences show sequence conservation restricted to tetrapods (Supplementary Fig. 13). However, one regulatory sequence (island 1) is shared by tetrapods and coelacanth, but not by ray-finned fish (Fig. 2b and Supplementary Fig. 14). When tested in a transient transgenic assay in mouse, the coelacanth sequence of island 1 was able to drive reporter expression in a limb-specific pattern (Fig. 2c). This suggests that island 1 was a lobe-fin developmental enhancer in the fish ancestor of tetrapods that was then coopted into the autopod enhancer of modern tetrapods. In this case, the autopod developmental regulation was derived from an ancestral lobe-finned fish regulatory element.

a, Organization of the mouse HOX-D locus and centromeric gene desert, flanked by the Atf2 and Mtx2 genes. Limb regulatory sequences (I1, I2, I3, I4, CsB and CsC) are noted. Using the mouse locus as a reference (NCBI and mouse genome sequencing consortium NCBI37/mm9 assembly), corresponding sequences from human, chicken, frog, coelacanth, pufferfish, medaka, stickleback, zebrafish and elephant shark were aligned. Alignment shows regions of homology between tetrapod, coelacanth and ray-finned fishes. b, Alignment of vertebrate cis-regulatory elements I1, I2, I3, I4, CsB and CsC. c, Expression patterns of coelacanth island I in a transgenic mouse. Limb buds are indicated by arrowheads in the first two panels. The third panel shows a close-up of a limb bud.
Changes in the urea cycle provide an illuminating example of the adaptations associated with transition to land. Excretion of nitrogen is a major physiological challenge for terrestrial vertebrates. In aquatic environments, the primary nitrogenous waste product is ammonia, which is readily diluted by surrounding water before it reaches toxic levels, but on land, less toxic substances such as urea or uric acid must be produced instead (Supplementary Fig. 15). The widespread and almost exclusive occurrence of urea excretion in amphibians, some turtles and mammals has led to the hypothesis that the use of urea as the main nitrogenous waste product was a key innovation in the vertebrate transition from water to land37.
With the availability of gene sequences from coelacanth and lungfish, it became possible to test this hypothesis. We used a branch-site model in the HYPHY package38, which estimates the ratio of synonymous (dS) to non-synonymous (dN) substitutions (ω values) among different branches and among different sites (codons) across a multiple-species sequence alignment. For the rate-limiting enzyme of the hepatic urea cycle, carbamoyl phosphate synthase I (CPS1), only one branch of the tree shows a strong signature of selection (P = 0.02), namely the branch leading to tetrapods and the branch leading to amniotes (Fig. 3); no other enzymes in this cycle showed a signature of selection. Conversely, mitochondrial arginase (ARG2), which produces extrahepatic urea as a byproduct of arginine metabolism but is not involved in the production of urea for nitrogenous waste disposal, did not show any evidence of selection in vertebrates (Supplementary Fig. 16). This leads us to conclude that adaptive evolution occurred in the hepatic urea cycle during the vertebrate land transition. In addition, it is interesting to note that of the five amino acids of CPS1 that changed between coelacanth and tetrapods, three are in important domains (the two ATP-binding sites and the subunit interaction domain) and a fourth is known to cause a malfunctioning enzyme in human patients if mutated39.

Branch lengths are scaled to the expected number of substitutions per nucleotide, and branch colours indicate the strength of selection (dN/dS or ω). Red, positive or diversifying selection (ω > 5); blue, purifying selection (ω = 0); yellow, neutral evolution (ω = 1). Thick branches indicate statistical support for evolution under episodic diversifying selection. The proportion of each colour represents the fraction of the sequence undergoing the corresponding class of selection.
The adaptation to a terrestrial lifestyle necessitated major changes in the physiological environment of the developing embryo and fetus, resulting in the evolution and specialization of extra-embryonic membranes of the amniote mammals40. In particular, the placenta is a complex structure that is critical for providing gas and nutrient exchange between mother and fetus, and is also a major site of haematopoiesis41.
