
Abstract
Neuroblastoma is a childhood tumour of the peripheral sympathetic nervous system. The pathogenesis has for a long time been quite enigmatic, as only very few gene defects were identified in this often lethal tumour1. Frequently detected gene alterations are limited to MYCN amplification (20%) and ALK activations (7%)2,3,4,5. Here we present a whole-genome sequence analysis of 87 neuroblastoma of all stages. Few recurrent amino-acid-changing mutations were found. In contrast, analysis of structural defects identified a local shredding of chromosomes, known as chromothripsis, in 18% of high-stage neuroblastoma6. These tumours are associated with a poor outcome. Structural alterations recurrently affected ODZ3, PTPRD and CSMD1, which are involved in neuronal growth cone stabilization7,8,9. In addition, ATRX, TIAM1 and a series of regulators of the Rac/Rho pathway were mutated, further implicating defects in neuritogenesis in neuroblastoma. Most tumours with defects in these genes were aggressive high-stage neuroblastomas, but did not carry MYCN amplifications. The genomic landscape of neuroblastoma therefore reveals two novel molecular defects, chromothripsis and neuritogenesis gene alterations, which frequently occur in high-risk tumours.
Similar content being viewed by others


Main
Neuroblastoma have a highly variable clinical outcome, with an excellent prognosis for stage 1 and 2 tumours, but a poor outcome for high-stage tumours. Stage 4S neuroblastoma are metastasized but nevertheless undergo spontaneous regression. Low-stage tumours are marked by numeric changes of chromosomal copy numbers, whereas high-stage tumours typically show structural chromosomal defects resulting in, for example, hemizygous deletions of the chromosomal regions 1p36 or 11q and gain of 17q (refs 1, 10–12). Age at diagnosis above 1.5 year is associated with high-stage tumours and poor outcome.
We performed whole-genome paired-end sequencing as used by Complete Genomics13 for 87 untreated primary neuroblastoma tumours of all stages (Supplementary Table 1) and their corresponding lymphocyte DNAs. All samples had a minimal tumour content of 80% as determined by immunohistochemical analysis. Genomes were sequenced at an average coverage of 50 and an average fully called genome fraction of 96.6% (Supplementary Table 2). Compared to the HG18 reference genome we obtained an average of 3,347,592 single-nucleotide variants (SNVs) per genome, in accordance with reported frequencies of interpersonal variants. CGAtools was used to compare tumour with lymphocyte genomes and provided a somatic score estimating the likelihood of mutations to be somatic (http://cgatools.sourceforge.net/docs/1.3.0/). Validation of 1,014 candidate somatic small mutations (SNVs, substitutions, insertions, deletions), including 763 SNVs, established a specificity of 88% and a sensitivity of 85% at a somatic score cut-off of 0.1 (Supplementary Fig. 1a). SNVs above this score and all validated SNVs with lower scores were used for further analyses (total 586 genes, Supplementary Table 3). The sequence data identified an average of 12 somatic candidate amino-acid-affecting mutations per tumour (Fig. 1a and Supplementary Fig. 1b). The frequency of somatic events strongly correlated to tumour stage where stage 1, 2 and 4S tumours have very few mutations compared to stage 3 and 4 tumours (analysis of variance (ANOVA) P = 7.6 × 10−6; Fig. 1b). In addition mutation frequencies were strongly correlated to overall survival (log-rank P = 9.8 × 10−7; Fig. 1c) and age at diagnosis (r = 0.53, P = 1.1 × 10−7; Fig. 1d), as was also observed in medulloblastoma14. Within high-stage neuroblastoma, MYCN amplification status did not correlate to mutation frequency (Supplementary Fig. 1c).

a, The number of amino-acid-changing mutations in 87 primary neuroblastoma (single nucleotide variants (SNVs) in red, deletions in grey, insertions in green and substitutions affecting more than 1 base pair (Sub) in blue). Numbers shown are events after CGAtools CallDiff with somatic scores >0.1 and not present in dbSNP130, nor in 46 reference genomes released by Complete Genomics. b, Average number of mutations per tumour stage (International Neuroblastoma Staging System (INSS) stage 1, n = 9; stage 2, n = 14; stage 3, n = 5; stage 4, n = 50 and stage 4S, n = 9). Boxes include 50% of data and error bars indicate extremes with a maximum of two times the box size. st., stage. c, Kaplan–Meier curves for tumours with high versus low frequency of mutations. The optimal cut-off level for the categories was determined by Kaplan scanning (see Supplementary Information and Methods). The significance (log-rank test) was corrected for the multiple testing (Bonferroni correction). Number of patients per group is shown in parentheses. d, Age at diagnosis (rank-order) versus the number of somatic variants. e, Average number of structural variations per tumour stage (INSS). Group sizes and the definition of the error bars as in Fig. 1b.
