
Abstract
A major challenge of biology is understanding the relationship between molecular genetic variation and variation in quantitative traits, including fitness. This relationship determines our ability to predict phenotypes from genotypes and to understand how evolutionary forces shape variation within and between species. Previous efforts to dissect the genotype–phenotype map were based on incomplete genotypic information. Here, we describe the Drosophila melanogaster Genetic Reference Panel (DGRP), a community resource for analysis of population genomics and quantitative traits. The DGRP consists of fully sequenced inbred lines derived from a natural population. Population genomic analyses reveal reduced polymorphism in centromeric autosomal regions and the X chromosome, evidence for positive and negative selection, and rapid evolution of the X chromosome. Many variants in novel genes, most at low frequency, are associated with quantitative traits and explain a large fraction of the phenotypic variance. The DGRP facilitates genotype–phenotype mapping using the power of Drosophila genetics.
Similar content being viewed by others



Main
Understanding how molecular variation maps to phenotypic variation for quantitative traits is central for understanding evolution, animal and plant breeding, and personalized medicine1,2. The principles of mapping quantitative trait loci (QTLs) by linkage to, or association with, marker loci are conceptually simple1,2. However, we have not yet achieved our goal of explaining genetic variation for quantitative traits in terms of the underlying genes; additive, epistatic and pleiotropic effects as well as phenotypic plasticity of segregating alleles; and the molecular nature, population frequency and evolutionary dynamics of causal variants. Efforts to dissect the genotype–phenotype map in model organisms3,4 and humans5,6,7 have revealed unexpected complexities, implicating many, novel loci, pervasive pleiotropy, and context-dependent effects.
Model organism reference populations of inbred strains that can be shared among laboratories studying diverse phenotypes, and for which environmental conditions can be controlled and manipulated, greatly facilitate efforts to dissect the genetic architecture of quantitative traits3,4. Measuring many individuals of the same homozygous genotype increases the accuracy of the estimates of genotypic value1 and the power to detect variants, and genotypes of molecular markers need only be obtained once. We constructed the Drosophila melanogaster Genetic Reference Panel (DGRP) as such a community resource. Unlike previous populations of recombinant inbred lines derived from limited samples of genetic variation, the DGRP consists of 192 inbred strains derived from a single outbred population. The DGRP contains a representative sample of naturally segregating genetic variation, has an ultra-fine-grained recombination map suitable for precise localization of causal variants, and has almost complete euchromatic sequence information.
Here, we describe molecular and phenotypic variation in 168 re-sequenced lines comprising Freeze 1.0 of the DGRP, population genomic inferences of patterns of polymorphism and divergence and their correlation with genomic features, local recombination rate and selection acting on this population, genome-wide association mapping analyses for three quantitative traits, and tools facilitating the use of this resource.
Molecular variation in the DGRP
We constructed the DGRP by collecting mated females from the Raleigh, North Carolina, USA, population, followed by 20 generations of full-sibling inbreeding of their progeny. We sequenced 168 DGRP lines using a combination of Illumina and 454 sequencing technology: 29 of the lines were sequenced using both platforms, 129 lines have only Illumina sequence, and 10 lines have only 454 sequence. We mapped sequence reads to the D. melanogaster reference genome, re-calibrated base quality scores, and locally re-aligned Illumina reads. Mean sequence coverage was 21.4× per line for Illumina sequences and 12.1× per line for 454 sequences (Supplementary Table 1). On average, we assayed 113.5 megabases (94.25%) of the euchromatic reference sequence with ∼22,000 read mapping gaps per line (Supplementary Table 2). We called 4,672,297 single nucleotide polymorphisms (SNPs) using the Joint Genotyper for Inbred Lines (JGIL; E.A.S., personal communication), which takes into account coverage and quality sequencing statistics, and expected allele frequencies after 20 generations of inbreeding from an outbred population initially in Hardy–Weinberg equilibrium. In cases where base calls were made by both technologies, concordance was 99.36% (Supplementary Table 3).
