
Abstract
Crop domestications are long-term selection experiments that have greatly advanced human civilization. The domestication of cultivated rice (Oryza sativa L.) ranks as one of the most important developments in history. However, its origins and domestication processes are controversial and have long been debated. Here we generate genome sequences from 446 geographically diverse accessions of the wild rice species Oryza rufipogon, the immediate ancestral progenitor of cultivated rice, and from 1,083 cultivated indica and japonica varieties to construct a comprehensive map of rice genome variation. In the search for signatures of selection, we identify 55 selective sweeps that have occurred during domestication. In-depth analyses of the domestication sweeps and genome-wide patterns reveal that Oryza sativa japonica rice was first domesticated from a specific population of O. rufipogon around the middle area of the Pearl River in southern China, and that Oryza sativa indica rice was subsequently developed from crosses between japonica rice and local wild rice as the initial cultivars spread into South East and South Asia. The domestication-associated traits are analysed through high-resolution genetic mapping. This study provides an important resource for rice breeding and an effective genomics approach for crop domestication research.
Similar content being viewed by others

Main
Cultivated rice (Oryza sativa L.), which is grown worldwide and is one of the most important cereals for human nutrition, is considered to have been domesticated from wild rice (Oryza rufipogon) thousands of years ago1,2,3,4. The differences between O. sativa and O. rufipogon are reflected in a wide range of morphological and physiological traits5,6,7,8,9. Despite the fact that rice is a major cereal and a model system for plant biology, the evolutionary origins and domestication processes of cultivated rice have long been debated. The puzzles about rice domestication include: (1) where the geographic origin of cultivated rice was, (2) which types of O. rufipogon served as its direct wild progenitor, and (3) whether the two subspecies of cultivated rice, indica and japonica, are derived from a single or multiple domestications.
A wide range of genetic and archaeological studies have been carried out to examine the phylogenetic relationships of rice, and investigate the demographic history of rice domestication10,11,12,13,14,15,16,17,18,19. Molecular phylogenetic analyses indicated that indica and japonica originated independently3,10,20. However, the well-characterized domestication genes in rice were found to be fixed in both subspecies with the same alleles, thus supporting a single domestication origin6,7,8,9,16. Recently, a demographic analysis of single-nucleotide polymorphisms (SNPs) detected from 630 gene fragments suggested a single domestication origin of rice17. Meanwhile, population genetics analyses of genome-wide data of cultivated and wild rice have tended to suggest that indica and japonica genomes generally appear to be of independent origin18,19, but many genomic segments bearing domestication alleles may have originated only once18. Despite these advances, wider sampling with population-scale whole-genome sequencing is needed to shed greater light on the evolutionary history of rice domestication. An in-depth investigation of the haplotype structure near the domestication sites will be critical for evaluating the direction of introgression. The specific ancestral population and the subsequent demographic event are yet to be identified.
Moreover, a comprehensive map of rice genome variation will facilitate genetic mapping of complex traits in rice. We recently collected diverse rice cultivars for sequencing, and carried out genome-wide association studies (GWAS) for many agronomic traits in cultivated rice21,22. Here we sequenced and analysed the genomes of 446 O. rufipogon accessions to investigate the phylogenetic relationships between cultivated and wild rice and identify the signatures of selection in rice domestication. This research also provides a robust foundation that will enable rice breeders to effectively exploit diverse genetic resources for rice improvement.
Analysis of wild rice populations
The strategy of this study is briefly described in Supplementary Fig. 1. The genus Oryza consists of 23 species23,24, and the wild rice O. rufipogon is believed to be the immediate progenitor of the cultivated rice O. sativa (Supplementary Fig. 2 and Supplementary Table 1). From large collections of wild rice germplasm maintained in China and Japan, we selected 446 diverse O. rufipogon accessions including both perennial and annual (also called as Oryza nivara) forms from Asia and Oceania, spanning the native geographic range of the species (Supplementary Tables 2 and 3). We sequenced these accessions with twofold genome coverage. After aligning the reads against the rice reference genome sequence, we identified a total of 5,037,497 non-singleton SNPs. Based on the SNP data, the sequence diversity (π) of O. rufipogon was estimated at ∼0.003, which is higher than that in O. sativa—the sequence diversity is 0.0024 for O. sativa, and 0.0016 and 0.0006 for indica and japonica, respectively21. Approximately 82% of SNPs (minor allele frequency > 0.05) segregating in O. rufipogon also segregate in O. sativa (Supplementary Table 4). This observation is consistent with previous suggestions that part of the genetic diversity in the progenitor would be lost because only a limited number of individuals were used during domestication11,25.