Only very few recurrent mutations were identified. ALK mutations were found in 6% of the tumours, in accordance with frequencies established in large neuroblastoma tumour series (Supplementary Table 4)2,3,4,5. Three tumours carried mutations in TIAM1, a known regulator of cytoskeleton organization and neuritogenesis15. In a parallel study we sequenced four primary neuroblastoma tumours as well as cell lines derived from these tumours and their metastases. This revealed that primary tumours are already heterogeneous for mutations and that the large majority of them were passenger or late mutations (J.J.M. et al., submitted). Together with the lack of recurrent mutations, our data indicate that neuroblastoma carry few early somatic tumour-driving mutations with amino-acid-changing consequences.
Analysis of the paired-end clones with discordant ends can be used to identify candidate structural rearrangements, which together with sequence coverage data can identify somatic structural variants (SVs). Comparison of tumour versus lymphocyte coverage generated ultra-high-resolution comparative genomic hybridization (CGH)-like profiles (Supplementary Fig. 2a). Analysis of the frequency of structural variations per chromosome revealed ten tumours with chromothripsis characteristics6 (see Methods). Chromothripsis is a localized shredding of a chromosomal region and subsequent random reassembly of the fragments. An extreme example of chromothripsis in chromosome 5 is shown in Fig. 2a and 2b (for other cases see Supplementary Fig. 2b). The neuroblastoma with chromothripsis were associated with a poor prognosis (log-rank test P = 7.1 × 10−3; Fig. 2c). They were found in 18% of the stage 3 and 4 neuroblastoma, but not in low-stage tumours (Fisher’s exact test P = 0.01). Accordingly, their prognostic impact is not independent of age and stage in multivariate analyses. Chromothripsis-related structural aberrations frequently affected genes involved in neuroblastoma pathogenesis and were associated with amplification of MYCN or CDK4 and loss of heterozygosity of 1p (Supplementary Fig. 2c). In one tumour, chromothripsis resulted in amplification and very strong overexpression of MYC (c-Myc) (Supplementary Fig. 2d). Chromosome 5 had undergone chromothripsis in three tumours, but no clear tumorigenic target on this chromosome was identified. To identify genetic defects that allowed chromothripsis and subsequent survival of the cell, we searched for defects in DNA damage response pathways in tumours with chromothripsis. The most extreme case of chromothripsis (N492, Fig. 2a and 2b) showed an inactivating deletion in FANCM and another chromothripsis tumour sample (N576) had a missense mutation in FAN1, predicted to be damaging by the polyphen2 program16. These findings might suggest involvement of inactivating events in the Fanconi anaemia signalling pathway to allow chromothripsis17.

a, Circos plot showing structural variations in sample N492. The inner ring represents the copy number variations (red, gain; green, loss) based on coverage of the tumour and lymphocyte genomes. The lines traversing the ring indicate inter- and intrachromosomal rearrangements identified by discordant mate pairs from paired-end reads. N492 is a chromothripsis sample with an extreme amount of junctions on chromosome 5. b, Circos plot of the affected chromosome 5 in sample N492. c, Kaplan–Meier curves of the overall survival for tumours with or without chromothripsis. Numbers of patients per group are shown between brackets.
Full genome paired-end sequencing allowed us to identify structural variants specifically perturbing single genes (see Methods and Supplementary Fig. 4 for selection procedure). We detected a total of 451 genes harbouring structural variants (306 genes without the events on chromothripsis chromosomes, Supplementary Tables 5 and 6). The structural variants often consisted of deletions of one or a few exons, inversions or translocations deleting part of a gene. One tumour showed an intrachromosomal rearrangement activating FOXR1 transcription (Supplementary Fig. 2e), which we recently identified as a recurrent but rare event in neuroblastoma18. Similar to the findings for amino-acid-changing mutations, there was a strong relation between the frequency of structural variations and the tumour stage (one-way ANOVA P = 0.03; Fig. 1e), which extends the well-established relationship between tumour stage and structural chromosomal defects in neuroblastoma10,11,12. Breakpoints identifying deletions were supported by changes in coverage plots. Most of the structural variants affected only one allele of a gene (Supplementary Table 5). This indicates that the tumour-driving mechanism of these defects is haploinsufficiency, possibly combined with epigenetic attenuation of the non-affected allele. On average, genes with structural variants resulting in loss of coverage indeed showed a reduced expression in tumours with these defects, as compared to tumours with normal alleles (Supplementary Fig. 2f). As an additional validation, we generated SNP arrays of 52 of the sequenced tumours. Although the SNP data have a much lower resolution than the sequence coverage plots, they supported the deletions and gains of sufficient size. This is especially evident on plots of chromothripsis samples (Supplementary Fig. 2g).
