
Abstract
Bananas (Musa spp.), including dessert and cooking types, are giant perennial monocotyledonous herbs of the order Zingiberales, a sister group to the well-studied Poales, which include cereals. Bananas are vital for food security in many tropical and subtropical countries and the most popular fruit in industrialized countries1. The Musa domestication process started some 7,000 years ago in Southeast Asia. It involved hybridizations between diverse species and subspecies, fostered by human migrations2, and selection of diploid and triploid seedless, parthenocarpic hybrids thereafter widely dispersed by vegetative propagation. Half of the current production relies on somaclones derived from a single triploid genotype (Cavendish)1. Pests and diseases have gradually become adapted, representing an imminent danger for global banana production3,4. Here we describe the draft sequence of the 523-megabase genome of a Musa acuminata doubled-haploid genotype, providing a crucial stepping-stone for genetic improvement of banana. We detected three rounds of whole-genome duplications in the Musa lineage, independently of those previously described in the Poales lineage and the one we detected in the Arecales lineage. This first monocotyledon high-continuity whole-genome sequence reported outside Poales represents an essential bridge for comparative genome analysis in plants. As such, it clarifies commelinid-monocotyledon phylogenetic relationships, reveals Poaceae-specific features and has led to the discovery of conserved non-coding sequences predating monocotyledon–eudicotyledon divergence.
Similar content being viewed by others

Main
Banana cultivars mainly involve M. acuminata (A genome) and Musa balbisiana (B genome) and are sometimes diploid but generally triploid5,6. We sequenced the genome of DH-Pahang, a doubled-haploid M. acuminata genotype (2n = 22), of the subspecies malaccensis that contributed one of the three acuminata genomes of Cavendish7. A total of 27.5 million Roche/454 single reads and 2.1 million Sanger reads were produced, representing 20.5× coverage of the 523-megabase (Mb) DH-Pahang genome size, as estimated by flow cytometry. In addition, 50× of Illumina data were used to correct sequence errors. The assembly consisted of 24,425 contigs and 7,513 scaffolds with a total length of 472.2 Mb, which represented 90% of the estimated DH-Pahang genome size. Ninety per cent of the assembly was in 647 scaffolds, and the N50 (the scaffold size above which 50% of the total length of the sequence assembly can be found) was 1.3 Mb (Supplementary Text and Supplementary Tables 1–3). We anchored 70% of the assembly (332 Mb) along the 11 Musa linkage groups of the Pahang genetic map. This corresponded to 258 scaffolds and included 98.0% of the scaffolds larger than 1 Mb and 92% of the annotated genes (Supplementary Text, Supplementary Table 4 and Supplementary Fig. 1).
We identified 36,542 protein-coding gene models in the Musa genome (Supplementary Tables 1 and 5). A total of 235 microRNAs from 37 families were identified, including only one of the eight microRNA gene (MIR) families found so far solely in Poaceae8 (Supplementary Tables 6 and 7).
Viral sequences related to the banana streak virus (BSV) dsDNA plant pararetrovirus were found to be integrated in the Pahang genome, with 24 loci spanning 10 chromosomes (Supplementary Text and Supplementary Fig. 2). They belonged to a badnavirus phylogenetic group that differed from the endogenous BSV species (eBSV) found in M. balbisiana9 and most of them formed a new subgroup (Supplementary Fig. 3). Importantly, all of the integrations were highly reorganized and fragmented and thus did not seem to be capable of forming free infectious viral particles, contrary to the eBSV described in M. balbisiana10.
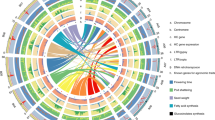
Transposable elements account for almost half of the Musa sequence (Supplementary Text and Supplementary Tables 1 and 8–10). Long terminal repeat retrotransposons represent the largest part, with Copia elements being much more abundant than Gypsy elements (25.7–11.6%) (Supplementary Fig. 4). No major recent wave of long terminal repeat retrotransposon insertions appears to have occurred in the Musa lineage. Fewer than 1% of the long terminal repeat retrotransposons are complete and their median date of insertion is around 4 Myr ago, corresponding to the half-life of this type of transposable element11 (Supplementary Fig. 5). Long interspersed elements (LINEs) represent 5.5% of the genome. The banana genome is exceptional in the composition of its class 2 element population, which represents only about 1.3% of the genome. The only superfamilies identified were hAT, followed by Harbinger and Mutator. Only the first family was significantly represented and had non-autonomous deletion derivatives. The superfamilies CACTA and Mariner, which have been found in high copy numbers in all angiosperm genomes studied so far, are absent from the banana genome. Gene-rich regions are mostly located on distal parts of chromosomes, as observed in other plant genomes (Fig. 1 and Supplementary Fig. 1). There is, however, a particularly sharp transition between gene-rich and transposable-element-rich regions. This observation is confirmed by the pattern observed after genomic in situ hybridization, which shows that transposable elements are typically concentrated around centromeres in Musa12 (Supplementary Fig. 6). The asymmetric transposable element distributions along the chromosomes indicated that chromosomes 1 and 2 are acrocentric in DH-Pahang (Fig. 1). Long terminal repeat retrotransposons are particularly abundant in centromeric and pericentromeric chromosome regions. Their accumulation in these regions, particularly for the oldest ones, suggests that they are preferentially eliminated from gene-rich regions13 (Supplementary Fig. 5). Remarkably, typical short tandem centromeric repeats were not found in Musa. However, one long interspersed element (named Nanica) identified in the unassembled reads was localized by fluorescence in situ hybridization in the centromeric region of all Musa chromosomes (Supplementary Fig. 7 and Supplementary Table 10).