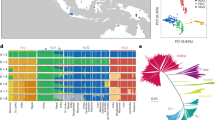
We investigated the population structure of the O. rufipogon accessions. On the basis of the neighbour-joining tree, as well as principal-component analysis (PCA), we classified the O. rufipogon species into three types, simply designated as Or-I, Or-II and Or-III in this study (Fig. 1a and Supplementary Fig. 3). We found that the rice population structure strongly correlated with geographic distribution (r2 = 0.2 between the first principal component and the longitude, and r2 = 0.3 between the second principal component and the latitude)26. Interestingly, the O. rufipogon accessions sampled from southern China mostly belong to the Or-III type (Fig. 1b and Supplementary Fig. 4). The level of population differentiation, FST, was estimated at 0.18 among the groups of O. rufipogon, which is much lower than that of O. sativa (∼0.55 on average21). The differentiation was not evenly distributed across the rice genome (Supplementary Fig. 5). We scanned the whole genome for highly differentiated loci and found 68 loci with FST > 0.3, which covered ∼3% of the complete rice genome (Supplementary Table 5). Of these loci, we found several known genes or quantitative trait loci (QTLs) that included DPL2 (hybrid incompatibility27) (Fig. 1c), OsSOC1 and Ghd7 (flowering time28,29,30) and qCTS12 (cold tolerance31), all of which have been reported to be closely related to indica–japonica differentiation in rice. Hence, the highly differentiated loci can provide important clues for searching the genes involved in local adaptation.

a, Neighbour-joining tree of 446 O. rufipogon accessions, which was calculated from ∼5 million SNPs, identifies the three groups of Or-I (red), Or-II (grey) and Or-III (blue). b, Geographic origins of wild rice accessions. c, The level of genetic differentiation (FST) in O. rufipogon population around the DPL2 gene that underlies indica–japonica hybrid incompatibility in rice. d, Regional Manhattan plots of GWAS for tiller angle in O. rufipogon population identify a known gene, PROG1, using a compressed mixed linear model. The genome-wide significance threshold (1 × 10−6) and the position of the peak SNP are indicated by a horizontal dash-dot line and a vertical red line, respectively.
Because O. rufipogon is an out-crossing species, it was expected to have a relatively high decay rate of linkage disequilibrium. We found that the decay rate of linkage disequilibrium in O. rufipogon, expressed as r2, dropped to half of its maximum value at ∼20 kilobases (kb) on average (Supplementary Fig. 6), which is much more rapid than that in O. sativa (∼123 kb and ∼167 kb in indica and japonica, respectively)21. To perform GWAS in O. rufipogon, we used the k-nearest neighbour algorithm for data imputation (Supplementary Table 6), and phenotyped the O. rufipogon population for two traits, leaf sheath colour and tiller angle. The strongest associations for sheath colour and tiller angle were found to be just around the known loci OsC1, for colouration32, and PROG1, for prostrate growth7,8 (Fig. 1d and Supplementary Figs 7–9). Through computational simulations we predicted that the mapping resolution of GWAS in O. rufipogon was approximately three times greater than that in O. sativa on average (Supplementary Figs 10–12). Hence, the wild rice population and accompanying comprehensive sequence resource should be of great utility for directly dissecting agronomic traits in rice.
Phylogenetic relationships of rice
We used whole-genome sequencing data for a large panel of accessions containing 446 O. rufipogon accessions and 1,083 O. sativa varieties to explore their phylogenetic relationships. The 1,083 diverse O. sativa varieties were collected throughout the world and sequenced with one-fold genome coverage (Supplementary Table 7). Of these, 950 genomes had previously been reported21,22, and the remaining 133 genomes, including many representative varieties (for example, aromatic rice), were sequenced and first reported in this study. A total of 7,970,359 non-singleton SNPs were identified from the 1,529 genome sequences. To determine the ancestral states of the SNPs, the close relatives of O. rufipogon and O. sativa were also sequenced with a total of approximately 50-fold genome coverage (Supplementary Table 8). Using genome sequences of these outgroups, we were able to identify the ancestral states of 6,119,311 SNPs out of the approximately 8 million SNPs (77%).
We used the genotype data set of the ∼8 million SNP sites from 1,529 genomes to infer genome-wide relationships. Both the phylogenetic tree and the PCA plots indicate that O. sativa indica and japonica are descended from Or-I and Or-III, respectively (Fig. 2a and Supplementary Fig. 13). We then investigated the detailed relationship between indica and Or-I and that between japonica and Or-III separately (Supplementary Figs 14–16). The level of genetic differentiation between indica and Or-I was modest (FST = 0.17), and indica contains approximately 75% of the genetic diversity in Or-I (Fig. 2b). A small number of indica and Or-I accessions seemed to be intermediate between cultivated and wild rice (Fig. 2a). In contrast, the japonica groups (temperate japonica, tropical japonica and aromatic) were all clustered together, and had descended from wild rice in southern China (sub-clade Or-IIIa). There was an obvious genetic distinction between japonica and Or-IIIa (Fig. 2a), which had a relatively high level of population differentiation (FST = 0.36). We found that only approximately 33% of the genetic diversity of Or-III persisted in japonica (Fig. 2b). The strong genetic bottleneck indicates that a small effective population from Or-III was used for domesticating japonica cultivars.

a, Phylogenetic tree of the full population (446 O. rufipogon accessions and 1,083 O. sativa varieties) calculated from ∼8 million SNPs in O. rufipogon and O. sativa. The double-layer rings indicate O. rufipogon (outer ring: Or-I, Or-II and Or-III are coloured in red, grey and blue, respectively) and O. sativa (inner ring: indica and japonica subspecies are in pink and sky blue, respectively). b, Illustration of genetic diversity and population differentiation in O. rufipogon and O. sativa. The size of the circles represents the level of genetic diversity (π) of the groups, and the FST values between the groups are indicated. ind, indica; jap, japonica. c, The spectrum of allele frequencies at the causal polymorphisms of Ghd7, DPL2 and GS3.
