
Abstract
Two African apes are the closest living relatives of humans: the chimpanzee (Pan troglodytes) and the bonobo (Pan paniscus). Although they are similar in many respects, bonobos and chimpanzees differ strikingly in key social and sexual behaviours1,2,3,4, and for some of these traits they show more similarity with humans than with each other. Here we report the sequencing and assembly of the bonobo genome to study its evolutionary relationship with the chimpanzee and human genomes. We find that more than three per cent of the human genome is more closely related to either the bonobo or the chimpanzee genome than these are to each other. These regions allow various aspects of the ancestry of the two ape species to be reconstructed. In addition, many of the regions that overlap genes may eventually help us understand the genetic basis of phenotypes that humans share with one of the two apes to the exclusion of the other.
Similar content being viewed by others

Main
Whereas chimpanzees are widespread across equatorial Africa, bonobos live only south of the Congo River in the Democratic Republic of Congo (Fig. 1a). As a result of their relatively small and remote habitat, bonobos were the last ape species to be described2 and are the rarest of all apes in captivity. As a consequence, they have, until recently, been little studied2. It is known that whereas DNA sequences in humans diverged from those in bonobos and chimpanzees five to seven million years ago, DNA sequences in bonobos diverged from those in chimpanzees around two million years ago. Bonobos are thus closely related to chimpanzees. Moreover, comparison of a small number of autosomal DNA sequences has shown that bonobo DNA sequences often fall within the variation of chimpanzees5.

a, Geographical distribution of bonobos and chimpanzees. b, D statistics for the admixture test between bonobos and three chimpanzee groups. Each pairwise comparison between one bonobo and two chimpanzee groups is depicted as one panel. Each point in a panel represents one bonobo individual compared with two chimpanzee individuals from different groups. Admixture between bonobo and chimpanzee is indicated by a Z-score greater than 4.4 or less than −4.4.
Bonobos and chimpanzees are highly similar to each other in many respects. However, the behaviour of the two species differs in important ways1. For example, male chimpanzees use aggression to compete for dominance rank and obtain sex, and they cooperate to defend their home range and attack other groups3. By contrast, bonobo males are commonly subordinate to females and do not compete intensely for dominance rank1. They do not form alliances with one another and there is no evidence of lethal aggression between groups3. Compared with chimpanzees, bonobos are playful throughout their lives and show intense sexual behaviour3 that serves non-conceptive functions and often involves same-sex partners4. Thus, chimpanzees and bonobos each possess certain characteristics that are more similar to human traits than they are to one another’s. No parsimonious reconstruction of the social structure and behavioural patterns of the common ancestor of humans, chimpanzees and bonobos is therefore possible. That ancestor may in fact have possessed a mosaic of features, including those now seen in bonobo, chimpanzee and human.
To understand the evolutionary relationships of bonobos, chimpanzees and humans better, we sequenced and assembled the genome of a female bonobo individual (Ulindi) and compared it to those of chimpanzees and humans. Compared with the 6× Sanger-sequenced chimpanzee genome6 (panTro2), the bonobo genome assembly has a similar number of bases in alignment with the human genome, a similar number of lineage-specific substitutions and similar indel error rates (Table 1 and Supplementary Information, sections 2 and 3), suggesting that the two ape genomes are of similar quality. Segmental duplications affect at least 80 Mb of the bonobo genome, according to excess sequence read-depth predictions. Owing to over-collapsing of duplications, only 14.6 Mb are present in the final assembly (Supplementary Information, section 4), a common error seen in assemblies from shorter-read technologies7. We used the finished chimpanzee sequence of chromosome 21 together with the human genome sequence to estimate an error rate of approximately two errors per 10 kb in the bonobo genome, with comparable qualities for the X chromosome and autosomes. The bonobo genome can therefore serve as a high-quality sequence for comparative genome analyses.
On average, the two alleles in single-copy, autosomal regions in the Ulindi genome are approximately 99.9% identical to each other, 99.6% identical to corresponding sequences in the chimpanzee genome and 98.7% identical to corresponding sequences in the human genome. A comprehensive analysis of the bonobo genome is presented in Supplementary Information. Here we summarize the most interesting results.