We have identified a region of the coelacanth HOX-A cluster that may have been involved in the evolution of extra-embryonic structures in tetrapods, including the eutherian placenta. Global alignment of the coelacanth Hoxa14–Hoxa13 region with the homologous regions of the horn shark, chicken, human and mouse revealed a CNE just upstream of the coelacanth Hoxa14 gene (Supplementary Fig. 17a). This conserved stretch is not found in teleost fishes but is highly conserved among horn shark, chicken, human and mouse despite the fact that the chicken, human and mouse have no Hoxa14 orthologues, and that the horn shark Hoxa14 gene has become a pseudogene. This CNE, HA14E1, corresponds to the proximal promoter-enhancer region of the Hoxa14 gene in Latimeria. HA14E1 is more than 99% identical between mouse, human and all other sequenced mammals, and would therefore be considered to be an ultra-conserved element42. The high level of conservation suggests that this element, which already possessed promoter activity, may have been coopted for other functions despite the loss of the Hoxa14 gene in amniotes (Supplementary Fig. 17bc). Expression of human HA14E1 in a mouse transient transgenic assay did not give notable expression in the embryo proper at day 11.5 (information is available online at the VISTA enhancer browser website; http://enhancer.lbl.gov/cgi-bin/imagedb3.pl?form=presentation&show=1&experiment_id=501&organism_id=1), which was unexpected as its location would predict that it would regulate axial structures caudally43. A similar experiment in chick embryos using the chicken HA14E1 also showed no activity in the anteroposterior axis. However, strong expression was observed in the extraembryonic area vasculosa of the chick embryo (Fig. 4a). Examination of a Latimeria BAC Hoxa14-reporter transgene in mouse embryos showed that the Hoxa14 gene is specifically expressed in a subset of cells in an extra-embryonic region at embryonic day 8.5 (Fig. 4b).

a, Chicken HA14E1 drives reporter expression in blood islands in chick embryos. A construct containing chicken HA14E1 upstream of a minimal (thymidine kinase) promoter driving enhanced green fluorescent protein (eGFP) was electroporated in HH4-stage chick embryos together with a nuclear mCherry construct. GFP expression was analysed at stage approximately HH11. The green aggregations and punctate staining are observed in the blood islands and developing vasculature. b, Expression of Latimeria Hoxa14-reporter transgene in the developing placental labyrinth of a mouse embryo. A field of cells from the labyrinth region of an embryo at embryonic day 8.5 from a BAC transgenic line containing coelacanth Hoxa9–Hoxa14 (ref. 49) in which the Hoxa14 gene had been supplanted with the gene for red fluorescence protein (RFP). Immunohistochemistry was used to detect RFP (brown staining in a small number of cells).
These findings suggest that the HA14E1 region may have been evolutionarily recruited to coordinate regulation of posterior HOX-A genes (Hoxa13, Hoxa11 and Hoxa10), which are known to be expressed in the mouse allantois and are critical for early formation of the mammalian placenta44. Although Latimeria does not possess a placenta, it gives birth to live young and has very large, vascularised eggs, but the relationship between Hoxa14, the HA14E1 enhancer and blood island formation in the coelacanth remains unknown.
The coelacanth lacks immunoglobulin-M
Immunoglobulin-M (IgM), a class of antibodies, has been reported in all vertebrate species that have been characterized so far, and is considered to be indispensable for adaptive immunity45. Interestingly, IgM genes cannot be found in coelacanth, despite an exhaustive search of the coelacanth sequence data, and even though all other major components of the immune system are present (Supplementary Note 12 and Supplementary Fig. 18). Instead, we found two IgW genes (Supplementary Figs 19–21); immunoglobulin genes that are found only in lungfish and cartilaginous fish and are believed to have originated in the ancestor of jawed vertebrates46 but subsequently lost in teleosts and tetrapods. IgM was similarly absent from the Latimeria RNA-seq data, although both IgW genes were found as transcripts. To characterize further the apparent absence of IgM, we screened large genomic L. menadoensis libraries exhaustively using a number of strategies and probes. We also carried out polymerase chain reaction (PCR) with degenerate primers that should universally amplify IgM sequences. The lack of IgM in Latimeria raises questions as to how coelacanth B cells respond to microbial pathogens and whether the IgW molecules can serve a compensatory function, even though there is no indication that the coelacanth IgW was derived from vertebrate IgM genes.