On the basis of our SNP data, most sequence variants observed between indica and japonica, which were estimated to have diverged hundreds of thousands of years ago20, already existed in the progenitor gene pools. However, the differentiation was enhanced during domestication, with FST expanding from 0.18 in O. rufipogon to 0.55 in O. sativa (Fig. 2b). The high level of genetic differentiation between indica and japonica has resulted from a combination of the modest differentiation present within their ancestor population and the recent domestication bottleneck (Fig. 2c). For example, at the causal SNP site for GS3 (a major QTL for grain shape33,34), the null allele mainly existed in the wild rice populations in the Guangxi and Guangdong provinces of southern China. This allele rapidly extended to become the major allele (∼98%) in the japonica population, thus generating a distinct phenotypic difference between indica and japonica. We screened all SNPs that were highly differentiated in frequency between indica and japonica, and found a total of 213,188 indica–japonica-differentiated SNPs. With regard to the differentiated SNPs, the differences present within the O. rufipogon, the domestication bottleneck in japonica and in indica were estimated to contribute ∼50%, ∼40% and ∼10% of total alterations in allele frequency, respectively (Supplementary Fig. 17).
We examined the ancestral states of indica–japonica-fixed SNPs, and found that 55% of SNPs of the ancestral alleles are fixed in indica and the other 45% are fixed in japonica. The dN/dS ratio (dN, number of non-synonymous substitutions per non-synonymous site; dS, number of synonymous substitutions per synonymous site) of the SNPs was calculated to be 0.34, which was almost equal to the average level of the total SNPs. In contrast, for the 9,595 SNPs that were fixed between O. rufipogon and O. sativa, the ancestral alleles of 93.3% SNPs are identical to O. rufipogon, indicating that O. rufipogon has retained more ancestral states than O. sativa, which also further supported our conclusion that O. rufipogon is likely to be the ancestral progenitor of O. sativa. The dN/dS ratio of the fixed SNPs between O. rufipogon and O. sativa is calculated to be 1.04, indicating positive selection during domestication (P < 0.001, chi-square test).
Screens and annotation of domestication loci
Selective signatures from domestication include a reduction in nucleotide diversity and altered allele frequency in the domestication loci19,25. We measured the ratio of the genetic diversity in wild rice to that in cultivated rice (πw/πc) across the rice genome, and determined the cutoff on the basis of permutation tests (Supplementary Information section 2). We performed whole-genome screening in indica, japonica and the full population using the diversity ratios (Fig. 3 and Supplementary Figs 18–20). In total we identified 60 loci in indica, 62 in japonica and 55 in the full population (Supplementary Tables 9–11). We noticed that many loci with strong signals of selection were nearly identical in both indica and japonica where FST between indica and japonica was extremely low, indicating that introduction of traits during domestication has in many cases involved introgression events. We noted that most well-characterized domestication genes, including Bh4 (hull colour9), PROG1 (tiller angle7,8), sh4 (seed shattering5,6), qSW5 (grain width35) and OsC1 (leaf sheath colour and apiculus colour32), were among the 55 loci detected in the full population (Fig. 3). Another three well-characterized domestication genes, qSH1 for seed shattering36, Waxy for grain quality37 and Rc for pericarp colour38,39, which showed strong selection signals in the japonica panel, were not fully shared in the indica population.

a, Whole-genome screening of domestication sweeps in the full population of O. rufipogon and O. sativa. The values of πw/πc are plotted against the position on each chromosome. The horizontal dashed line indicates the genome-wide threshold of selection signals (πw/πc > 3). b–d, A large-scale high-resolution mapping for fifteen domestication-related traits was performed in an O. rufipogon × O. sativa population. The domestication sweeps overlapped with characterized domestication-related QTLs are shown in dark red, and the loci with known causal genes are shown in red. Among them, three strong selective sweeps were found to be associated with grain width (b), grain weight (c) and exserted stigma (d), respectively. In b–d, the likelihood of odds (LOD) values from the composite interval mapping method are plotted against position on the rice chromosomes. Grey horizontal dashed line indicates the threshold (LOD > 3.5).
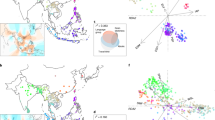
We investigated the genetic patterns around the well-characterized domestication loci, which were quite different from those at the whole-genome scale. According to phylogenetic trees calculated from SNPs on these loci, most cultivars were clustered together, and the Or-III population from southern China tended to be the closest ancestral progenitor of all the cultivars (Supplementary Fig. 21). These patterns were much clearer when expanded to the total regions of the 55 domestication-related loci (Fig. 4a). The indica cultivars were generally close to Or-I across the genome, but were closer to Or-IIIa than to Or-I at the 55 selected loci (Supplementary Fig. 22). We further calculated the genetic distances between cultivated rice and the wild rice populations from each geographic sampling region through analysis of the genomic regions around the 55 domestication-related loci (Supplementary Fig. 23). Our genetic approach showed that the middle area of the Pearl River in Guangxi province, southern China, was probably the place of the first development of cultivated rice (Fig. 4b–d), although an archaeological finding had identified the Lower Yangtze region in eastern China as one of the centres of rice cultivation14,15. These results suggest a model in which japonica was first domesticated from Or-III in southern China, and was subsequently crossed to local wild rice in South East Asia and South Asia, thus generating indica after many cross-differentiation-selection cycles (Fig. 4e). Furthermore, we performed computational simulations to generate in silico data sets under various demographic scenarios (Supplementary Fig. 24), using the forward simulator SFS_CODE40 (Supplementary Information section 5). We calculated the genetic distances of indica and japonica with different clades of O. rufipogon for both the real and simulated data (Supplementary Fig. 25), which showed that there were significant differences between the real data and the simulated ones in all the scenarios except for the model proposed here (P < 0.01, rank sum test), providing further support for the model that we propose.