Distribution of genes and transposable elements (left) and paralogous relationships between the 11 chromosomes indicated with 12 distinct colours corresponding to the 12 Musa α/β ancestral blocks (right). LINEs, long interspersed elements.
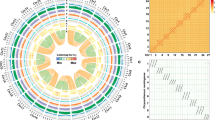
Whole-genome duplications (WGDs) have played a major role in angiosperm genome evolution14; the first evidence of a WGD event in the Musa lineage was reported by Lescot et al.15. We uncovered a complex pattern of paralogous relationships between the 11 Musa chromosomes (Supplementary Text and Supplementary Fig. 8). Most paralogous gene clusters shared relationships with three other clusters, suggesting that two WGDs (denoted as α and β) occurred (Supplementary Fig. 9). Based on Ks and synteny relationships, duplicated gene clusters were tentatively assembled into 12 Musa ancestral blocks representing the ancestral genome before the α/β duplications (Figs 1 and 2 and Supplementary Figs 10–12). The duplicated segments included in the Musa ancestral blocks cover 222 Mb (67% of the anchored assembly) and contain 26,829 genes (80% of the anchored genes) (Supplementary Table 11). The Ks distribution among pairs of paralogous gene clusters dated the two WGDs at a similar period around 65 Myr ago (Supplementary Fig. 13), consistent with the WGDs that occurred in many different plant lineages near the Cretaceous–Tertiary boundary14 (Fig. 3). Additional paralogous relationships between the 12 Musa ancestral blocks displaying higher Ks values suggested that an additional, more ancient duplication event (denoted as γ) occurred around 100 Myr ago (Fig. 3 and Supplementary Figs 10, 11, 13 and 14).

a, Paralogous relationships between chromosome segments from Musa α/β ancestral blocks 2 (red) and 8 (green). The 12 Musa α/β ancestral blocks are shown in different colours on the circle. b, Orthologous relationships of Musa ancestral blocks 2 and 8 with rice ancestral blocks ρ2, ρ5 and σ6. We did not observe a one-to-one relationship between, for instance, Musa α/β ancestral block 2 and one ρ ancestral block, which suggests that the γ and σ duplications are two separate events. c, Representation of the deduced WGD event.

Boxes indicate WGD events. Green boxes indicate WGD events analysed in this paper. All nodes have 100% bootstrap support in a maximum likelihood analysis. Branch lengths (synonymous substitution rate) are indicated. The timing of the β WGD event relative to the Musaceae/Zingiberaceae split remains to be clarified.
In the grass lineage, it is well established that one WGD (denoted as ρ) occurred around 50–70 Myr ago, after Poales separated from other monocotyledon orders16,17. Evidence was reported on an additional WGD (denoted as σ) earlier in the monocotyledon lineage, but after its divergence from the eudicotyledons18. Our comparison of the Musa ancestral blocks with the Poaceae ρ and σ ancestral blocks as defined by Tang et al.18 revealed that genes from segments of different ρ blocks (corresponding to one σ block) have orthologous relationships with the same Musa regions, showing that the σ Poaceae event is not shared with Musa. Reciprocally, genes from Musa α/β paralogous segments have orthologous relationships with the same ρ and σ regions, showing that the earliest duplication (γ) we identified in the Musa lineage is not shared with Poaceae (Fig. 2 and Supplementary Fig. 15).
Independent phylogenomic analyses performed on 3,553 gene families, including genes mapped to syntenic ancestral blocks, generated further evidence (98.7–77.6% of the gene trees, Supplementary Text) that the three rounds of palaeopolyploidization identified in the Musa genome and the two previously reported in the Poaceae lineage occurred independently after the Poales and Zingiberales divergence estimated at 109–123 Myr ago19 (Fig. 3 and Supplementary Fig. 16).