To identify relevant genes and pathways that contribute to neuroblastoma pathogenesis, we generated one list of all genes with amino-acid changing mutations (n = 586), mutations in splice junctions (n = 37) and structural variations (n = 451). The total of 1,041 genes with alterations were analysed by two approaches. First, we analysed the most frequently affected genes (Supplementary Table 7). Four genes belonged to the MYCN amplicons (MYCN, MYCNOS, DDX1 and NBAS) and except for MYCN probably play no role in pathogenesis. Three genes, PTPRD, ODZ3 and ATRX, showed structural variants in five tumours each (Fig. 3a and Supplementary Fig. 3a) and 61 genes showed alterations in two to four tumours (Supplementary Table 7). A conservative randomization, taking the length of all genes and the structure of our data set into account, showed that the chance of finding three genes affected in five or more tumours is <2.11 × 10−4 (see Methods). This strongly indicates that at least the defects in PTPRD, ATRX and ODZ3 did not accumulate due to for example, the genomic length of the genes, but that they were selected for during the process of tumorigenesis.

a, Coverage plots displaying the structural variations in the ATRX gene. The dots indicate summed coverage for bins of 1,000 base pairs of the tumour genome, normalized to the coverage in corresponding normal tissue. The intron-exon structure of ATRX is shown in red (dark red are exons). b, ATRX mRNA expression of 70 tumours as measured on Affymetrix full-exon arrays. Tumours with ATRX deletions are encircled. Coloured tracks below figure, from top to bottom: age at diagnosis (green < 1.5 year, red > 1.5 year); survival (red, dead; dark green, alive >5 year; light green, alive <2 year); MYCN amplification (red, yes; green, no); stage (light green, stage 1; dark green, stage 2; brown, stage 3; red, stage 4; blue, stage 4S).
The X-chromosome-encoded ATRX gene was affected by structural variants in five tumours (Fig. 3a). In two male patients this resulted in complete inactivation of the gene. Frequent ATRX defects were recently found in pancreatic neuroendocrine tumours19. ATRX is a chromatin remodelling protein involved in exchange of H3.3 in GC-rich repeats and mutations of this gene are associated with X-linked mental retardation20. Exon mRNA profiles of part of the sequenced series showed that the three samples included with ATRX structural variations had the lowest ATRX mRNA expression of all samples and showed a specific collapse of the signal in the deleted regions, illustrating the inactivating nature of the ATRX defects (Fig. 3b and Supplementary Fig. 3b).
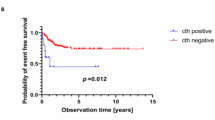
ODZ3 and PTPRD were also hit by structural variations in five tumours each (Supplementary Fig. 3a). One tumour showed homozygous inactivation of ODZ3 (see legends of Supplementary Fig. 3a). In addition, ODZ2 and ODZ4, two highly homologous members of the conserved ODZ family, were together affected three times. PTPRD and ODZ genes encode transmembrane receptors expressed in the developing nervous system and localizing to axons and axonal growth cones21. Targeted silencing of ODZ homologues in Drosophila, Caenorhabditis elegans and mouse caused severe axon guidance defects9. Overexpression of ODZ2 in neuroblastoma cells enhanced neuritogenesis22. PTPRD is a member of the LAR subfamily of receptor protein tyrosine phosphatases. Transgenic mouse models strongly implicate the LAR subfamily receptors in neuritogenesis8. PTPRD defects in neuroblastoma were reported previously23. Low expression of ODZ3 and of PTPRD as assessed by mRNA profiling were both associated with a poor prognosis (log-rank P = 3.1 × 10−4 and P = 5.7 × 10−3, respectively; Supplementary Fig. 3a). Interestingly, CSMD1, which showed structural variants in three tumours, is also a transmembrane protein expressed on nerve growth cones7. As the frequencies of PTPRD and ODZ3 defects exclude that they were found by chance, we propose that the function of these genes and of ODZ2, ODZ4 and CSMD1 in neuronal growth cones might hold a clue to their function in neuroblastoma pathogenesis.
The second analysis that we performed for the list for 1,041 affected genes was a gene ontology study to identify enrichment of genes with defects in specific molecular processes. The gene ontology category ‘regulation of GTPase activity’ was the most significantly enriched group (Bonferroni corrected for multiple testing: P = 6.7 × 10−4; see supplementary methods and Supplementary Table 9). This finding urged us to further investigate GTPase-regulating genes in the list. TIAM1 was mutated in three tumours (see Supplementary Table 4). It functions as a guanine nucleotide exchange factor (GEF) for the small GTPase Rac and is, together with Rac, central to regulation of cellular polarity and neuritogenesis24,25. The W1285S* mutation creates a premature stop-codon in the carboxy-terminal pleckstrin homology domain required for Rac activation, whereas the other mutations were predicted to be damaging by polyphen2 analysis16. Rac is activated by GEFs and inactivated by GTPase activating proteins (GAPs)26 (Fig. 4). We identified a total of eight alterations in six GEFs specific for Rac (including TIAM1), but none in GAPs specific for Rac (Supplementary Tables 7, 8 and 10 for functional consequences). Whereas activation of Rac1 stimulates neuritogenesis, activation of its small GTPase antagonist RhoA promotes axon retraction and growth cone collapse (Fig. 4a)15. Strikingly, we detected seven alterations in five GAPs for RhoA, but only one GEF specific for RhoA (ARHGEF12) showed a translocation with unknown functional consequences (Fig. 4a, Supplementary Tables 7, 8 and 10 for functional consequences). The bias for inactivation of GAPs for Rho and GEFs for Rac is highly significant (one-sided Fischer’s exact test: P < 0.0007). This indicates that alterations in GTPase-regulating genes specifically function to activate Rho or inactivate Rac, which both tip the balance in Rac/Rho signalling towards inhibition of neuritogenesis. Of note, transgenic mice with ATRX mutations causing mental retardation in humans showed abnormal dendritic spine formation with increased TIAM1 phosphorylation and Rac1 signalling27.