a, Phylogenetic tree of 446 O. rufipogon accessions and 1,083 O. sativa varieties calculated from SNPs in the overall regions of the 55 major domestication sweeps. b, Geographic locations of 62 O. rufipogon accessions, whose phylogenetic positions during domestication are indicated. Colour index represents the average of the genetic distance of O. rufipogon accessions to all cultivated rice accessions. Two major rivers in southern China are labelled in grey in the map. c, The average distance of O. rufipogon accessions from different countries to all cultivars. The distance was estimated by simple matching distance of SNPs around the Bh4 locus or all SNPs within the 55 domestication sweeps. d, The average distance of O. rufipogon accessions from different provinces in southern China to all cultivars. e, Schematics of the origin of cultivated rice. The aus and aromatic rice are minor groups of rice accessions with small geographic distributions.
To investigate why these chromosomal loci were selected during rice domestication, QTL mapping of domestication-related traits was performed using a population that was developed from a cross between O. sativa indica Guangluai-4 and O. rufipogon (Or-IIIa accession W1943). Using a sequencing-based genotyping method41, we sequenced 271 lines in the population with 0.5× coverage for each, and constructed an ultra-dense genotype map. A total of 15 domestication-related traits were phenotyped, with 58 QTLs detected using a composite interval mapping method (Supplementary Fig. 26 and Supplementary Tables 12, 13). For ten QTLs for which causal genes have been reported, almost all the peak signals were within 200 kb from the known genes, indicating that a high mapping resolution was achieved (Supplementary Fig. 27).
Of the 58 QTLs detected, 32 QTLs were located within domestication sweeps (Fig. 3b–d and Supplementary Figs 28–30). The overlap of the QTLs for domestication traits with domestication sweeps is very significant (P < 0.001, chi-square test). Among them, the QTLs for traits like exserted stigma (controlling mating system) and grain size (controlling output yield) show stronger signals than those for shattering (sh4) and plant architecture (PROG1). Mating systems have been proposed to have a fundamental part in crop domestication42,43. We identified three major loci and two minor loci responsible for exserted stigma, and found that all the five QTLs were located within domestication sweeps.
Identification of domestication-associated variants
Characterization of domestication-associated genes and their genetic variation relies on high-quality genome sequences of both the cultivated species and its immediate progenitor44,45. Hence, we further sequenced the O. rufipogon accession W1943 with 100-fold genome coverage and carried out de novo genome assembly. The total length of the assembly is 406 megabases (Mb) and the N50 length of the initial contigs was 16 kb. After aligning the assembly against the reference genome of cultivated rice, we identified 2,621,077 SNPs, 619,132 small indels and 140,075 structural variants of large size (Supplementary Table 14). We examined the sequence variants for their potential effects on protein coding, and identified a total of 128,010 non-synonymous SNPs and 49,236 sequence variants with large effect. Moreover, we surveyed the allele information at all the polymorphic sites across all rice accessions, and constructed a comprehensive sequence-variant frequency spectrum map.
We investigated the patterns of sequence polymorphism to detect variants that affect gene coding and have differential frequency in different populations. For the domestication loci with well-characterized genes, this approach allowed us to narrow down the causative polymorphism to a limited number of sequence variants (Supplementary Figs 31–34). For the domestication loci for which known genes have not yet been identified, we found a total of 273 novel functional variants within 204 genes that showed high differentiation in allele frequencies between cultivated and wild rice (Supplementary Table 15). Moreover, we identified 305 sequence variants in the promoter regions with a differential frequency between cultivated and wild rice (Supplementary Table 16), and 1,120 functional variants that were fixed in either japonica or indica panel (Supplementary Tables 17 and 18).
Discussion
Our study has provided new insights into how and where rice was likely to be domesticated, and we have identified a set of domestication sweeps and putative causal genes. Such endeavours will be enhanced by continuing improvements in assembly and annotation of wild rice genome sequences, generation of functional genomics data sets of wild rice46, advances in mapping of rice domestication traits47, and biological follow-up of the putative causal genes. The understanding of past domestication, including the selections on critical traits and the recent rapid speciation, will further guide future breeding efforts48. Moreover, the great diversity in the wild rice populations, which have much more natural allelic variation than domesticated rice, will further facilitate breeding to modify crops in the post-domestication era.
Methods Summary
The cultivated and wild rice accessions were all from large collections of rice accessions preserved at the China National Rice Research Institute in Hangzhou, China, and the National Institute of Genetics in Mishima, Japan. The DNA samples were sequenced on the Illumina Genome Analyzer IIx or HiSeq2000.