Discussion
Since its discovery, the coelacanth has been referred to as a ‘living fossil’, owing to its morphological similarities to its fossil ancestors1. However, questions have remained as to whether it is indeed evolving slowly, as morphological stasis does not necessarily imply genomic stasis. In this study, we have confirmed that the protein-coding genes of L. chalumnae show a decreased substitution rate compared to those of other sequenced vertebrates, even though its genome as a whole does not show evidence of low genome plasticity. The reason for this lower substitution rate is still unknown, although a static habitat and a lack of predation over evolutionary timescales could be contributing factors to a lower need for adaptation. A closer examination of gene families that show either unusually high or low levels of directional selection indicative of adaptation in the coelacanth may provide information on which selective pressures acted, and which pressures did not act, to shape this evolutionary relict (Supplementary Note 13 and Supplementary Fig. 22).
The vertebrate land transition is one of the most important steps in our evolutionary history. We conclude that the closest living fish to the tetrapod ancestor is the lungfish, not the coelacanth. However, the coelacanth is critical to our understanding of this transition, as the lungfish have intractable genome sizes (estimated at 50–100 Gb)47. Here we have examined vertebrate adaptation to land through coelacanth whole-genome analysis, and have shown the potential of focused analysis of specific gene families involved in this process. Further study of these changes between tetrapods and the coelacanth may provide important insights into how a complex organism like a vertebrate can markedly change its way of life.
Methods Summary
A full description of methods, including information on sample collection, sequencing, assembly, annotation, all sequence analysis and functional validation, can be found in the Supplementary Information.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
Genome assemblies, transcriptomes and mitochondrial DNA sequences have been deposited in GenBank/EMBL/DDBJ. The L. chalumnae genome assembly has been deposited under the accession number AFYH00000000. The L. chalumnae transcriptome has been deposited under the accession number SRX117503 and the P. annectans transcriptomes have been deposited under the accession numbers SRX152529, SRX152530 and SRX152531. The P. annectans mitochondrial DNA sequence was deposited under the accession number JX568887. All animal experiments were approved by the MIT Committee for Animal Care.
References
Smith, J. L. B. A living fish of mesozoic type. Nature 143, 455–456 (1939)
Nulens, R., Scott, L. & Herbin, M. An Updated Inventory of All Known Specimens of the Coelacanth, Latimeria Spp. Smithiana Vol. 3 (South African Institute for Aquatic Biodiversity, 2010)
Erdmann, M. V., Caldwell, R. L. & Kasim Moosa, M. Indonesian 'king of the sea' discovered. Nature 395, 335 (1998)
Smith, J. L. B. Old Fourlegs: the Story of the Coelacanth (Longmans, Green, 1956)
Zhu, M. et al. Earliest known coelacanth skull extends the range of anatomically modern coelacanths to the Early Devonian. Nature Commun. 3, 772 (2012)
Zimmer, C. At the Water's Edge: Fish with Fingers, Whales with Legs, and How Life Came Ashore but then Went Back to Sea (Free Press, 1999)
Zardoya, R. & Meyer, A. The complete DNA sequence of the mitochondrial genome of a “living fossil,” the coelacanth (Latimeria chalumnae). Genetics 146, 995–1010 (1997)
Amemiya, C. T. et al. Complete HOX cluster characterization of the coelacanth provides further evidence for slow evolution of its genome. Proc. Natl Acad. Sci. USA 107, 3622–3627
Larsson, T. A., Larson, E. T. & Larhammar, D. Cloning and sequence analysis of the neuropeptide Y receptors Y5 and Y6 in the coelacanth Latimeria chalumnae. Gen. Comp. Endocrinol. 150, 337–342 (2007)
Noonan, J. P. et al. Coelacanth genome sequence reveals the evolutionary history of vertebrate genes. Genome Res. 14, 2397–2405 (2004)
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011)
Bogart, J. P., Balon, E. K. & Bruton, M. N. The chromosomes of the living coelacanth and their remarkable similarity to those of one of the most ancient frogs. J. Hered. 85, 322–325 (1994)
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008)
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotech. 29, 644–652 (2011)
Pallavicini, A. et al. Analysis of the transcriptome of the Indonesian coelacanth Latimeria menadoensis. BMC Genomics (in the press)
Schultze, H. P. & Trueb, L. Origins of the Higher Groups of Tetrapods: Controversy and Consensus. (Comstock Publishing Associates, 1991)
Meyer, A. & Dolven, S. I. Molecules, fossils, and the origin of tetrapods. J. Mol. Evol. 35, 102–113 (1992)