Resolution of the Zingiberales relationship relative to Poales and Arecales (palms) has been problematic (see, for example, Givnish et al.20), but our analysis of 93 single-copy nuclear genes suggested that the palms are more closely related to Zingiberales (including Musa) than to Poales (Fig. 3, Supplementary Text and Supplementary Fig. 17). Phylogenomic and synteny analyses indicated that the palms do not, however, share any of the Poales or Zingiberales WGDs discussed here (Supplementary Figs 17 and 18). Moreover, our Ks analyses of date-palm gene models21 indicated that the palm genome had its own WGD event (Supplementary Fig. 19).
Most (65.4%) of the genes included in the Musa α/β ancestral blocks are singletons and only 10% are retained in four copies, in agreement with the loss of most gene-duplicated copies after WGD22. The most retained gene ontology categories corresponded to genes involved in transcription regulation (transcription factor activity), signal transduction including small GTPase-mediated signal transduction and protein kinases, and translational elongation (Supplementary Text and Supplementary Tables 12–14). This might be explained by the gene balance hypothesis23, which suggests that genes involved in multiproteic complexes or regulatory genes are dosage sensitive and thus are more prone to be co-retained or co-lost after WGD24. With 3,155 genes, the number of Musa transcription factors identified is among the highest of all sequenced plant genomes (Supplementary Table 15 and 16).
Comparison of Musa, rice, sorghum, Brachypodium, date palm (Phoenix dactylifera) and Arabidopsis proteomes revealed 7,674 gene clusters in common to all six species, thus representing ancestral gene families (Fig. 4). Interestingly, many specific clusters (2,809 in our setting) proved specific to Poaceae, suggesting a high level of gene divergence and diversification within the grass lineage. Specific Musa clusters (759) were enriched in genes encoding transcription factors (for example, Myb and AP2/ERF families), defence-related proteins, enzymes of cell-wall biosynthesis and enzymes of secondary metabolism (Supplementary Table 17).

Numbers of clusters are provided in the intersections. The total number of sequences for each species is provided under the species name (total number of sequences/total number of clustered sequences).
We compared the distribution of GC3 content (G or C in the third codon position) in Musa coding sequences with those of rice, ginger (Zingiber officinale) and date palm because this distribution was shown to be bimodal in Poaceae and unimodal in all analysed eudicotyledons25. In Musa, a GC-rich peak was apparent but less distinct from the GC-poor one (Supplementary Text, Supplementary Figs 20–23 and Supplementary Table 18), which confirms preliminary evidence that placed Musa in an intermediate position15. This feature was shared with ginger (Zingiberales) and contrasts with the unimodal GC distribution of date-palm coding sequences (Supplementary Fig. 21).
Plant conserved non-coding sequences (CNSs)—a type of phylogenetic footprint—are enriched in known transcription factors or other cis-acting binding sites, and are usually clustered around regulatory genes, supporting their functionality26. Starting with a collection of 16,978 CNSs conserved in Poaceae, we used the Musa genome to identify the 116 most deeply conserved regulatory binding sequences in the commelinid monocotyledon lineage (Supplementary Text, Supplementary Tables 19 and 20, and Supplementary Fig. 24). Deeply conserved CNSs in commelinids were frequently found located 5′ to genes encoding transcription factors, and were significantly enriched in WRKY motifs (Supplementary Table 21). After WGD, genes associated with deeply conserved CNSs were found to be retained as duplicates more often than genes with less deeply conserved CNSs (Supplementary Table 22). The banana genome also served as a stepping-stone to finding CNSs conserved beyond monocotyledons, including 18 CNSs that were found in this study to be conserved in the expected syntenic position in eudicotyledons as well (Supplementary Table 23). This evolutionary distance is not unusual for vertebrate CNSs (detectable after more than 400 million years of divergence)27 but it surpasses the findings of previous plant whole-genome surveys26. Plant deeply conserved CNSs are therefore rare but do exist, and are short compared with those of animals27, and must be at least as old as monocotyledon–eudicotyledon divergence (more than 130 million years of divergence).