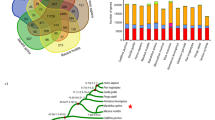
We identified and validated experimentally a total of 704 kb of DNA sequences that occur in bonobo-specific segmental duplications. They contain three partially duplicated genes (CFHR2, DUS2L and CACNA1B) and two completely duplicated genes (CFHR4 and DDX28). However, bonobos and chimpanzees share the majority of segmental duplications, and they carry approximately similar numbers of bases in lineage-specific duplications (Fig. 2a).

a, Venn diagram showing segmental duplications in the human (H), chimpanzee (C) and bonobo (B) genomes. Each number of megabases refers to the total amount of sequence that occurs in segmental duplications (Supplementary Information, section 4). b, Accumulation of different retrotransposon classes on each lineage.
As in other mammals, transposons, that is, mobile genetic elements, make up approximately half of the bonobo genome (Supplementary Information, section 6). In agreement with previous results6, we find that Alu insertions accumulated about twice as fast on the human lineage as on the bonobo and chimpanzee lineages (Fig. 2b). We identified two previously unreported Alu subfamilies in bonobos and chimpanzees, designated AluYp1, which is present in 5 copies in the human genome and in 54 and 114 copies in the bonobo and chimpanzee genomes, respectively, and AluYp2, which is absent from humans and present in 24 and 37 copies, respectively, in the two apes. We found that, as in mice8, African-ape-specific L1 insertions are enriched near genes involved in neuronal activities or cell adhesion and are depleted near genes encoding transcription factors or involved in nucleic-acid metabolism (Supplementary Information, section 6). In humans, L1 retrotransposition has been shown to occur preferentially in neuronal precursor cells and has been speculated to contribute to functional diversity in the brain9. The tendency of new L1 integrants to accumulate near neuronal genes on evolutionary timescales may mimic the somatic variation found in the brain.
To investigate whether bonobos and chimpanzees exchanged genes subsequent to their separation, we used a test (the D statistic10) to investigate the extent to which the bonobo genomes might be closer to some chimpanzees than to others (Supplementary Information, section 10). To this end, we generated Illumina shotgun sequences from two western, seven eastern, and seven central chimpanzees (Fig. 1a) and from three bonobos (Supplementary Information, section 5). We then used alignments of sets of four genomes, each consisting of two chimpanzees, the bonobo and the human, and tested for an excess of shared derived alleles between bonobo and one chimpanzee as compared with the other chimpanzee. We observe no significant difference between the numbers of shared derived alleles (Fig. 1b). There is thus no indication of preferential gene flow between bonobos and any of the chimpanzee groups tested. Such a complete separation contrasts with reports of hybridization between many other primates11. It is, however, consistent with the suggestion that the formation of the Congo River 1.5–2.5 million years ago created a barrier to gene flow that allowed bonobos and chimpanzees to evolve different phenotypes over a relatively short time.
Because the population split between bonobo and chimpanzee occurred relatively close in time to the split between the bonobo–chimpanzee ancestor (Pan ancestor) and humans, not all genomic regions are expected to show the pattern in which DNA sequences from bonobos and chimpanzees are more closely related to each other than to humans. Previous work using very low-coverage sequencing of ape genomes has suggested that less than 1% of the human genome may be more closely related to one of the two apes than the ape genomes are to one another12. To investigate the extent to which such so-called incomplete lineage sorting (ILS) exists between the three species, we used the bonobo genome and a coalescent hidden Markov model (HMM) approach13 to analyse non-repetitive parts of the bonobo, chimpanzee6, human14 and orang-utan15 genomes. This showed that 1.6% of the human genome is more closely related to the bonobo genome than to the chimpanzee genome, and that 1.7% of the human genome is more closely related to the chimpanzee than to the bonobo genome (Fig. 3a).