The SNP site frequency distribution (Fig. 1a) is characterized by a majority of low frequency variants. The numbers of SNPs vary by chromosome and site class (Fig. 1b). Linkage disequilibrium8 decays to r2 = 0.2 on average within 10 base pairs on autosomes and 30 base pairs on the X chromosome (Fig. 1c and Supplementary Fig. 1). This difference is expected because the population size of the X chromosome is three quarters that of autosomes, and the X chromosome can experience greater purifying selection because of exposure of deleterious recessive alleles in hemizygous males. There is little evidence of global population structure in the DGRP (Fig. 1d and Supplementary Fig. 2). The rapid decline in linkage disequilibrium locally and lack of global population structure are favourable for genome-wide association mapping.

a, Site frequency spectrum. b, Numbers of SNPs per site class. c, Decay of linkage disequilibrium (r2) with physical distance for the five major chromosome arms. d, Lack of population structure. The red curve depicts the ranked eigenvalues of the genetic covariance matrix in decreasing order with respect to the marginal variance explained; the blue curve shows their cumulative sum as a fraction of the total with respect to cumulative variance explained. The partitioning of total genetic variance is balanced among the eigenvectors. The principal eigenvector explains < 1.1% of the total genetic variance.
Not all SNPs are fixed within individual DGRP lines (Supplementary Table 4). The expected inbreeding coefficient (F) after 20 generations of full-sibling inbreeding1 is F = 0.986; therefore, we expect some SNPs to remain segregating by chance. Segregating SNPs can also arise from new mutations, or if natural selection opposes inbreeding, due to true overdominance for fitness at individual loci or associative overdominance due to complementary deleterious alleles that are closely linked or in segregating inversions.
We identified 390,873 microsatellite loci, 105,799 of which were polymorphic (Supplementary Table 5); 36,810 transposable element insertion sites and 197,402 total insertions (Supplementary Table 6). On average, each line contained 1,175 transposable element insertions (Supplementary Table 6), although most transposable element insertion sites (25,562) were present in only one line (Supplementary Table 7). We identified 149 transposable element families. The number of copies per family varied greatly from an average of 315.7 INE-1 elements per line to an average of 0.003 Gandalf-Dkoe-like elements per line (Supplementary Table 8).
Wolbachia pipientis is a maternally inherited bacterium found in insects, including Drosophila, and can affect reproduction9. We assessed Wolbachia infection status in the DGRP lines to account for it in analyses of genotype–phenotype associations, and found 51.2% of lines harbouring sufficient Wolbachia DNA to imply infection (Supplementary Table 9).
Polymorphism and divergence
We used the DGRP Illumina sequence data and genome sequences from Drosophila simulans and Drosophila yakuba10 to perform genome-wide analyses of polymorphism and divergence, assess the association of these parameters with genomic features and the recombination landscape, and infer the historical action of selection on a much larger scale than had been possible previously11,12,13,14,15,16. We computed polymorphism (π and θ, refs 17 and 18) and divergence (k, ref. 19) for the whole genome, by chromosome arm (X, 2L, 2R, 3L, 3R), by chromosome region (three regions of equal size in Mb — telomeric, middle and centromeric), in 50-kbp non-overlapping windows, and by site class (synonymous and non-synonymous sites within coding sequences, and intronic, untranslated region (UTR) and intergenic sites) (Supplementary Tables 10 and 11).
Averaged over the entire genome, π = 0.0056 and θ = 0.0067, similar to previous estimates from North American populations16,20. Average polymorphism on the X chromosome (πX = 0.0040) is reduced relative to the autosomes (πA = 0.0060) (X/A ratio = 0.67, Wilcoxon test P = 0), even after correcting for the X/A effective population size (X4/3 = 0.0054, Wilcoxon test P < 0.00002; Supplementary Table 10). Autosomal nucleotide diversity is reduced on average 2.4-fold in centromeric regions relative to non-centromeric regions, and at the telomeres (Fig. 2a and Supplementary Table 10), whereas diversity is relatively constant along the X chromosome. Thus, πX > πA in centromeric regions, but πA > πX in other chromosomal regions (Fig. 2a and Supplementary Table 10).

a, Nucleotide polymorphism (π). The solid curves give the recombination rate (cM Mb−1). b, Divergence (k) for D. simulans (light green) and D. yakuba (dark green). c, Polymorphism to divergence ratio (Pol/Div), estimated as 1 − [(π0-fold/π4-fold)/(k0-fold/k4-fold)]. An excess of 0-fold divergence relative to polymorphism (k0-fold/k4-fold) > (π0-fold/π4-fold) is interpreted as adaptive fixation whereas an excess of 0-fold polymorphism relative to divergence (π0-fold/π4-fold) > (k0-fold/k4-fold) indicates that weakly deleterious or nearly neutral mutations are segregating in the population.