The reference Musa genome sequence represents a major advance in the quest to unravel the complex genetics of this vital crop, whose breeding is particularly challenging. Having access to the entire Musa gene repertoire is a key to identifying genes responsible for important agronomic characters, such as fruit quality and pest resistance. Bananas are exported green and then ripened by application of ethylene. RNA-Seq analysis indicated strong transcriptional reprogramming in mature green banana fruits after ethylenic treatment (Supplementary Text, Supplementary Tables 24–26 and Supplementary Fig. 25). Transcription factors were particularly involved with 597 differentially regulated genes. Various modifications confirmed the biochemistry of the banana ripening process28, such as highly upregulated genes encoding cell-wall modifying enzymes, three downregulated starch synthase genes and one upregulated β-amylase gene. Two WGD-derived paralogous vacuolar invertase genes involved in sucrose conversion displayed opposite expression profiles, suggesting subfunctionalization and possible contribution to the soluble sugar balance in ripening bananas (Supplementary Fig. 26). The race against pathogen evolution is particularly critical in clonally propagated crops such as banana. Up to 50 pesticide treatments a year are required in large plantations against black leaf streak disease, a recent pandemy caused by Mycosphaerella fijiensis3. Moreover, outbreaks of a new race of the devastating Panama disease fungus (Fusarium oxysporum f. sp. cubense) are spreading in Asia4. Among defence-related genes, those encoding nucleotide-binding site leucine-rich repeat proteins were found to be little represented in the Musa sequence (89 genes) (Supplementary Table 27). RNA-Seq analysis showed that receptor-like kinase genes were upregulated in a partially resistant interaction with M. fijiensis (Supplementary Text, Supplementary Table 28 and Supplementary Fig. 27). Interestingly, direct links between basal plant immunity triggered by receptor-like kinase proteins and quantitative trait loci for partial resistance have been recently established in several plant species (see, for example, Poland et al.29). In addition, we showed that DH-Pahang is highly resistant to the new broad-range Fusarium oxysporum race 4 (Supplementary Text and Supplementary Fig. 28), thus conferring additional specific value to the DH-Pahang sequence.
The Musa genome sequence reported here bridges a large gap in genome evolution studies. As such, it sheds new light on the monocotyledon lineage. Several Poaceae-specific characteristics could be highlighted, boosting prospects for analysing the emergence of this very successful family. The Musa genome also enabled identification of deeply conserved CNS within commelinid monocotyledons and between monocotyledons and eudicotyledons, representing an invaluable resource for detecting novel motifs with a gene regulation function. We detected three rounds of polyploidization in the Musa lineage, which were followed by gene loss and chromosome rearrangements, resulting in little synteny conservation between lineages (Supplementary Figs 29 and 30) and over-retention of some gene classes, thus providing ample opportunities for independent diversification. In particular, transcription factor families are strikingly expanded in Musa compared with other plant genomes and probably contribute to specific aspects of banana development.
The Musa genome sequence is therefore an important advance towards securing food supplies from new generations of Musa crops, and provides an invaluable stepping-stone for plant gene and genome evolution studies.
Methods Summary
Sanger (ABI 3730xl sequencers) and Roche/454 (GSFLX pyrosequencing platform) reads were assembled with Newbler. Scaffolds were anchored to Pahang linkage groups using 652 markers (SSR and DArT). Protein-coding gene model prediction on the repeat-masked sequence was done with the GAZE30 computational framework by combining ab initio gene predictions, protein similarity, existing banana and monocotyledon transcript information and banana RNA-Seq data. A reference library of Musa transposable elements was built based on sequence similarity at the protein and nucleic acid levels and on searches for transposable-element structural signatures. The library was used with the REPET package (http://urgi.versailles.inra.fr/Tools/REPET) to screen the Musa assembly and quantify repeats.
RNA-Seq differential gene expression analysis was performed using Illumina GAIIx 76 bases reads that were mapped to the DH-Pahang sequence using SOAP2 (http://soap.genomics.org.cn/).
Online Methods
Plant material and DNA preparation
Doubled-haploid Pahang (DH-Pahang, ITC1511) was obtained from wild M. acuminata subspecies malaccensis accession ‘Pahang’ through anther culture and spontaneous chromosome doubling31. Genome sizes were estimated by flow cytometry according to Marie and Brown32. High molecular weight DNA was prepared from the youngest fully expanded leaf of DH-Pahang as described in Piffanelli et al.33 with minor modifications (Supplementary Text).
Genome sequencing
The genome was sequenced using a Whole Genome Shotgun strategy combining Sanger, Roche/454 GSFLX and Illumina GAIIx technologies. Sanger sequencing was performed with the ABI 3730xl on 10-kilobase (kb) inserts and on two BAC libraries generated with the HindIII and BamHI restriction enzymes resulting in 2.0 million 10-kb fragment-ends and about 90,500 BAC-ends. A total of 27.5 million reads were obtained using Roche/454 GSFLX.
Genome assembly and automatic error corrections with Solexa/Illumina reads
All reads were assembled with Newbler version MapAsmResearch-03/15/2010. From the initial 29,620,875 reads, 87.8% were assembled. We obtained 24,425 contigs that were linked into 7,513 scaffolds. The contig N50 (the contig size above which 50% of the total length of the sequence assembly is included) was 43.1 kb, and the scaffold N50 was 1.3 Mb. The cumulative scaffold size was 472.2 Mb, about 10% smaller than the estimated genome size of 523 Mb. Sequence quality of scaffolds from the Newbler assembly was improved as described previously34, by automatic error corrections with Solexa/Illumina reads (50-fold genome coverage), which have a different bias in error type compared with 454 reads. To validate the assembly, we built a unigene set corresponding to 15,017 isotigs that were obtained from the assembly with Newbler (version MapAsmResearch-03/15/2010) of Roche/454 GSFLX reads from six different complementary DNA (cDNA) libraries (829,587 reads, Supplementary Text). The unigenes were aligned with the assembly using the BLAT algorithm35 with default parameters, and the best match was kept for each unigene. The assembly covers a very large proportion of the euchromatin of the M. acuminata genome, as 99% of the set of 15,017 unigenes was recovered in the DH-Pahang genome assembly.