Brinkmann, H., Venkatesh, B., Brenner, S. & Meyer, A. Nuclear protein-coding genes support lungfish and not the coelacanth as the closest living relatives of land vertebrates. Proc. Natl Acad. Sci. USA 101, 4900–4905 (2004)
Lartillot, N. & Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109 (2004)
Takezaki, N., Rzhetsky, A. & Nei, M. Phylogenetic test of the molecular clock and linearized trees. Mol. Biol. Evol. 12, 823–833 (1995)
Tajima, F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135, 599–607 (1993)
Bejerano, G. et al. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 441, 87–90 (2006)
Voss, S. R. et al. Origin of amphibian and avian chromosomes by fission, fusion, and retention of ancestral chromosomes. Genome Res. 21, 1306–1312 (2011)
Smith, J. J. & Voss, S. R. Gene order data from a model amphibian (Ambystoma): new perspectives on vertebrate genome structure and evolution. BMC Genomics 7, 219 (2006)
Inoue, J. G., Miya, M., Venkatesh, B. & Nishida, M. The mitochondrial genome of Indonesian coelacanth Latimeria menadoensis (Sarcopterygii: Coelacanthiformes) and divergence time estimation between the two coelacanths. Gene 349, 227–235 (2005)
Holder, M. T., Erdmann, M. V., Wilcox, T. P., Caldwell, R. L. & Hillis, D. M. Two living species of coelacanths? Proc. Natl Acad. Sci. USA 96, 12616–12620 (1999)
Canapa, A. et al. Composition and phylogenetic analysis of vitellogenin coding sequences in the Indonesian coelacanth Latimeria menadoensis. J. Exp. Zool..B 318, 404–416 (2012)
The Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005)
Zhang, J. et al. Loss of fish actinotrichia proteins and the fin-to-limb transition. Nature 466, 234–237 (2010)
Jovelin, R. et al. Evolution of developmental regulation in the vertebrate FgfD subfamily. J. Exp. Zool.B 314, 33–56 (2010)
Braasch, I. & Postlethwait, J. H. The teleost agouti-related protein 2 gene is an ohnolog gone missing from the tetrapod genome. Proc. Natl Acad. Sci. USA 108, E47–E48 (2011)
Navratilova, P. et al. Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev. Biol 327, 526–540 (2009)
Xie, X. et al. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc. Natl Acad. Sci. USA 104, 7145–7150 (2007)
Jones, F. C. et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 (2012)
Shubin, N., Tabin, C. & Carroll, S. Deep homology and the origins of evolutionary novelty. Nature 457, 818–823 (2009)
Montavon, T. et al. A regulatory archipelago controls Hox genes transcription in digits. Cell 147, 1132–1145 (2011)
Wright, P. A. Nitrogen excretion: three end products, many physiological roles. J. Exp. Biol. 198, 273–281 (1995)
Kosakovsky Pond, S. L. et al. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 28, 3033–3043 (2011)
Häberle, J. et al. Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations. Hum. Mutat. 32, 579–589 (2011)
Carroll, R. L. Vertebrate Paleontology and Evolution (W.H. Freeman and Company, 1988)
Gekas, C. et al. Hematopoietic stem cell development in the placenta. Int. J. Dev. Biol. 54, 1089–1098 (2010)
Bejerano, G. et al. Ultraconserved elements in the human genome. Science 304, 1321–3125 (2004)
Wellik, D. M. Hox patterning of the vertebrate axial skeleton. Dev. Dyn. 236, 2454–2463 (2007)
Scotti, M. & Kmita, M. Recruitment of 5′ Hoxa genes in the allantois is essential for proper extra-embryonic function in placental mammals. Development 139, 731–730 (2012)
Bengtén, E. et al. Immunoglobulin isotypes: structure, function, and genetics. Curr. Top. Microbiol. Immunol. 248, 189–219 (2000)
Ota, T., Rast, J. P., Litman, G. W. & Amemiya, C. T. Lineage-restricted retention of a primitive immunoglobulin heavy chain isotype within the Dipnoi reveals an evolutionary paradox. Proc. Natl Acad. Sci. USA 100, 2501–2506 (2003)
Gregory, T. R. The Evolution of the Genome 1–71 (Elsevier Academic, 2004)
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463 (2005)
Smith, J. J., Sumiyama, K. & Amemiya, C. T. A living fossil in the genome of a living fossil: Harbinger transposons in the coelacanth genome. Mol. Biol. Evol. 29, 985–993 (2012)
Acknowledgements
Acquisition and storage of Latimeria chalumnae samples was supported by grants from the African Coelacanth Ecosystem Programme of the South African National Department of Science and Technology. Generation of the Latimeria chalumnae and Protopterus annectens sequences by the Broad Institute of the Massachusetts Institute of Technology (MIT) and Harvard University was supported by grants from the National Human Genome Research Institute (NHGRI). K.L.T. is the recipient of a EURYI award from the European Science Foundation. We would also like to thank the Genomics Sequencing Platform of the Broad Institute for sequencing the L. chalumnae genome and L. chalumnae and P. annectens transcriptomes, S. Ahamada, R. Stobbs and the Association pour le Protection de Gombesa (APG) for their help in obtaining coelacanth samples, Y. Zhao for the use of data from Rana chensinensis, and L. Gaffney, C. Hamilton and J. Westlund for assistance with figure preparation.