a, Schematic description of ILS states and percentage of bases assigned to each state. b, Effective population sizes and split times inferred from ILS and based on a molecular clock with a mutation rate of 10−9 yr−1. Myr, million years. We note that other estimates of mutation rates will correspondingly affect the estimates of the split times. c, Overlap between predicted ILS transposons and the closest HMM ILS assignments within 100 bp of a transposon insertion. d, Proportion of ILS in exons, introns and across the whole genome, counted within ∼1-Mb segments of alignment (Supplementary Information, section 8). e, Proportion of ILS dependent on recombination rates. Errors, 95% confidence interval.
To test this result independently, we analysed transposon integrations, which occur so rarely in ape and human genomes that the chance of two independent insertions of the same type of transposon at the same position and in the same orientation in different species is exceedingly low. We identified 991 integrations of transposons absent from the orang-utan genome but present in two of the three species bonobo, chimpanzee and human. Of these, 27 are shared between the bonobo and human genomes but are absent from the chimpanzee genome, and 30 are shared between the chimpanzee and human genomes but are absent from the bonobo genome, suggesting that approximately 6% (95% confidence interval, 4.1–7.0%) of the genome is affected by ILS among the three species. The HMM estimation of ILS is further supported by the fact that the HMM tree topology assignments tend to match the ILS status of the neighbouring transposons (P = 7.2 × 10−6 and 0.025 for bonobo–human and chimpanzee–human ILS, respectively; Fig. 3c and Supplementary Information, section 6). We conclude that more than 3% of the human genome is more closely related to either bonobos or chimpanzees than these are to each other.
Such regions of ILS may influence phenotypic similarities that humans share with one of the apes but not the other. In fact, about 25% of all genes contain regions of ILS (Supplementary Information, section 8), and genes encoding membrane proteins and proteins involved in cell adhesion have a higher fraction of bases assigned to ILS than do other genes. Amino-acid substitutions that are fixed in the apes and show ILS may be particularly informative about phenotypic differences. We identified 18 such amino-acid substitutions shared between humans and bonobos and 18 shared between chimpanzees and humans (Supplementary Information, section 12). These are candidates for further study. An interesting example is the gene encoding the trace amine associated receptor 8 (TAAR8), a member of a family of G-coupled protein receptors that in the mouse detect volatile amines in urine that may provide social cues16. Although this gene seems to be pseudogenized independently on multiple ape lineages, humans and bonobos share a single amino-acid change in the first extracellular domain and carry the longest open reading frames (of 342 and 256 amino acids, respectively; open reading frames in all other apes, <180 amino acids) (SI 12). Further work is needed to clarify if TAAR8 is functional in humans and apes.
The ILS among bonobos, chimpanzees and humans opens the possibility of gauging the genetic diversity and, hence, the population history of the Pan ancestor. We used the HMM to estimate the effective population size of the Pan ancestor to 27,000 individuals (Fig. 3b), which is almost three times larger than that of present-day bonobos (Supplementary Information, section 9) and humans17 but is similar to that of central chimpanzees5,18,19. We also estimated a population split time between bonobos and chimpanzees of one million years, which is in agreement with most previous estimates18,19.
Differences in female and male population history, for example, with respect to reproductive success and migration rates, are of special interest in understanding the evolution of social structure. To approach this question in the Pan ancestor, we compared the inferred ancestral population sizes of the X chromosome and the autosomes. Because two-thirds of X chromosomes are found in females whereas autosomes are split equally between the two sexes, a ratio between their effective population sizes (X/A ratio) of 0.75 is expected under random mating. The X/A ratio in the Pan ancestor, corrected for the higher mutation rate in males, is 0.83 (0.75–0.91) (Fig. 4 and Supplementary Information, section 8). Similarly, we estimated an X/A ratio of 0.85 (0.79–0.93) for present-day bonobos using Ulindi single nucleotide polymorphisms in 200-kb windows (Supplementary Information, section 9). Under the assumption of random mating, this would mean that on average two females reproduce for each reproducing male. The difference in the variance of reproductive success between the sexes certainly contributes to this observation, as does the fact that whereas bonobo females often move to new groups upon maturation, males tend to stay within their natal group20. Because both current and ancestral X/A ratios are similar to each other and also to some human groups (Fig. 4), this suggests that they may also have been typical for the ancestor shared with humans.