Construction of the Pahang genetic map and sequence anchoring
A genetic map was specifically developed for scaffold anchoring and orientation. A total of 2,454 single sequence repeats (SSR) markers and 1,008 polymorphic diversity array technology (DArT) markers were analysed including 1,411 new SSRs defined on sequence contigs and scaffolds. The map used for anchoring was built with 589 SSR and 63 DArT markers that were genotyped on 180 individuals of the Pahang self progeny. Data were analysed using JoinMap 4 (Plant Research International). The 652 markers anchored 258 scaffolds along the 11 linkage groups of the genetic map. Orientation of scaffolds was possible when two or more separated genetic markers were present on the same scaffold. All these data were used to generate 11 banana pseudo-chromosomes with 100Ns inserted between neighbouring scaffolds (Supplementary Fig. 1 and Supplementary Table 4).
Gene prediction
The following resources were integrated to automatically build Musa acuminata gene models using GAZE30: ab initio gene predictions from Geneid36, SNAP37 and FGENESH38; Genewise39 alignments of the UniProt40 database; Est2genome41 alignments of full-length cDNAs from six tissue samples of DH-Pahang and a collection of 6,888,879 monocotyledon messenger RNAs from the EMBL database and finally Gmorse models42 derived from RNA-Seq reads (Supplementary Text). MicroRNAs were predicted based on comparison using the Plant MicroRNA Database (http://bioinformatics.cau.edu.cn/PMRD/).
Identification of integrated pararetrovirus sequences
Viral integrants in the DH-Pahang genome were detected with a BLASTN analysis using either full-length BSV sequences or a 540-base-pair fragment of the RT/RNase H region of the badnaviruses genome (Supplementary Text).
Identification, classification and distribution of Musa transposable elements
Musa transposable elements were identified based on sequence similarity at the protein and nucleic-acid levels using BLASTP and TBLASTN43 and by de novo identification based on transposable-element structural signatures. Repeats from 1,832,094 remaining unassembled reads were characterized with a BLASTN ‘walking’ approach44. The obtained reference Musa transposable-element library was used with REPET45 to screen the assembly and quantify repeats (Supplementary Text). Insertion dates of full-length long terminal repeat retrotransposons were determined as described in Ma et al.46 with a substitution rate of 9 × 10−9 per site per year, which is twofold higher than that determined for Musa genes by Lescot et al.15.
Identification of Musa WGDs and comparative genome analyses
For the identification of Musa WGD, an all-against-all comparison of Musa proteins was done using the GenomeQuest BLAST package (LASSAP47) and retaining ten best hits for each gene. Clusters of paralogues composed of at least 20 genes with a maximal distance of 40 genes between syntenic genes were built with an in-house perl script, using a single linkage clustering with a Euclidian distance based on the gene index order in each chromosome. These clusters were refined using Synmap (http://synteny.cnr.berkeley.edu/CoGe/SynMap.pl) with the BLASTZ algorithm, an average distance expected between syntenic genes of 10, a maximum distance between two matches of 30, a minimum number of aligned gene pairs of 10 and a quota-align ratio of 3 to 3 (Supplementary Text).
For comparative genome analyses, orthologous gene-pairs were identified using predicted proteomes of M. acuminata, O. sativa (IRGPS/RAP, build 4), Vitis vinifera (http://www.genoscope.cns.fr/externe/Download/Projets/Projet_ML/data/12X/annotation/) and Phoenix dactylifera (draft sequence version 3, http://qatar-weill.cornell.edu/research/datepalmGenome/download.html). Alignments were performed using BLASTP (e value 1 × 10−5) and retaining best hits. Syntenic clusters of genes were built using a single linkage clustering with a Euclidian distance. Dot-plots were performed using an in-house perl program allowing the painting of paralogous and orthologous gene clusters. Circle diagrams were made with Circos48.
To calculate the number of synonymous substitutions per site (Ks), ClustalW49 alignments of paralogous and orthologous protein sequences were used to guide nucleic coding sequence alignments with PAL2NAL50. Ks values were calculated using the Yang–Nielson method implemented in PAML51.