Online Methods
Sampling and sequencing
The cultivated and wild rice accessions were all from large collections of rice accessions preserved at the China National Rice Research Institute in Hangzhou, China, and the National Institute of Genetics in Mishima, Japan. The accessions were selected on the basis of the germplasm database records of phenotypic data and sampling localities to maximize genetic and geographic diversity. The collection was maintained by selfing in the laboratories. For each accession, genomic DNA from a single plant was used for sequencing, and seeds derived from the same plant were used for following field trials. In total, the genomes of 1,083 O. sativa accessions, 446 O. rufipogon accessions and 15 accessions of outgroup species were sequenced on the Illumina Genome Analyzer IIx generating 73-bp (or 117-bp) paired-end reads, each to approximately onefold (for O. sativa accessions), twofold (for O. rufipogon accessions) or threefold (for outgroup species) coverage. The detailed information, including geographic origin and sequencing coverage of the rice accessions, was listed in Supplementary Tables 2, 7 and 8. Library construction and sequencing of these accessions were performed as described21. One representative accession of O. rufipogon, W1943, was sequenced on the Illumina HiSeq2000, generating 100-bp paired-end reads with 100-fold genome coverage. An amplification-free method of library preparation49 was used in deep sequencing of the rice accession, which reduced the incidence of duplicate sequences, thus facilitating genome assembly and variation analysis.
Read alignment and SNP calling
The paired-end reads of all the rice accessions were aligned against the rice reference genome (IRGSP 4.0) using the software Smalt (version 0.4) with the parameters of ‘-pair 50, 700’ and ‘-mthresh 50’. SNPs were called using the Ssaha Pileup package (version 0.5) with detailed procedure described previously21. Genotypes of the rice accessions, including 1,083 O. sativa accessions, 446 O. rufipogon accessions and 15 accessions of outgroup species, were further called at the SNP sites from the Ssaha Pileup outputs. The genotype calls in 15 accessions of outgroup species were used to determine the ancestral states of SNPs in O. sativa and O. rufipogon. SNPs in coding regions, which were defined based on the gene models in the RAP-DB (release 2), were then annotated to be synonymous or non-synonymous for calculating the non-synonymous/synonymous ratio and dN/dS ratio. The genotype data set of the 1,529 rice accessions (1,083 O. sativa accessions and 446 O. rufipogon accessions) was generated on the basis of the calls in each rice accession. Seven sets of genome sequences, which included bacterial-artificial-chromosome-based Sanger sequences and high-coverage resequencing data, were used to assess the accuracy of the genotype data sets (Supplementary Table 6). The wild rice accessions with sequencing coverage >9 were selected to investigate the heterozygosity based on the overlapped reads that were aligned onto the reference sequence. For each accession, the proportion of heterozygosity genotypes was calculated at the polymorphic sites (Supplementary Table 3).
Population genetics analysis
The software Haploview was used to calculate linkage disequilibrium with default settings, using SNPs with information in 446 O. rufipogon accessions50. Pairwise r2 was calculated for all the SNPs and then averaged across the whole genome. The matrix of pairwise genetic distance derived from simple SNP-matching coefficients was used to construct phylogenetic trees using the software PHYLIP51 (version 3.66). The software TreeView and MEGA5 were used for visualizing the phylogenetic trees. Principal component analysis of the SNPs was performed using the software EIGENSOFT52. The sequence diversity statistics (π) and the population-differentiation statistics (FST) were computed using a 100-kb window. The value of π was calculated for each group in O. rufipogon and O. sativa, respectively, and the ratio of π in the full population (or each clade) of O. rufipogon to that in the full population (or corresponding subspecies) of O. sativa was used to detect selective sweeps. The genomic regions where both O. rufipogon and O. sativa show a low level of genetic diversity were excluded for further analysis. To adopt appropriate thresholds to reduce the false-positive rate but also retain true selection signals, thresholds were chosen on the basis of both whole-genome permutation tests and signals at known loci. Permutation tests were performed to estimate the genome-wide type I error rate and determine the threshold to call selective sweeps (see Supplementary Information section 2 for details)53. The method cross-population extended haplotype homozygosity (XP-EHH) was also tested for detecting selective sweeps using the software xpehh54 (http://hgdp.uchicago.edu/Software/) (Supplementary Fig. 20). The genetic distance between two clades was computed based on the matrix of pairwise genetic distance, where the distance of all pairs of accessions from the two clades were retrieved and averaged. A custom Perl script was developed to plot all O. rufipogon accessions, using the public geographic information of world borders from the ‘Thematic Mapping’ data set (version 0.3). The computational simulations under different demographic scenarios were performed using the program SFS_CODE40.
Planting, crossing and phenotyping
For the O. rufipogon population, approximately five seeds for each accession from the collection of wild rice were germinated and planted in the experimental field (in Sanya, China at N 18.65°, E 109.80°) from March 2011. The leaf sheath colour was observed and scored directly and the tiller angle was measured for each plant. The mapping population of 210 backcross inbred lines (BILs) and 61 chromosome segment substitution lines (CSSLs) was derived from a cross between O. sativa ssp. indica cv. Guangluai-4 and O. rufipogon accession W1943. The BILs were developed by one generation of backcross to Guangluai-4 followed with six generations of self-fertilization. The CSSLs were developed by five generations of backcross to Guangluai-4 followed with three generations of self-fertilization. Phenotyping was conducted in the experimental field (in Shanghai, China at N 31.13°, E 121.28°) from May to October, 2011. The fifteen traits that we phenotyped for this study include germination rate, tiller angle, heading date, stigma colour, the degree of stigma exsertion, plant height, panicle length, the degree of shattering, awn length, grain number per panicle, grain length, grain width, grain weight per 1,000 grains, hull colour and pericarp colour. The degree of stigma exsertion was scored based on the observation of ∼20 randomly sampled spikelets of each line, on a scale of 1–3 (no, incomplete or complete exertion). Seed germination rate was measured by using mature seeds which were placed in a plastic Petri dishes kept at 30 °C in the dark for 48 h5. Other traits were phenotyped and scored as described previously21,22,29.