The X/A ratios for Ulindi (bonobo), an African human and a European human were inferred from heterozygosity, and that for the Pan ancestor was inferred from ILS. The low X/A ratio for the European has been suggested to be due to demographic effects connected to migrating out of Africa30. Errors, 95% confidence interval (Supplementary Information, sections 8 and 9).
Because factors that reduce the effective population size, in particular positive and negative selection, will decrease the extent of ILS, the distribution of ILS across the genome allows regions affected by selection in the Pan ancestor to be identified. In agreement with this, we find that exons show less ILS than introns (Fig. 3d and Supplementary Information, section 8). We also find that recombination rates are positively correlated with ILS (Fig. 3e), probably because recombination uncouples regions from neighbouring selective events. Unlike positive and negative selection, balancing selection is expected to increase ILS. In agreement with this, we find that ILS is most frequent in the major histocompatibility complex (MHC), which encodes cell-surface proteins that present antigens to immune cells (Supplementary Information, section 10) and is known to contain genes that evolve under balancing selection21.
To identify regions affected by selective sweeps in the Pan ancestor, we isolated long genomic regions devoid of ILS. The largest such region is 6.1 Mb long and is located on human chromosome 3. This region contains a cluster of tumour suppressor genes22, has an estimated recombination rate of 10% of the human genome average23 and has been found to evolve under strong purifying selection in humans24. The diversity in the region, corrected for mutation rate, is lower than in neighbouring regions in chimpanzee but not in bonobos (Fig. 5a), and parts of the region show signatures of positive selection in humans10,25,26. Apparently this region evolves in unique ways that may involve both strong background selection and several independent events of positive selection among apes and humans.

a, Diversity in chimpanzee and bonobo around the region on chromosome 3 devoid of ILS. b, Regions where bonobos fall outside the variation of chimpanzee upstream of the MHC. The MHC region is not plotted because the SNP density is sparse there as a result of duplications. Five regions among the 50 longest regions are shown in yellow. Red points show posterior probabilities >0.8.
The fact that the chimpanzee diversity encompasses bonobos for most regions of the genome can be exploited to identify regions that have been positively selected in chimpanzees after their separation from bonobos, because in such regions bonobos will fall outside the chimpanzee variation. We implemented a search for such regions, which is similar to a test previously applied to humans to detect selective sweeps since their split from Neanderthals10 (Homo neanderthalensis), in an HMM that uses coalescent simulations for parameter training, the chimpanzee resequencing data and the megabase-wide average of the human recombination rates (Supplementary Information, section 7). Because the size of a region affected by a selective sweep will be larger the faster fixation was reached, the intensity of selection will correlate positively with genetic length. We therefore ranked the regions according to genetic length and further corrected for the effect of background selection24. The highest-ranking region contains an miRNA, miR-4465, that has not yet been functionally characterized. Four of the ten highest-ranking regions contain no protein- or RNA-coding genes, and may thus contain structural or regulatory features that have been subject to selection. Notably, four of these ten regions are on chromosome 6, and two of these four are within 2 Mb of the MHC (Fig. 5b). This suggests that the MHC and surrounding genomic regions have been a major target of positive selection in chimpanzees, presumably as a result of infectious diseases. Indeed, chimpanzees have experienced a selective sweep that targeted MHC class-I genes and reduced allelic diversity across a wide region surrounding the MHC27, perhaps caused by the HIV-1/SIVCPZ retrovirus27,28.
The bonobo genome shows that more than 3% of the human genome is more closely related to either bonobos or chimpanzees than these are to each other. This can be used to illuminate the population history and selective events that affected the ancestor of bonobos and chimpanzees. In addition, about 25% of human genes contain parts that are more closely related to one of the two apes than the other. Such regions can now be identified and will hopefully contribute to the unravelling of the genetic background of phenotypic similarities among humans, bonobos and chimpanzees.