Phylogenomic analysis
To infer the timing of genome duplication events relative to speciation events, all annotated Musa genes were sorted based on best BLASTP hit into the gene family clusters circumscribed by Jiao et al.52 and the PlantTribes database53 (http://fgp.bio.psu.edu/tribedb/), including sequenced eudicotyledons and monocotyledons, along with transcriptome assemblies for other non-grass monocotyledons (Supplementary Text). Gene family clusters were queried for Sorghum18 and Musa orthologues mapping to syntenic blocks, and maximum likelihood gene trees were estimated for these gene families using the GTR+GAMMA model of molecular evolution in RAxML54. The estimation of divergence times was performed on maximum likelihood trees based on concatenated MAFFT55 alignments for 93 gene families that included only one gene from each of the sequenced genomes (Supplementary Text).
Comparative analysis of gene families
The Musa proteome was globally compared with O. sativa (RGAP version 6.0), S. bicolor (JGI version 1.4), B. distachyon (JGI version 1.0), P. dactylifera (draft sequence version 3, http://qatar-weill.cornell.edu/research/datepalmGenome/download.html) and A. thaliana (TAIR version 9) proteomes filtered of transposable elements and alternative splicing. An all-against-all comparison was performed using BLASTP (1 × 10−10) followed by clustering with OrthoMCL56 (inflation 1.5). Analysis of species-specific sets was made with a Fisher’s exact test (P < 0.0001) on InterPro (version 28) domains. For analyses of specific gene families, the 36,542 Musa protein sequences were inserted in the plant proteome clustering of the GreenPhyl database57. Transcription factor families were mostly retrieved based on InterPro domains, using the IPR2genomes tool in GreenphylDB57 (Supplementary Text). Kinases and nucleotide-binding site proteins were retrieved using hidden markov models (hmmsearch version 3) to search for corresponding Pfam domains (Supplementary Text).
Identification of CNSs
Pan-grass CNSs conserved between rice, sorghum and Brachypodium were prepared using an automated pipeline58. The obtained 16,978 CNSs were used to query Musa using BLATSN (e value < 0.001) following a manual or a semi-automated procedure depending on CNS size (Supplementary Text and Supplementary Fig. 24). The resulting set of CNSs was extensively analysed using GEvo59 (http://synteny.cnr.berkeley.edu/CoGe/GEvo.pl) and the MSU Rice Genome Browser60 (http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/) to remove false positives (Supplementary Table 19). Adding rice and sorghum homeologues, Brachypodium and maize orthologues and Arabidopsis ‘best hit orthologues’ to GEvo panels enabled the identification of 18 CNSs conserved deeply throughout the plant kingdom.
Transcriptome sequencing
For RNA-Seq analyses (Supplementary Text), cDNA libraries were sequenced using 76-base length read chemistry in a single-flow cell on the Illumina GA IIx. Reads were mapped against the automatic annotated transcripts with SOAPaligner/Soap2 (2.20, http://soap.genomics.org.cn/) and only the unique mapped reads were kept. RNA-seq data were statistically analysed with the R packages baySeq version 1.6.0 (ref. 61) and DESeq version 1.5.6 (ref. 62).
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The final assembly and annotation are deposited in DDBJ/EMBL/ GenBank under accession numbers CAIC01000001–CAIC01024424 (contigs), HE806462–HE813974 (scaffolds) and HE813975–HE813985 (chromosomes). Genome sequence and annotation can be obtained and viewed at http://banana-genome.cirad.fr.
Change history
08 August 2012
The affiliation details for author M.D. were amended; the shaded area denoting the monocotyledons and eudicotyledons in Fig. 3 was corrected.