a, Diagram of a neurite growth cone depicting the function of the proteins encoded by genes with genomic aberrations in neuritogenesis. Red proteins have defects (for references see Supplementary Table 8). Rac and Rho small GTPases cycle between an inactive GDP-bound and active GTP-bound conformation, transducing signals from a wide variety of membrane receptors. They are activated by GEFs and inactivated by GAPs. Guanine nucleotide dissociation inhibitors (GDIs) sequester GDP-bound GTPases. Proteins with aberrations in more than one tumour are marked with an asterisk (*). b, Diagram of genetic defects and clinical parameters of all 87 sequenced neuroblastoma. Each vertical lane summarizes one tumour. Patients are sorted by the presence of genomic aberrations in neuritogenesis genes (Neuritogenesis, n = 19), by MYCN amplification (MYCN, n = 23), and by INSS stage (high to low). Clinical and molecular genetic characteristics are shown for each tumour as tracks; INSS stage (green, stage 1 and 2; red, stage 3 and 4; orange, stage 4S); Survival (red, death; green, alive), Age group (green, <1.5 year; red, ≥1.5 year), ALK (red, mutated; green, wild type), Chromothripsis (red, yes; green, no). Middle panel, amino-acid-changing mutations and structural variations are indicated for all genes having two or more events and that are involved in neuritogenesis (red, mutated or structural variant; grey, not affected).
We conclude that alterations with significant frequencies (PTPRD and ODZ genes) affect transmembrane receptors that function in neuronal growth cone guidance and maintenance. In addition gene ontology analysis of the 1,041 genes showed significant enrichment of GTPase-regulating genes. Alterations in GEFs for Rac and GAPs for Rho significantly deviate from a random distribution, implicating inhibition of Rac1 and activation of RhoA in impairing neuritogenesis in neuroblastoma (Fig. 4a).
From these findings we propose that defects in neuritogenesis-regulating genes form an important category of tumour-driving events in neuroblastoma. For a preliminary analysis of tumours with these defects, we selected the genes with recurrent defects in tumours that function in neuronal growth cones (PTPRD, ODZ3, ODZ2, CSMD1) or regulation of these processes through Rac/Rho signalling (TIAM1, DLC1, ARHGAP10, ATRX). The 19 tumours with defects in these genes were almost all stage 3 and 4 tumours diagnosed above 1.5 year of age with an aggressive clinical course. Only few of them showed amplification of MYCN (Fig. 4b). Consistent with their occurrence in high-stage neuroblastoma, defects in neuritogenesis genes did not show an independent prognostic power in multivariate analysis with the clinical parameters age and stage.
Here we report the first whole-genome sequence study of a comprehensive series of neuroblastoma including both low- and high-stage tumours. Low-stage neuroblastoma lacked recurrent gene alterations (mutations and structural variations), raising the question whether they are primarily driven by chromosomal imbalances and the consequent gene dosage effects. Tumours with defects in genes functioning in neuritogenesis or growth cone guidance mostly are aggressive high-stage tumours without MYCN amplification. Interestingly, there is indication that MYCN also inhibits neuritogenesis in neuroblastoma. MYCN downregulates the mRNA expression of the chromosome 1p36 gene CDC42, which resulted in inhibition of neuritogenesis of neuroblastoma cells28. CDC42 is a small GTPase protein with a function similar to Rac1. Rac1 and CDC42 both regulate the Par3–Par6 complex (also known as PARD3–PARD6A) involved in cell polarization and growth cone development and which has been shown to drive neuroblastoma cell differentiation25. In light of our data demonstrating genomic alterations in Rho/Rac signalling, it is tempting to postulate that the Par3–Par6 complex is a recurrent target for inactivation in neuroblastoma. Intriguingly, the block in neuritogenesis of neuroblastoma is probably not absolute. Retinoic acid can induce neuronal outgrowth in many neuroblastoma cell lines, which all are derived from high-stage tumours. Retinoic acid is also used in long-term neuroblastoma treatment protocols to prevent recurrences29. It is currently unknown which tumours will clinically respond to retinoic acid therapy. Deletions of NF1 were previously implicated in retinoic acid resistance of neuroblastoma cell lines30. The identification of genomic alterations in a range of genes mediating neuritogenesis now allows investigation of therapeutic modalities to surmount these defects.