Genes on the X chromosome evolve faster (kX = 0.140) than autosomal genes (kA = 0.126) (X/A ratio = 1.131, Wilcoxon test P = 0) (Fig. 2b and Supplementary Table 10). Divergence is more uniform (coefficient of variation (CV)k = 0.2841) across chromosome arms than is polymorphism (CVπ = 0.4265). The peaks of divergence near the centromeres could be attributable to the reduced quality of alignments in these regions. Patterns of divergence are similar regardless of the outgroup species used (Fig. 2b and Supplementary Table 11).
The pattern of polymorphism and divergence by site class is consistent within and among chromosomes (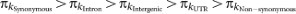 ), in agreement with previous studies on smaller data sets12,15 (Supplementary Figs 3 and 4 and Supplementary Table 11). Polymorphism levels between synonymous and non-synonymous sites differ by an order of magnitude. Variation and divergence patterns within the site classes generally follow the same patterns observed overall, with reduced polymorphism for all site classes on the X chromosome relative to autosomes, increased X chromosome divergence relative to autosomes for all but synonymous sites, decreased polymorphism in centromeric regions, and greater variation among regions and arms for polymorphism than for divergence. Other diversity measures and more detailed patterns at different window-sizes for each chromosome arm can be accessed from the Population Drosophila Browser (popDrowser) (Table 1 and Methods).
), in agreement with previous studies on smaller data sets12,15 (Supplementary Figs 3 and 4 and Supplementary Table 11). Polymorphism levels between synonymous and non-synonymous sites differ by an order of magnitude. Variation and divergence patterns within the site classes generally follow the same patterns observed overall, with reduced polymorphism for all site classes on the X chromosome relative to autosomes, increased X chromosome divergence relative to autosomes for all but synonymous sites, decreased polymorphism in centromeric regions, and greater variation among regions and arms for polymorphism than for divergence. Other diversity measures and more detailed patterns at different window-sizes for each chromosome arm can be accessed from the Population Drosophila Browser (popDrowser) (Table 1 and Methods).
Recombination landscape
Evolutionary models of hitchhiking and background selection21,22 predict a positive correlation between polymorphism and recombination rate. This expectation is realized in regions where recombination is less than 2 cM Mb−1 (Spearman’s ρ = 0.471, P = 0), but recombination and polymorphism are independent in regions where recombination exceeds 2 cM Mb−1 (Spearman’s ρ = −0.0044, P = 0.987) (Fig. 2a and Supplementary Table 12). The average rate of recombination of the X chromosome (2.9 cM Mb−1) is greater than that of autosomes (2.1 cM Mb−1), which may account for the low overall X-linked correlation between recombination rate and π. The lack of correlation between recombination and divergence (Supplementary Table 12) excludes mutation associated with recombination as the cause of the correlation. We assessed the independent effects of recombination rate, divergence, chromosome region and gene density on nucleotide variation of autosomes and the X chromosome (Supplementary Table 13). Recombination is the major predictor of polymorphism on the X chromosome and autosomes; however, the significant effect of autosomal chromosome region remains after accounting for variation in recombination rates between centromeric and non-centromeric regions.
Selection regimes
We used the standard23 and generalized12,24,25 McDonald Kreitman tests (MKT) to scan the genome for evidence of selection. These tests compare the ratio of polymorphism at a selected site with that of a neutral site to the ratio of divergence at a selected site to divergence at a neutral site. The standard MKT is applied to coding sequences, and synonymous and non-synonymous sites are used as putative neutral and selected sites, respectively. The generalized MKT is applied to non-coding sequences and uses fourfold degenerate sites as neutral sites. Using polymorphism and divergence data avoids confounding inference of selection with mutation rate differences, and restricting the tests to closely linked sites controls for shared evolutionary history26,27,28. We infer adaptive divergence when there is an excess of divergence relative to polymorphism, and segregation of slightly deleterious mutations when there is an excess of polymorphism over divergence. Estimates of α, the proportion of adaptive divergence, are biased downwards by low frequency, slightly deleterious mutations29,30. Rather than eliminate low frequency variants31, we incorporated information on the site frequency distribution to the MKT test framework to obtain estimates of the proportion of sites that are strongly deleterious (d), weakly deleterious (b), neutral (f) and recently neutral (γ) at segregating sites, as well as unbiased estimates of α (Supplementary Methods).