Author information
Authors and Affiliations
Contributions
Author Contributions J.A., C.T.A., A.M. and K.L.T. planned and oversaw the project. R.A.D. and C.T.A. provided blood and tissues for sequencing. C.T.A. and M.L. prepared the DNA for sequencing. I.M., S.G., D.P., F.J.R., T.S. and D.B.J. assembled the genome. N.R.S. and C.T.A. prepared RNA from L. chalumnae and P. annectens, and L.F. and J.Z.L. made the L. chalumnae RNA-seq library. A.C., M.B., M.A.B., M.F., F.B., G.S., A.M.F., A.P., M.G., G.D.M., J.T.-M. and E.O. sequenced and analysed the L. menadoensis RNA-seq library. B.A., S.M.J.S., S.W., M.S.C. and M.Y. annotated the genome. W.H. and C.P.P. carried out the annotation and analysis of long non-coding RNAs. P.F.S., S.H., A.N., H.T. and S.J.P. annotated non-coding RNAs. M.G., G.D.M., A.P., M.R. and C.T.A. compared L. chalumnae and L. menadoensis sequences. H.B., D.B. and H.P. carried out the phylogenomic analysis. T.Ma. and A.M. performed the gene relative-rate analysis. A.C., J.G., S.P., B.P., P.v.H. and U.H. carried out the analysis, annotation and statistical enrichment of L. chalumnae specific gene duplications. N.F. and A.M. analysed the homeobox gene repertoires. G.L.B. and A.L.E. analysed the chaperone genes. D.C., S.F., O.S., J.-N.V., M.S. and A.M. analysed transposable elements. J.J.S. analysed large-scale rearrangements in vertebrate genomes. I.B., J.H.P., N.F. and S.K. analysed genes lost in tetrapods. T.Mi. analysed actinodin and pectoral-fin musculature. C.O. and M.S. analysed selection in urea cycle genes. A.P.L. and B.V. carried out the conserved non-coding element analysis. I.S., N.R., V.R., N.S. and C.J.T. carried out the analysis of autopodial CNEs. K.S., T.S.-S. and C.T.A. examined the evolution of a placenta-related CNE. N.R.S., G.W.L., M.G.M., T.O. and C.T.A. performed the IgM analysis. J.A., C.T.A., A.M. and K.L.T. wrote the paper with input from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Notes 1-13, Supplementary Figures 1-22, Supplementary Tables 1-24 and Supplementary references. (PDF 5244 kb)
Supplementary Data
This zipped file contains Supplementary Data files 1-7. (ZIP 2702 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
Amemiya, C., Alföldi, J., Lee, A. et al. The African coelacanth genome provides insights into tetrapod evolution. Nature 496, 311–316 (2013). https://doi.org/10.1038/nature12027
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12027
This article is cited by
-
Effects of indole-3-butyric acid supplementation in diets containing high soybean meal on growth, intestinal inflammation, and intestinal flora of pearl gentian grouper (Epinephelus fuscoguttatus♀ × Epinephelus lanceolatus♂)
Aquaculture International (2024)
-
The chromosome-level genome and key genes associated with mud-dwelling behavior and adaptations of hypoxia and noxious environments in loach (Misgurnus anguillicaudatus)
BMC Biology (2023)
-
Conceptual and empirical bridges between micro- and macroevolution
Nature Ecology & Evolution (2023)
-
Accelerated body size evolution in upland environments is correlated with recent speciation in South American freshwater fishes
Nature Communications (2023)
-
Origin and evolutionary malleability of T cell receptor α diversity
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