Methods Summary
All neuroblastoma samples were derived from primary tumours of untreated patients. Tumour material was obtained during surgery and a portion was immediately frozen in liquid nitrogen. We used leukocytes derived from peripheral blood for isolation of constitutional DNA. High-molecular-weight DNA was extracted from tumour tissue and leukocytes using standard procedures. Fifteen microgram of DNA was subjected to paired-end whole-genome sequencing according to the Complete Genomics technology. Initial data analyses were performed using the CGAtools v1.3.0 package (http://cgatools.sourceforge.net/docs/1.3.0/) and for subsequent analysis and figure preparation we used the R2 bioinformatic platform (http://r2.amc.nl) and PERL scripts. More details on mutation analysis, coverage analysis, analysis of structural variants and the Circos plots is given in the Supplementary Information. Gene expression was assayed on Affymetrix EXON ST 1.0 GeneChips. SNP genotyping microarray analysis details are described in the supplementary information. Mutation validations were performed using Sanger sequencing (Supplementary Information). Statistical analysis (gene ontology enrichment using the Cytoscape BINGO plugin, DAVID functional ontology cluster analysis and analysis for gene length enrichment) are described in the Methods.
Online Methods
Small variants
Variant selection procedure: Potential somatic variants were determined with the CallDiff method with somatic output within the CGAtools v1.3.0 package, maintained by Complete Genomics (http://cgatools.sourceforge.net/docs/1.3.0/). Every tumour or cell line sample was compared to its matched blood sample across the whole genome. The somatic score which is calculated tries to tease apart true somatic mutations from false somatic mutations (http://cgatools.sourceforge.net/docs/1.3.0/). The somatic output files were then filtered to those regions where coding sequences are defined for the UCSC refflat annotation (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/refFlat.txt 2 August 2010), thereby removing discontinued genes from the original annotation of NCBI36.3. Subsequently, gene symbol, amino acid change and effect categories were extracted from gene-GSXXX-ASM.tsv files for all genomes and added as annotation to the somatic output results. Those variants that could not be annotated (new/updated genes) were annotated with custom PERL scripts where possible. All variants were annotated for their presence in dbSNP130 (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/snp130.txt), as well as the presence within 37 public HapMap genomes released by complete genomics (ftp://ftp2.completegenomics.com/). SIFT (http://sift.jcvi.org/www/SIFT_chr_coords_submit.html NCBI36) and polyphen2 (http://genetics.bwh.harvard.edu/pph2/bgi.shtml NCBI36) scores were determined to assess potential impact for all the SNP variants. Variants, which are reported in dbSNP130, that were found in any of the normal blood samples or that were found within the public genomes from Complete Genomics were removed from the data set. Finally, variants which were found in genes that are not expressed in neuroblastoma, but do show expression in a series of 500 normal samples were removed (See Affymetrix expression analysis).
Somatic small variants trim-down
CallDiff with somatic output was performed on all of the 87 tumour/lymphocyte pairs and processed. The number of events (split by deletion/insertion/substitution/SNP) were counted for the complete genome. Next, the somatic output files were filtered on those parts of the genome that are covered by amino-acid-encoding regions(based on the coding sequence (CDS) within the refflat file from the UCSC). In the next step, only those variants were kept that have an impact on the coding sequence (non-silent), do not occur in any of our normal samples, nor in any of the publicly available genomes from Complete Genomics, nor are present in dbSNP130. Finally, we filtered for the somatic score to be ≥0.1 (as determined by plotting of the Sanger sequence validation results as a function of somatic scores and total scores).
Sanger sequencing
High-molecular-weight DNA was extracted from tumour tissue and leukocytes using standard procedures31. Primers for PCR amplification were automatically designed by custom PERL scripts that execute the Primer3 software. PCR was performed using 20 ng of genomic DNA. Sanger sequencing was performed on a capillary sequencer using standard procedures.
Complete Genomics comparative genomic hybridization (cgCGH) procedure
First we determine the summed coverage (uniqueSequenceCoverage) in windows of 1,000 base pairs (measured on the reference genome) for the normal and the tumour sample (coverageRefScore files). Here we take the Integer(position/1,000) as the bin for any position and keep track of the sum. Then we determine the total coverage sums of the genome and normalize to this value, to remove differences in total coverage between samples. Subsequently we determine the log(tumour/genome)/log(2) for every bin (1,000-bp window) and obtain a cgCGH profile that expresses the somatic changes of the respective tumour. As the profile is normalized to its own normal material, the most prominent sources of bias such as GC-content, and also per-person copy number variation characteristics are corrected for. We feed these results to the DNAcopy algorithm as provided in R BioConductor to segment the information into blocks of similar characteristics and use these segments boundaries to store the information in an efficient way. cgCGH data was visualized within the embedded genome browser of R2 (http://r2.amc.nl).