Imputation and association analysis
For the genotype data set in O. rufipogon, genotypes of 446 O. rufipogon accessions were called specifically at the ∼5 million SNP sites that were polymorphic in the O. rufipogon population. In the panel for GWAS, only the SNPs that have a minor allele frequency (MAF) of more than 5% and contain genotype calls of more than 100 accessions were left for subsequent imputation. The k-nearest neighbour algorithm-based imputation method was used for inferring missing calls21. The specificity of the genotype data set before and after imputation was assessed using three sets of genome sequences (Supplementary Table 6). Association analysis was conducted using the compressed mixed linear model55. The top five principle components were used as fixed effects and the matrix of genetic distance was used to model the variance–covariance matrix of the random effect. Permutation tests were used to define the threshold of association signals of the GWAS in the wild rice population. A total of 20 permutation analyses were performed (10 independent permutation tests for each of the two traits, sheath colour and tiller angle), which resulted in two ‘association signals’ with the thresholds we set53. Hence, there were an average of 0.1 false positives (that is, totally two false positives in 20 permutation tests) in a single whole-genome scanning analysis. Simulation tests were used to compare the performance of GWAS between the populations of cultivated and wild rice.
Genotyping and linkage analysis
Genomic DNA of each line in the mapping population was sequenced on the Illumina Genome Analyzer IIx, each to approximately 0.5× coverage. Both parents of the population, Guangluai-4 and W1943, were sequenced with at least 20× genome coverage, in a previous work21 and in this study, respectively. SNP identification between parents was conducted as described previously41. Genotype calling, recombination breakpoint determination and bin map construction was performed using the software SEG-Map (http://www.ncgr.ac.cn/software/SEG/). QTL analysis of the fifteen traits was conducted with the composite interval mapping (CIM) method implemented in the software Windows QTL Cartographer56 (version 2.5) with a window size of 10 cM and a step size of 2 cM. QTL with LOD value higher than 3.5 were called, of which the location was described according to its LOD peak location. The phenotypic effect (r2) of each QTL was computed using Windows QTL Cartographer. QTLs located within selective sweep regions were further used to associate the selected regions with their biological functions. It needs to be noted that we adopted a stringent threshold in the QTL calling (LOD > 3.5), and the genomic regions with LOD ranging from 2.5 to 3.5 may include many minor QTLs (the threshold was set to 2.5 in most studies).
Genome assembly and contig anchoring
The genome of W1943 was assembled by using a custom pipeline integrating Phusion2 (clustering the raw reads into different groups)57 and Phrap (then assembling all the reads in each group to generate contigs)58. The N50 length of the entire assembly was calculated for the initial contigs with small contigs of <200 bp excluded. All the full-length complementary DNA sequences46 of W1943 were aligned with the final assembly of W1943 genome sequence using the software GMAP59 (version 6) with the parameters ‘-K 15000’ and ‘-k 0.97’. The resulting contigs from whole-genome de novo assembly were anchored to the rice reference genome sequence (IRGSP4.0) using the software MUMmer60 (version 3).
Genome annotation and variant detection
Gene models of the genome of the wild rice W1943 were predicted using the software Fgenesh that was set for a monocot model61 (version 2.0). The resulting proteome of W1943 was compared with protein sequences in Rice Genome Annotation Project (version 7.0) using BLASTP with a cutoff of a minimum of 95% identity. Sequence variants, including SNPs, indels and imbalanced substitutions, were called using the diffseq program in the EMBOSS package62. Indels of large size were called from the alignment results of MUMmer. Effects of the sequence variants were predicted according to the gene models of Nipponbare in the RAP-DB (release 2) across the rice genome. For indels in genic regions and SNPs with large effect around the domestication loci, the effects were mainly based on the reference gene models.
Population-scale sequence comparison
The sequence reads of 1,083 O. sativa accessions, 446 O. rufipogon accessions and 15 outgroup accessions were then aligned against assembled genome sequences of W1943 using the same parameters with those against the reference Nipponbare genome sequences. Genotypes of each accession were called at all sequence variant sites (including SNPs, indels and imbalanced substitutions that were detected from assembled sequences), based on the alignment outputs against the two genome sequences. The allele frequencies at the sequence variant sites were calculated for each clade of O. sativa and O. rufipogon. In each clade, variant sites with information of less than 10 accessions (less than 2 for the outgroups) were then excluded for computing allele frequencies, namely no data available.
Accession codes
Primary accessions
European Nucleotide Archive
Data deposits
DNA sequencing data are deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) under accession numbers ERP001143, ERP000729 and ERP000106. De novo assembly and genome annotation of wild rice W1943, the genotype dataset of 1,529 rice accessions and the imputed dataset of 446 O. rufipogon accessions for GWAS are available at the Rice Haplotype Map Project database (http://www.ncgr.ac.cn/RiceHap3).