Deleterious and neutral sites
Averaged over the entire genome, we infer that 58.5% of the segregating sites are neutral or nearly neutral, 1.9% are weakly deleterious and 39.6% are strongly deleterious. However, these proportions vary between the X chromosome and autosomes, site classes and chromosome regions (Supplementary Tables 14–16 and Fig. 3). Non-synonymous sites are the most constrained (d = 77.6%), whereas in non-coding sites d ranges from 29.1% in 5′ UTRs to 41.3% in 3′ intergenic regions. The inferred pattern of selection differs between autosomal centromeric and non-centromeric regions: d is reduced and f is increased in centromeric regions for all site categories (Fig. 3). We observe an excess of polymorphism relative to divergence in autosomal centromeric regions, even after correcting for weakly deleterious mutations, implying a relaxation of selection from the time of separation of D. melanogaster and D. yakuba. Because selection coefficients depend on the effective population size32 (Ne), this could occur if the recombination rate has specifically diminished in centromeric regions during the divergence between D. melanogaster and D. yakuba; or with an overall reduction of Ne associated with the colonization of North American habitats33,34. In the latter case, we expect a genome-wide signature of an excess of low-frequency polymorphisms and of polymorphism relative to divergence, exacerbated in regions of low recombination. We indeed find an excess of low-frequency polymorphism relative to neutral expectation as indicated by the negative estimates of Tajima’s D statistic35 (D = −0.686 averaged over the whole genome and D = −0.997 in autosomal centromeric regions). In contrast, the X chromosome does not show a differential pattern of selection in the centromeric region, has a lower fraction of relaxation of selection, fewer neutral alleles, and a higher percentage of strongly deleterious alleles for all site classes and regions (Fig. 3 and Supplementary Tables 14–16).

The selection regimes are strongly deleterious (d, dark blue), weakly deleterious (b, blue), recently neutral (γ, white) and old neutral (f − γ, light blue). Each chromosome arm has been divided in three regions of equal size (in Mb): centromere, middle and telomere.
Transposable element insertions are thought to be largely deleterious. There are more singleton insertions in regions of high recombination (≥ 2 cM Mb−1) and more insertions shared in multiple lines in regions of low recombination (< 2 cM Mb−1) (Fisher’s exact test P = 0), and comparison of observed and expected site occupancy spectra reveals an excess of singleton insertions (P = 0, Supplementary Fig. 5).
Adaptive fixation
We find substantial evidence for positive selection in autosomal non-centromeric regions and the X chromosome (Fig. 2c and Supplementary Tables 15 and 17). We estimated α by aggregating all sites in each region analysed to avoid underestimation by averaging across genes36 in comparisons of chromosomes, regions and site classes. We also computed the direction of selection, DoS37, which is positive with adaptive selection, zero under neutrality and negative when weakly deleterious or new nearly neutral mutations are segregating. Estimates of α from the standard and generalized MKT indicate that on average 25.2% of the fixed sites between D. melanogaster and D. yakuba are adaptive, ranging from 30% in introns to 7% in UTR sites (Supplementary Fig. 6). Estimates of DoS and α are negative for non-synonymous and UTR sites in the autosomal centromeres, consistent with underestimating the fraction of adaptive substitutions in regions of low recombination because weakly deleterious or nearly neutral mutations are more common than adaptive fixations. The majority of adaptive fixation on autosomes occurs in non-centromeric regions (Fig. 2c). We find over four times as many adaptive fixations on the X chromosome relative to autosomes. The pattern holds for all site classes, in particular non-synonymous sites and UTRs, as well as individual genes, and is not solely due to the autosomal centromeric effect (Supplementary Table 15 and Supplementary Figs 6 and 7). Finally, when we consider DoS in recombination environments above and below 2 cM Mb−1, we find greater adaptive propensity in genes whose recombination context is ≥ 2 cM Mb−1 (Wilcoxon test, P = 0; Supplementary Fig. 8).