References
Lescot, T. The genetic diversity of banana in figures. FruiTrop 189, 58–62 (2011)
Perrier, X. et al. Multidisciplinary perspectives on banana (Musa spp.) domestication. Proc. Natl Acad. Sci. USA 108, 11311–11318 (2011)
De Lapeyre de Bellaire, L., Fouré, E., Abadie, C. & Carlier, J. Black leaf streak disease is challenging the banana industry. Fruits 65, 327–342 (2010)
Dita, M. A., Waalwijk, C., Buddenhagen, I. W., Souza, M. T. & Kema, G. H. J. A molecular diagnostic for tropical race 4 of the banana fusarium wilt pathogen. Plant Pathol. 59, 348–357 (2010)
Simmonds, N. W. The Evolution of the Bananas 101–131 (Longman, 1962)
D’Hont, A., Paget-Goy, A., Escoute, J. & Carreel, F. The interspecific genome structure of cultivated banana, Musa spp. revealed by genomic DNA in situ hybridization. Theor. Appl. Genet. 100, 177–183 (2000)
Raboin, L. M. et al. Diploid ancestors of triploid export banana cultivars: molecular identification of 2n restitution gamete donors and n gamete donors. Mol. Breed. 16, 333–341 (2005)
Cuperus, J. T., Fahlgren, N. & Carrington, J. C. Evolution and functional diversification of MIRNA genes. Plant Cell 23, 431–442 (2011)
Gayral, P. & Iskra-Caruana, M. L. Phylogeny of banana streak virus reveals recent and repetitive endogenization in the genome of its banana host (Musa spp.). J. Mol. Evol. 69, 65–80 (2009)
Iskra-Caruana, M. L., Baurens, F. C., Gayral, P. & Chabannes, M. A four-partner plant–virus interaction: enemies can also come from within. Mol. Plant Microbe Interact. 23, 1394–1402 (2010)
Ma, J., Devos, K. M. & Bennetzen, J. L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004)
Jeridi, M. et al. Homoeologous chromosome pairing between the A and B genomes of Musa spp. revealed by genomic in situ hybridization. Ann. Bot. (Lond.) 108, 975–981 (2011)
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009)
Van de Peer, Y., Fawcett, J. A., Proost, S., Sterck, L. & Vandepoele, K. The flowering world: a tale of duplications. Trends Plant Sci. 14, 680–688 (2009)
Lescot, M. et al. Insights into the Musa genome: syntenic relationships to rice and between Musa species. BMC Genomics 9, 58 (2008)
Paterson, A. H. et al. Comparative genome analysis of monocots and dicots, toward characterization of angiosperm diversity. Curr. Opin. Biotechnol. 15, 120–125 (2004)
Salse, J. et al. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell 20, 11–24 (2008)
Tang, H., Bowers, J. E., Wang, X. & Paterson, A. H. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl Acad. Sci. USA 107, 472–477 (2010)
Magallon, S. & Castillo, A. Angiosperm diversification through time. Am. J. Bot. 96, 349–365 (2009)
Givnish, T. J. et al. Assembling the tree of the monocotyledons: plastome sequence phylogeny and evolution of Poales. Ann. Mo. Bot. Gard. 97, 584–616 (2010)
Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nature Biotechnol. 29, 521–527 (2011)
Schnable, J. C., Springer, N. M. & Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl Acad. Sci. USA 108, 4069–4074 (2011)
Birchler, J. A., Riddle, N. C., Auger, D. L. & Veitia, R. A. Dosage balance in gene regulation: biological implications. Trends Genet. 21, 219–226 (2005)
Veitia, R. A., Bottani, S. & Birchler, J. A. Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet. 24, 390–397 (2008)
Carels, N. & Bernardi, G. Two classes of genes in plants. Genetics 154, 1819–1825 (2000)
Freeling, M. & Subramaniam, S. Conserved noncoding sequences (CNSs) in higher plants. Curr. Opin. Plant Biol. 12, 126–132 (2009)
Woolfe, A. et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3, e7 (2005)
Fils-Lycaon, B. et al. Acid invertase as a serious candidate to control the balance sucrose versus (glucose plus fructose) of banana fruit during ripening. Sci. Hortic. (Amsterdam) 129, 197–206 (2011)
Poland, J. A., Bradbury, P. J., Buckler, E. S. & Nelson, R. J. Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc. Natl Acad. Sci. USA 108, 6893–6898 (2011)
Howe, K. L., Chothia, T. & Durbin, R. GAZE: a generic framework for the integration of gene-prediction data by dynamic programming. Genome Res. 12, 1418–1427 (2002)
Bakry, F., Assani, A. & Kerbellec, F. Haploid induction: androgenesis in Musa balbisiana . Fruits 63, 45–49 (2008)
Marie, D. & Brown, S. C. A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biol. Cell 78, 41–51 (1993)
Piffanelli, P., Vilarinhos, A., Safar, J., Sabau, X. & Dolezel, J. Construction of bacterial artificial chromosome (BAC) libaries of banana (Musa acuminata and Musa balbisiana). Fruits 63, 375–379 (2008)
Aury, J. M. et al. High quality draft sequences for prokaryotic genomes using a mix of new sequencing technologies. BMC Genomics 9, 603 (2008)
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002)
Parra, G., Blanco, E. & Guigo, R. GeneID in Drosophila . Genome Res. 10, 511–515 (2000)
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004)
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000)
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004)
Bairoch, A. et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 33, D154–D159 (2005)
Mott, R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 13, 477–478 (1997)
Denoeud, F. et al. Annotating genomes with massive-scale RNA sequencing. Genome Biol. 9, R175 (2008)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Argout, X. et al. The genome of Theobroma cacao . Nature Genet. 43, 101–108 (2010)