Methods Summary
We generated a total of 86 Gb of DNA sequence from Ulindi, a female bonobo who lives in Leipzig Zoo (Supplementary Information, section 1). All sequencing was done on the 454 sequencing platform and included 10 Gb of paired-end reads from clones of insert sizes of 3, 9 and 20 kb. The genome was assembled using the open-source Celera Assembler software29 (Supplementary Information, section 2). In addition, we sequenced 19 bonobo and chimpanzee individuals on the Illumina GAIIx platform to about one-fold genomic coverage per individual (Supplementary Information, section 5). Supplementary Information provides a full description of our methods.
Accession codes
Primary accessions
Sequence Read Archive
Data deposits
The bonobo genome assembly has been deposited with the International Nucleotide Sequence Database Collaboration (DDBJ/EMBL/GenBank) under the EMBL accession number AJFE01000000. 454 shotgun data of Ulindi have been made available through the NCBI Sequence Read Archive under study ID ERP000601; Illumina sequences of 19 chimpanzee and bonobo individuals are available under study ID ERP000602.
References
Boesch, C., Hohmann, G. & Marchant, L. Behavioural Diversity in Chimpanzees and Bonobos (Cambridge Univ. Press, 2002)
de Waal, F. & Lanting, F. Bonobo: the Forgotten Ape (Univ. California Press, 1997)
Hare, B., Wobber, V. & Wrangham, R. The self-domestication hypothesis: evolution of bonobo psychology is due to selection against aggression. Anim. Behav. 83, 573–585 (2012)
Kano, T. The Last Ape: Pygmy Chimpanzee Behavior and Ecology (Stanford Univ. Press, 1992)
Fischer, A. et al. Bonobos fall within the genomic variation of chimpanzees. PLoS ONE 6, e21605 (2011)
The. Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005)
Alkan, C., Sajjadian, S. & Eichler, E. E. Limitations of next-generation genome sequence assembly. Nature Methods 8, 61–65 (2011)
Akagi, K., Li, J., Stephens, R. M., Volfovsky, N. & Symer, D. E. Extensive variation between inbred mouse strains due to endogenous L1 retrotransposition. Genome Res. 18, 869–880 (2008)
Baillie, J. K. et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479, 534–537 (2011)
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010)
Arnold, M. L. & Meyer, A. Natural hybridization in primates: one evolutionary mechanism. Zoology 109, 261–276 (2006)
Caswell, J. L. et al. Analysis of chimpanzee history based on genome sequence alignments. PLoS Genet. 4, e1000057 (2008)
Hobolth, A., Christensen, O. F., Mailund, T. & Schierup, M. H. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 3, e7 (2007)
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001)
Locke, D. P. et al. Comparative and demographic analysis of orang-utan genomes. Nature 469, 529–533 (2011)
Liberles, S. D. & Buck, L. B. A second class of chemosensory receptors in the olfactory epithelium. Nature 442, 645–650 (2006)
Takahata, N. Allelic genealogy and human evolution. Mol. Biol. Evol. 10, 2–22 (1993)
Hey, J. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 27, 921–933 (2010)
Wegmann, D. & Excoffier, L. Bayesian inference of the demographic history of chimpanzees. Mol. Biol. Evol. 27, 1425–1435 (2010)
Eriksson, J. et al. Y-chromosome analysis confirms highly sex-biased dispersal and suggests a low male effective population size in bonobos (Pan paniscus). Mol. Ecol. 15, 939–949 (2006)
Gyllensten, U. B. & Erlich, H. A. Ancient roots for polymorphism at the HLA-DQ alpha locus in primates. Proc. Natl Acad. Sci. USA 86, 9986–9990 (1989)
Hesson, L. B., Cooper, W. N. & Latif, F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 26, 7283–7301 (2007)
Kong, A. et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103 (2010)
McVicker, G., Gordon, D., Davis, C. & Green, P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 5, e1000471 (2009)
Voight, B. F., Kudaravalli, S., Wen, X. & Pritchard, J. K. A map of recent positive selection in the human genome. PLoS Biol. 4, e72 (2006)
Wang, E. T., Kodama, G., Baldi, P. & Moyzis, R. K. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc. Natl Acad. Sci. USA 103, 135–140 (2006)