Circos plots
Comparisons of somatic structural variants between tumour and lymphocyte genomes were performed with the JunctionDiff and Junction2Event tool from CGAtools (http://cgatools.sourceforge.net/docs/1.4.0/). These somatic events were filtered with the following criteria: events annotated as artefacts, footprints smaller than 70 bases, less than 10 discordant mate pairs, under-represented repeats, and presence in a set of v2.0 baseline genomes (as provided at the website of Complete Genomics (B36baseline-junctions.tsv)). cgCGH profiles and the remaining events were plotted with the Circos program (http://www.circos.ca).
Chromothripsis
Genomes were annotated as having chromothripsis-like characteristics when the sum of intra-chromosomal somatic junctions (as reported by JunctionDiff and filtered as above) within a single chromosome was larger or equal to 20. Focused amplified regions (cgCGH scores ≥3) within a chromosome were excluded from this sum. Using these characteristics, we annotated 10 out of the 87 patients as chromothripsis-like. Nine out of these patients were diagnosed with stage 4 neuroblastoma (P = 0.0392 stage4 versus rest) and all 10 were present in high-stage neuroblastoma (P = 0.0116). Eight of these patient have died of the disease (P = 0.0413; log-rank P = 7.1 × 10−3).
Affymetrix expression analysis (expressed genes)
To assess whether genes containing variants are expressed in neuroblastoma, we make use of a panel of neuroblastoma tumours (also including 53 of the sequenced tumours), classical neuroblastoma cell lines as well as recently generated patient derived cell lines (n = 119 in total).All samples were derived from primary tumours of untreated patients. Material was obtained during surgery and immediately frozen in liquid nitrogen. The original sources for classical neuroblastoma cell lines can be found in ref. 32. Total RNA of neuroblastoma samples was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. RNA concentration and quality were determined using the RNA 6000 nano assay on the Agilent 2100 Bioanalyzer (Agilent Technologies). Fragmentation of complementary RNA, hybridization to hg-u133 plus 2.0 microarrays and scanning were carried out according to the manufacturers protocol (Affymetrix). The data were deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE16476.
Affymetrix expression data from adult tumours were derived from the Expression Project for Oncology (ExpO) database from the International Genomics Consortium (IGC) (http://www.intgen.org/expo.cfm). Expression data on normal tissues was downloaded from GEO (GSE7307). The expression data were normalized with the MAS5.0 algorithm within the GCOS program of Affymetrix. Target intensity was set to 100. If more than one probe set was available for one gene the probe set with the highest expression was selected, considered that the probe set was correctly located on the gene of interest. For 101 patients (60 of the sequenced tumours), we have also generated Affymetrix Exon array data. The z-score of expression within this data set was used as a independent validation of structural variants annotated as deletions, where possible. All data were analysed using our in-house-developed R2 web application, which is freely accessible at http://r2.amc.nl.
SNP array analysis
SNP arrays were processed according to the manufacturer’s recommendations with the Infinium II assay on Human370- and Human660-quad arrays containing >370.000 and >660,000 markers, respectively, and run on the Illumina BeadStation (Swegene Centre for Integrative Biology, Lund University — SCIBLU, Sweden) according to the manufacturer’s recommendations. Raw data were processed using Illumina’s BeadStudio software suite (Genotyping module 3.0), producing report files containing normalized intensity data and SNP genotypes. Subsequently, log2 ratio and B-allele frequency data were imported into the R2 web application for detailed analysis and comparison with the CGH and expression data.
Selection procedure somatic small variants table
We ran CallDiff with somatic output on all of the 87 tumour/lymphocyte pairs and processed the somatic output files as described in the small variants section earlier. Due to the low validation percentages of substitutions and insertions, these were removed from the table. Variants which were tested by Sanger sequencing and were all-reference or all-variant were removed from the table. Variants with somatic scores lower than 0.1 or of types insertion/substitution, but which were validated by Sanger sequencing, were maintained in the table. In addition, we determined the presence of somatic splice-site variants in the two bases surrounding exons as defined by the UCSC refflat data.
Selection procedure somatic structural variant table
Comparisons between tumour and lymphocyte genomes were performed with the JunctionDiff and Junction2Event tool from CGAtools. These somatic events were filtered with the following criteria: events annotated as artefacts, footprints smaller than 70 bases, less than 10 discordant mate pairs, under-represented repeats, and presence in a set of baseline genomes (as provided at the website of Complete Genomics (B36baseline-junctions.tsv)). Of the remaining entries, we kept the following events: exon_bites where both ends of a junction are within the same gene, and in addition affect exonic sequence. Breaks by inversion, where both ends of a junction land within a gene, thereby damaging both genes, but leaving the genes in between unaffected. Potential fusion genes which are strand-matched, where both ends of a junction land within a gene, and the resulting end product fits in terms of orientation of both genes. Regions (deletions/(tandem) duplications) of up to 1 megabase, containing up to five genes which are expressed in neuroblastoma.