References
Oka, H. I. Origin of cultivated rice. (Japan Scientific Societies Press, 1988)
Khush, G. S. Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 35, 25–34 (1997)
Cheng, C. et al. Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Mol. Biol. Evol. 20, 67–75 (2003)
Fuller, D. Q. et al. Consilience of genetics and archaeobotany in the entangled history of rice. Archaeol. Anthropol. Sci. 2, 115–131 (2010)
Li, C., Zhou, A. & Sang, T. Genetic analysis of rice domestication syndrome with the wild annual species, Oryza nivara. New Phytol. 170, 185–194 (2006)
Li, C., Zhou, A. & Sang, T. Rice domestication by reducing shattering. Science 311, 1936–1939 (2006)
Jin, J. et al. Genetic control of rice plant architecture under domestication. Nature Genet. 40, 1365–1369 (2008)
Tan, L. et al. Control of a key transition from prostrate to erect growth in rice domestication. Nature Genet. 40, 1360–1364 (2008)
Zhu, B. F. et al. Genetic control of a transition from black to straw-white seed hull in rice domestication. Plant Physiol. 155, 1301–1311 (2011)
Londo, J. P., Chiang, Y. C., Hung, K. H., Chiang, T. Y. & Schaal, B. A. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc. Natl Acad. Sci. USA 103, 9578–9583 (2006)
Caicedo, A. L. et al. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet. 3, e163 (2007)
Kovach, M. J., Sweeney, M. T. & McCouch, S. R. New insights into the history of rice domestication. Trends Genet. 23, 578–587 (2007)
Sang, T. & Ge, S. The puzzle of rice domestication. J. Integr. Plant Biol. 49, 760–768 (2007)
Zong, Y. et al. Fire and flood management of coastal swamp enabled first rice paddy cultivation in east China. Nature 449, 459–462 (2007)
Fuller, D. Q. et al. The domestication process and domestication rate in rice: spikelet bases from the Lower Yangtze. Science 323, 1607–1610 (2009)
Zhang, L. B. et al. Selection on grain shattering genes and rates of rice domestication. New Phytol. 184, 708–720 (2009)
Molina, J. et al. Molecular evidence for a single evolutionary origin of domesticated rice. Proc. Natl Acad. Sci. USA 108, 8351–8356 (2011)
He, Z. et al. Two evolutionary histories in the genome of rice: the roles of domestication genes. PLoS Genet. 7, e1002100 (2011)
Xu, X. et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nature Biotechnol. 30, 105–111 (2012)
Ma, J. & Bennetzen, J. L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl Acad. Sci. USA 101, 12404–12410 (2004)
Huang, X. et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genet. 42, 961–967 (2010)
Huang, X. et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nature Genet. 44, 32–39 (2012)
Ge, S., Sang, T., Lu, B. R. & Hong, D. Y. Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc. Natl Acad. Sci. USA 96, 14400–14405 (1999)
Vaughan, D. A., Morishima, H. & Kadowaki, K. Diversity in the Oryza genus. Curr. Opin. Plant Biol. 6, 139–146 (2003)
Doebley, J. F., Gaut, B. S. & Smith, B. D. The molecular genetics of crop domestication. Cell 127, 1309–1321 (2006)
Novembre, J. & Stephens, M. Interpreting principal component analyses of spatial population genetic variation. Nature Genet. 40, 646–649 (2008)
Mizuta, Y., Harushima, Y. & Kurata, N. Rice pollen hybrid incompatibility caused by reciprocal gene loss of duplicated genes. Proc. Natl Acad. Sci. USA 107, 20417–20422 (2010)
Tadege, M. et al. Reciprocal control of flowering time by OsSOC1 in transgenic Arabidopsis and by FLC in transgenic rice. Plant Biotechnol. J. 1, 361–369 (2003)
Wang, L. et al. Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor. Appl. Genet. 122, 327–340 (2011)
Xue, W. et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nature Genet. 40, 761–767 (2008)
Andaya, V. C. & Tai, T. H. Fine mapping of the qCTS12 locus, a major QTL for seedling cold tolerance in rice. Theor. Appl. Genet. 113, 467–475 (2006)
Saitoh, K., Onishi, K., Mikami, I., Thidar, K. & Sano, Y. Allelic diversification at the C (OsC1) locus of wild and cultivated rice: nucleotide changes associated with phenotypes. Genetics 168, 997–1007 (2004)
Fan, C. et al. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 112, 1164–1171 (2006)
Takano-Kai, N. et al. Evolutionary history of GS3, a gene conferring grain length in rice. Genetics 182, 1323–1334 (2009)
Shomura, A. et al. Deletion in a gene associated with grain size increased yields during rice domestication. Nature Genet. 40, 1023–1028 (2008)
Konishi, S. et al. An SNP caused loss of seed shattering during rice domestication. Science 312, 1392–1396 (2006)
Wang, Z. Y. et al. The amylose content in rice endosperm is related to the post-transcriptional regulation of the waxy gene. Plant J. 7, 613–622 (1995)
Sweeney, M. T., Thomson, M. J., Pfeil, B. E. & McCouch, S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18, 283–294 (2006)
Sweeney, M. T. et al. Global dissemination of a single mutation conferring white pericarp in rice. PLoS Genet. 3, e133 (2007)
Hernandez, R. D. A flexible forward simulator for populations subjects to selection and demography. Bioinformatics 24, 2786–2787 (2008)
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076 (2009)
Chen, K. Y., Cong, B., Wing, R., Vrebalov, J. & Tanksley, S. D. Changes in regulation of a transcription factor lead to autogamy in cultivated tomatoes. Science 318, 643–645 (2007)