de Groot, N. G. et al. AIDS-protective HLA-B*27/B*57 and chimpanzee MHC class I molecules target analogous conserved areas of HIV-1/SIVcpz. Proc. Natl Acad. Sci. USA 107, 15175–15180 (2010)
Yohn, C. T. et al. Lineage-specific expansions of retroviral insertions within the genomes of African great apes but not humans and orangutans. PLoS Biol. 3, e110 (2005)
Miller, J. R. et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24, 2818–2824 (2008)
Gottipati, S., Arbiza, L., Siepel, A., Clark, A. G. & Keinan, A. Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nature Genet. 43, 741–743 (2011)
Acknowledgements
The sequencing effort was made possible by the ERC (grant 233297, TWOPAN) and the Max Planck Society. We thank D. Reich and L. Vigilant for comments; the 454 Sequencing Center, the MPI-EVA sequencing group, M. Kircher, M. Rampp and M. Halbwax for technical support; the staff of Zoo Leipzig (Germany), the Ngamba Island Chimpanzee Sanctuary (Entebbe, Uganda), the Tchimpounga Chimpanzee Rehabilitation Center (Pointe-Noire, Republic of Congo) and the Lola ya Bonobo bonobo sanctuary (Kinshasa, Democratic Republic of Congo) for providing samples; and A. Navarro, E. Gazave and C. Baker for performing the ArrayCGH hybridizations. The ape distribution layers for Fig. 1a were provided by UNEP-WCMC and IUCN.2008 (IUCN Red List of Threatened Species, Version 2011.2, http://www.iucnredlist.org). The National Institutes of Health provided funding for J.R.M., B.W., S.K., G.S. (2R01GM077117-04A1), J.C.M. (Intramural Research Program of the National Human Genome Research Institute) and E.E.E. (HG002385). E.E.E is an Investigator of the Howard Hughes Medical Institute. T.M.-B. was supported by a Ramón y Cajal grant (MICINN-RYC 2010) and an ERC Starting Grant (StG_20091118); D.E.S., K.A. and S.H. were supported by the Ohio State University Comprehensive Cancer Center, the Ohio Supercomputer Center (#PAS0425) and the Ohio Cancer Research Associates (GRT00024299); and G.L. was supported by a Wellcome Trust grant (090532/Z/09/Z). The US National Science Foundation provided an International Postdoctoral Fellowship (OISE-0754461) to J.M.G. The Danish Council for Independent Research | Natural Sciences (grant no. 09-062535) provided funding for K.M. and M.H.S.
Author information
Authors and Affiliations
Contributions
K.P., K.M., I.H., K.A., J.R.M., B.W., S.K., G.S., C.K., R.W., J.R.K., J.C.M., S.J.M, C.P.P., G.L., S.H., A.H., J.D., E.K., C. Alkan, S.S., C.R.C., M.V., T.M.-B., E.E.E., N.P., M.S., J.M.G., A.F., S.E.P., M.L., D.E.S., T.M., M.H.S., A.M.A., J.K. and S.P. analysed genetic data. C. André, R.A., L.M. and J.J. provided samples. K.P., J.K. and S.P. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
C.K., R.W. and J.R.K. are employees of Roche/454 Life sciences, which developed the technology used for the sequencing of the genome studied here.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data sections 1-12, which include Supplementary Figures, Supplementary Tables and Supplementary References (see Contents for details). (PDF 12082 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
Prüfer, K., Munch, K., Hellmann, I. et al. The bonobo genome compared with the chimpanzee and human genomes. Nature 486, 527–531 (2012). https://doi.org/10.1038/nature11128
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature11128
This article is cited by
-
The development of human causal learning and reasoning
Nature Reviews Psychology (2024)
-
CEGA: a method for inferring natural selection by comparative population genomic analysis across species
Genome Biology (2023)
-
Humans recognize affective cues in primate vocalizations: acoustic and phylogenetic perspectives
Scientific Reports (2023)
-
Human-specific genetics: new tools to explore the molecular and cellular basis of human evolution
Nature Reviews Genetics (2023)
-
Between-group variation in production of pant-grunt vocalizations by wild bonobos (Pan paniscus)
Behavioral Ecology and Sociobiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