Combination of small, splice and structural somatic variants table
The small variant, structural variant and splice-site somatic tables were merged on the gene symbol, and unique tumour IDs were counted to obtain a final list of recurrent gene affecting variants.
Kaplan scan
Kaplan scanning was performed within R2 (http://r2.amc.nl). In short, for each gene or other numerical characteristic, R2 calculates the optimal cut-off expression level dividing the patients in a good and bad prognosis cohort. Samples within a data set are sorted according to the expression of the investigated gene and divided into two groups on the basis of a cut-off expression value. All cut-off expression levels and their resulting groups are analysed for survival, with the provision that minimal group number is 8 (or any other user-defined value) samples. For each cut-off level and grouping, the log-rank (as described in ref. 33) significance of the projected survival is calculated. The best P value and corresponding cut-off value is selected. This cut-off level is reported and used to generate a Kaplan–Meier graph. The graph depicts the log-rank significance (‘raw P’), as well as a P value corrected for the multiple testing (Bonferroni correction) of cut-off levels for each gene.
Statistical analysis for gene ontology gene enrichment
To investigate the possible role of the mutated genes we used the BinGO plugin34 for the network visualization tool Cytoscape35. This tool assesses enrichment of gene ontology categories for a set of genes. The P-value assigned to the overrepresentation for a specific category is calculated through a hypergeometric test. Results are corrected for multiple testing. We used the set of 1,041 genes having a mutation or structural variation in one or more neuroblastoma tumours. In the more informative gene ontology categories (filter set to less than 500 genes per category); the ‘regulation of GTPase activity’ gene ontology category seemed to be the most significant. The DAVID online functional clustering tool36 confirmed this analysis for the biological process gene ontology branch; the third cluster in the list contained the GTPase regulation category with an enrichment score of 1.44.
Statistical significance of genes affected by amino-acid-changing mutations and structural variations
We analysed the likelihood of finding defects in specific genes, or of finding defects in equal to/more than the number of affected patients. We therefore used the following randomization strategy: we started by counting the number of genes affected by structural variations, for every sample in our data set. Next, the genomic footprints (bases from start to the end of the RefSeq on the HG18 genome (UCSC refflat August 2010)) were determined and merged to the largest possible span for every gene symbol. These stretches were fused to create an artificial sequence, with the length of all gene symbols combined. Within this artificial sequence, random nucleotide numbers were chosen and traced back to the corresponding gene symbols. This was done for each tumour sample separately, with the number of randomly selected nucleotides being identical to the number of structural defects identified in the actual (sequenced) tumour. An identical strategy was used for the CDS affecting amino-acid-changing mutations, where the length of the largest CDS (NCBI RefSeq data set August 2010) per gene symbol has been used, to generate the artificial sequence.
In this way, we could generate artificial data sets, that most closely reflect the complexity of our sequenced sample set, including the spread in events, as well as the genomic footprints (size)/lengths of the CDS of all the different genes within the genome. We generated 109,000 artificial data sets, after which we created histograms to assess the null distributions for all genes, as well as the likelihood of finding combinations of recurrent events. The chance of finding any combination of three or more genes affected in five or more patients within our data set was 0.000211. Of note, this is a conservative estimation, as our real data set also included four frequently affected genes in the MYCN amplicon. The chance of affecting seven genes in five or more patients is much lower than 10−5. In addition, the chances of finding ATRX, ODZ3 or PTPRD in five or more patients was <10−5, 6.42 × 10−5 and 0.00534, respectively.
Statistical analysis for Rac/Rho GEF/GAP specificity
To ascertain whether the ratio of mutations/structural variations in Rac-specific GEFs and Rho-specific GAPs was significantly different from expectance, we performed Fisher’s exact test on the number of mutations in GAP and GEF with specificity for only Rac or Rho in our data set (one-sided Fisher’s exact test, P = 0.0007). We also pooled events in Rac-specific GEFs and Rho-specific GAPs together and tested whether they possessed more events than the pool of Rac-specific GAPs and Rho-specific GEFs (one-sided Fisher’s exact test, P = 0.026).
Accession codes
Primary accessions
Gene Expression Omnibus
Data deposits
mRNA expression array data are deposited in the Gene Expression Omnibus at NCBI, under accession number GSE16476. The Sequence data has been deposited at the European Genome-phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under accession number EGAS00001000222.