Flutre, T., Duprat, E., Feuillet, C. & Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6, e16526 (2011)
Ma, J., Devos, K. M. & Bennetzen, J. L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004)
Glemet, E. & Codani, J. J. LASSAP, a LArge Scale Sequence compArison Package. Comput. Appl. Biosci. 13, 137–143 (1997)
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009)
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007)
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006)
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007)
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011)
Wall, P. K. et al. PlantTribes: a gene and gene family resource for comparative genomics in plants. Nucleic Acids Res. 36, D970–D976 (2008)
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006)
Katoh, K., Asimenos, G. & Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64 (2009)
Li, L., Stoeckert, C. J., Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003)
Rouard, M. et al. GreenPhylDB v2.0: comparative and functional genomics in plants. Nucleic Acids Res. 39, D1095–D1102 (2011)
Woodhouse, M. R. et al. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 8, e1000409 (2010)
Lyons, E. & Freeling, M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 53, 661–673 (2008)
Ouyang, S. et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 35, D883–D887 (2007)
Hardcastle, T. & Kelly, K. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11, 422 (2010)
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010)
Acknowledgements
This work was mainly supported by French National Research Agency, Commissariat à l’Energie Atomique and Centre de coopération Internationale en Recherche Agronomique pour le Développement. The Generation Challenge program supported DArT genotyping, and Stichting Het Groene Woudt part of BAC-end sequencing. We thank M. Teixeira Souza for authorizing early access to BAC-End sequences, L. Baudouin and T. Hardcastle for their help with the Bayseq analysis, O. Inizan, T. Flutre and F. Choulet for their help in transposable-element mapping. We thank the SouthGreen Bioinformatics Platform (http://southgreen.cirad.fr) for providing us with computational resources. We thank D. Manley for his help with the English in this paper.
Author information
Authors and Affiliations
Contributions
J.M.A., F.C.B., F.C., O.G., B.N. and S.B. contributed equally to this work. J.P., K.L., J.M.A. and M.B. performed sequencing and assembly. S.Br. performed genome size evaluation. F.C.B., N.R. and G.K. built or gave access to BAC libraries or BAC-end sequences. B.N., S.Bo., F.D., M.Co., J.Len., C.D.S., G.D., M.Ro., N.Y., F.C.B. and V.G. performed protein coding gene annotation. F.C., F.C.B., C.C., M.S., G.D., R.H., R.R., P.F., A.K., C.Je., F.B., S.J., M.R.G. and A.M.R. performed plant material development, ploidy analysis, DNA extraction, markers development, genotyping, genetic mapping, anchoring. K.J. performed gene GC content analyses. S.Bo., O.G., T.W., E.H., P.H.H., J.B., M.R.G., D.Burt., A.D.H., M.J., C.P., J.D., O.P., J.Len., G.D. and N.Y. performed transposable-element analysis. O.G., F.D., A.D.H., J.M.A., G.D., F.C.B., E.L., S.Bo. and O.J. performed WGD analyses based on synteny conservation. J.L.M., S.A., M.R.M., A.D.H., O.G. performed phylogenomic analyses of WGD. N.Y., M.Ro., J.Len., S.Bo., C.Jo., A.D., F.D., M.Ru. and A.Alm. performed gene family analyses. J.Lec., X.A., G.D. and S.Bo. performed transfer RNA and microRNA analyses. F.C.B. and M.Ch. performed endogenous virus analyses. D.Burg. and M.F. performed CNS analyses. N.Y., C.Jo., C.D.S., A.Alb., F.C., D.M.M., M.D., C.W., G.K., M.S., performed RNA extraction, phenotyping and/or transcriptomic analyses. A.D.H., N.Y., O.G., F.C., F.C.B., F.D., J.M.A., J.C.G., P.W., S.Bo., F.Q. and J.W. wrote or revised the paper. A.D.H., N.Y. and P.W. conceived and coordinated the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data 1-18 (see Contents for details), Supplementary References, Supplementary Figures 1-30, Supplementary Tables 1-19, 21-23, 27 (see separate zipped for file for Supplementary Tables 20, 24-26 and 28). (PDF 9831 kb)
Supplementary Tables
This zipped file contains Supplementary Tables 20, 24-26 and 28. (ZIP 1200 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
D’Hont, A., Denoeud, F., Aury, JM. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012). https://doi.org/10.1038/nature11241
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature11241
This article is cited by
-
Origin and evolution of the triploid cultivated banana genome
Nature Genetics (2024)
-
Genetics and evolutionary insights from Solanaceae genome sequences
Plant Systematics and Evolution (2024)
-
Lipid metabolism and MAPK-ICE1 cascade play crucial roles in cold tolerance of banana
Horticulture Advances (2024)
-
Metapangenomics of wild and cultivated banana microbiome reveals a plethora of host-associated protective functions
Environmental Microbiome (2023)
-
Three R2R3-MYB transcription factors from banana (Musa acuminata) activate structural anthocyanin biosynthesis genes as part of an MBW complex
BMC Research Notes (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