Rieseberg, L. H. & Blackman, B. K. Speciation genes in plants. Ann. Bot. 106, 439–455 (2010)
Gan, X. et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011)
Schneeberger, K. et al. Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc. Natl Acad. Sci. USA 108, 10249–10254 (2011)
Lu, T. et al. Collection and comparative analysis of 1888 full-length cDNAs from wild rice Oryza rufipogon Griff. W1943. DNA Res. 15, 285–295 (2008)
Tang, H., Sezen, U. & Paterson, A. H. Domestication and plant genomes. Curr. Opin. Plant Biol. 13, 160–166 (2010)
Morrell, P. L., Buckler, E. S. & Ross-Ibarra, J. Crop genomics: advances and applications. Nature Rev. Genet. 13, 85–96 (2012)
Kozarewa, I. et al. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nature Methods 6, 291–295 (2009)
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005)
Felsenstein, J. PHYLIP: phylogeny inference package (version 3.2). Cladistics 5, 164–166 (1989)
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genet. 38, 904–909 (2006)
Churchill, G. A. & Doerge, R. W. Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971 (1994)
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007)
Zhang, Z. et al. Mixed linear model approach adapted for genome-wide association studies. Nature Genet. 42, 355–360 (2010)
Wang, S., Basten, C. J. & Zeng, Z. B. Windows QTL Cartographer 2.5. (Department of Statistics, North Carolina State Univ., 2007)
Mullikin, J. C. & Ning, Z. The phusion assembler. Genome Res. 13, 81–90 (2003)
de la Bastide, M. & McCombie, W. R. Assembling genomic DNA sequences with PHRAP. Curr. Protoc. Bioinformatics 17, 11.4.1–11.4.15 (2007)
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005)
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004)
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000)
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000)
Acknowledgements
We thank the China National Rice Research Institute for providing all cultivated rice germplasm and Chinese wild rice accessions. The rest of the wild rice accessions were distributed from the Rice Collection of National Institute of Genetics jointly supported by the National Bioresource Project, MEXT, and Systems Functional Genetics Project of the Transdisciplinary Research Integration Center, ROIS, Japan. We thank Z. Ning, Y. Minobe and A. Osbourn for their advice and assistance in this work. This work was supported by the Ministry of Science and Technology of China (2011CB100205, 2012AA10A302 and 2012AA10A304), the Ministry of Agriculture of China (2011ZX08009-002 and 2011ZX08001-004), the National Natural Science Foundation of China (31121063) and the Chinese Academy of Sciences (to B.H.).
Author information
Authors and Affiliations
Contributions
B.H. conceived the project and its components. X.H. and B.H. designed studies and contributed to the original concept of the project. X.W., X.Y. and Q.X. contributed the collection of rice cultivars and Chinese wild rice accessions. N.K., H.F., T.K., T.M., A.F. and A.T. contributed the collection of other wild rice accessions and analysed geographical distributions of wild rice. Z.-X.W., A.W., Y.W., L.F., Qilin Z., C.L., G.D. and Q.Q. contributed in phenotyping of rice. Z.-X.W., A.W. and L.F. contributed in phenotyping of the backcross inbred line population and genetic mapping of domestication-related traits. W.L., Y.G., Y.L., C.Z., D.F., Q.W. and Q.F. performed the genome sequencing. X.H., Y.Z., K.L., C.Z., T.H., L.Z. and T.L. performed genome data analysis. Qiang Z. and H.L. performed de novo genome assembly. X.H. performed evolutionary study and genome annotation. X.H., Y.Z. and K.L performed GWAS, population genetics, and statistical analyses. J.L. contributed to functional analyses. X.H. and B.H. analysed whole data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Text 1-8, Supplementary References and Supplementary Tables 1, 3-6, and 8114 (see separate zipped file for Supplementary Tables 2, 7, 15, 16, 17 and 18. (PDF 458 kb)
Supplementary Figures
This file contains Supplementary Figures 1-34. (PDF 7906 kb)
Supplementary Tables
This file contains Supplementary Tables 2, 7 and 15-18. (ZIP 268 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Huang, X., Kurata, N., Wei, X. et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012). https://doi.org/10.1038/nature11532
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature11532
This article is cited by
-
Genomic insight into the origin, domestication, dispersal, diversification and human selection of Tartary buckwheat
Genome Biology (2024)
-
OsVPE2, a Member of Vacuolar Processing Enzyme Family, Decreases Chilling Tolerance of Rice
Rice (2024)
-
Genomic variation in weedy and cultivated broomcorn millet accessions uncovers the genetic architecture of agronomic traits
Nature Genetics (2024)
-
A mediator of OsbZIP46 deactivation and degradation negatively regulates seed dormancy in rice
Nature Communications (2024)
-
Dysfunction of duplicated pair rice histone acetyltransferases causes segregation distortion and an interspecific reproductive barrier
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