References
Maris, J. M. Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211 (2010)
Mossé, Y. P. et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935 (2008)
George, R. E. et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978 (2008)
Chen, Y. et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974 (2008)
Janoueix-Lerosey, I. et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967–970 (2008)
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011)
Kraus, D. M. et al. CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J. Immunol. 176, 4419–4430 (2006)
Sun, Q. L. et al. Growth cone steering by receptor tyrosine phosphatase delta defines a distinct class of guidance cue. Mol. Cell. Neurosci. 16, 686–695 (2000)
Zheng, L. et al. Drosophila Ten-m and filamin affect motor neuron growth cone guidance. PLoS ONE 6, e22956 (2011)
Chen, Q. R. et al. cDNA array-CGH profiling identifies genomic alterations specific to stage and MYCN-amplification in neuroblastoma. BMC Genomics 5, 70 (2004)
Vandesompele, J. et al. Multicentre analysis of patterns of DNA gains and losses in 204 neuroblastoma tumors: how many genetic subgroups are there? Med. Pediatr. Oncol. 36, 5–10 (2001)
Janoueix-Lerosey, I. et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. 27, 1026–1033 (2009)
Drmanac, R. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81 (2010)
Parsons, D. W. et al. The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439 (2011)
Leeuwen, F. N. et al. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 139, 797–807 (1997)
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nature Methods 7, 248–249 (2010)
Kee, Y. & D'Andrea, A. D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 24, 1680–1694 (2010)
E. E & Santo, a. l. Oncogenic activation of FOXR1 by 11q23 intrachromosomal deletion-fusions in neuroblastoma. Oncogene advance online publication. 10.1038/onc.2011.344 (22 August 2011)
Jiao, Y. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–1203 (2011)
Law, M. J. et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143, 367–378 (2010)
Arregui, C. O., Balsamo, J. & Lilien, J. Regulation of signaling by protein-tyrosine phosphatases: potential roles in the nervous system. Neurochem. Res. 25, 95–105 (2000)
Rubin, B. P. et al. Teneurins: a novel family of neuronal cell surface proteins in vertebrates, homologous to the Drosophila pair-rule gene product Ten-m. Dev. Biol. 216, 195–209 (1999)
Stallings, R. L. et al. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 66, 3673–3680 (2006)
Matsuo, N. et al. Roles of STEF/Tiam1, guanine nucleotide exchange factors for Rac1, in regulation of growth cone morphology. Mol. Cell. Neurosci. 24, 69–81 (2003)
Nishimura, T. et al. PAR-6–PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nature Cell Biol. 7, 270–277 (2005)
Bar-Sagi, D. & Hall, A. Ras and Rho GTPases: a family reunion. Cell 103, 227–238 (2000)
Shioda, N. et al. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J. Neurosci. 31, 346–358 (2011)
Valentijn, L. J. et al. Inhibition of a new differentiation pathway in neuroblastoma by copy number defects of N-myc, Cdc42, and nm23 genes. Cancer Res. 65, 3136–3145 (2005)
Matthay, K. K. et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J. Clin. Oncol. 27, 1007–1013 (2009)
Hölzel, M. et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 142, 218–229 (2010)
Molenaar, J. J., van Sluis, P., Boon, K., Versteeg, R. & Caron, H. N. Rearrangements and increased expression of cyclin D1 (CCND1) in neuroblastoma. Genes Chromosom. Cancer 36, 242–249 (2003)
Thiele, C. J. In Human Cell Culture vol. 1 (ed. Masters, J. ). 21–53 (1998)
Bewick, V., Cheek, L. & Ball, J. Statistics review 12: survival analysis. Crit. Care 8, 389–394 (2004)
Maere, S., Heymans, K. & Kuiper, M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 (2005)
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P.-L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 (2011)
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57 (2008)
Acknowledgements
The research in this paper was supported by grants from the Villa Joep Foundation, KIKA, Tom Voûte Fund, and the Netherlands Cancer Foundation. We thank R. Tearle and G. Tyrelle (Complete Genomics) for expert support and R. Volckmann, N. Hasselt, T. van Groningen, E. Dolman, K. Drabek and I. Dokter for their help.
Author information
Authors and Affiliations
Contributions
J.J.M.: project coordination, data analysis, preparation of manuscript; J.K.: supervision bioinformatics, bioinformatic data analysis, preparation of manuscript; D.A.Z., P.M.: bioinformatic analysis; P.v.S.: sample preparation and wet lab validation management; L.J.V., M.H., J.v.N., B.A.W.: data analysis and validation; I.v.d.P.: sample preparation and validation; J.v.A., M.E.E., F.H., A.L., L.S., P.S.: data validation; M.M.v.N., H.N.C.: sample and patient data organisation/contribution; I.Ø.: SNP profiling, E.M.W., E.E.S.: data analysis and pathway concepts; R.V.: project supervision, data analysis, preparation of manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Figures
This file contains Supplementary Figures 1-14 with legends. (PDF 2259 kb)
Supplementary Tables
This file contains Supplementary Tables 1-10. (XLS 576 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
Molenaar, J., Koster, J., Zwijnenburg, D. et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593 (2012). https://doi.org/10.1038/nature10910
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10910
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