To understand the global patterns of divergence and constraint across functional classes of genes, we examined the distributions of ω (dN/dS, the ratio of non-synonymous to synonymous divergence) and DoS across gene ontology (GO) categories. The 4.9% GO categories with significantly elevated DoS include the biological process categories of behaviour, developmental process involved in reproduction, reproduction and ion transport (Supplementary Table 18). Recombination context is the major determinant of variation in DoS (Supplementary Table 19) whereas GO category is as important as recombinational context for predicting variation in ω (Supplementary Table 19).
GO categories enriched for positive DoS values differ from those associated with high values of ω (Supplementary Table 18), indicating that positive selection does not occur necessarily on genes with high ω values. If adaptive substitutions are common, high values of ω reflect the joint contributions of neutral and adaptive substitutions. Further, equating high constraint (low ω) with functional importance overlooks the functional role of adaptive changes15. Unlike ω, DoS takes into account the constraints inferred from the current polymorphism, distinguishing negative, neutral and adaptive selection.
Genome-wide genotype-phenotype associations
We measured resistance to starvation stress, chill coma recovery time and startle response38 in the DGRP. We found considerable genetic variation for all traits, with high broad sense heritabilities. We also found variation in sex dimorphism for starvation resistance and chill coma recovery with cross-sex genetic correlations significantly different from unity (Supplementary Tables 20–22).
We performed genome-wide association analyses for these traits, using the 2,490,165 SNPs and 77,756 microsatellites for which the minor allele was represented in four or more lines, using single-locus analyses pooled across sexes and separately for males and females. At P < 10−5 (P < 10−6), we find 203 (32) SNPs and 2 (0) microsatellites associated with starvation resistance; 90 (7) SNPs and 4 (2) microsatellites associated with startle response; and 235 (45) SNPs and 5 (3) microsatellites associated with chill coma recovery time (Fig. 4a, Supplementary Fig. 9 and Supplementary Tables 23 and 24). The minor allele frequencies for most of the associated SNPs are low, and there is an inverse relationship between effect sizes and minor allele frequency (Supplementary Fig. 10).

a, Genome-wide association results for significant SNPs. The lower triangle depicts linkage disequilibrium (r2) among SNPs, with the five major chromosome arms demarcated by black lines. The upper panels give the significance threshold (−log(p), uncorrected for multiple tests), the effect in phenotypic standard deviation units, and the minor allele frequency (MAF). b, c, Partial least squares regressions of phenotypes predicted using SNP data on observed phenotypes. The blue dots represent the predicted and observed phenotypes of lines that were not included in the initial study. b, Females (r2 = 0.81); c, males (r2 = 0.85).
The DGRP is a powerful tool for rapidly reducing the search space for molecular variants affecting quantitative traits from the entire genome to candidate polymorphisms and genes. Although we cannot infer which of these polymorphisms are causal due to linkage disequilibrium between SNPs in close physical proximity as well as occasional spurious long range linkage disequilibrium (Fig. 4a and Supplementary Fig. 9), the candidate gene lists are likely to be enriched for causal variants. The majority of associations are in computationally predicted genes or genes with annotated functions not obviously associated with the three traits. However, genes previously associated with startle response39 (Sema-1a and Eip75B) and starvation resistance40 (pnt) were identified in this study; and a SNP in CG3213, previously identified in a Drosophila obesity screen41, is associated with variation in starvation resistance. Several genes associated with quantitative traits are rapidly evolving (psq, Egfr; Supplementary Tables 17 and 23) or are plausible candidates based on SNP or gene ontology annotations (Supplementary Table 23).
Predicting phenotypes from genotypes
We used regression models to predict trait phenotypes from SNP genotypes and estimate the total variance explained by SNPs. The latter cannot be done by summing the individual contributions of the single marker effects because markers are not completely independent, and estimates of effects of single markers are biased when more than one locus affecting the trait segregates in the population. We derived gene-centred multiple regression models to estimate the effects of multiple SNPs simultaneously. In all cases 6–10 SNPs explain from 51–72% of the phenotypic variance and 65–90% of the genetic variance (Supplementary Tables 25 and 26 and Supplementary Figs 11–13). We also derived partial least square regression models using all SNPs for which the single marker effect was significant at P < 10−5. These models explain 72–85% of the phenotypic variance (Fig. 4b, c and Supplementary Fig. 14).
Discussion
The DGRP lines, sequences, variant calls, phenotypes and web tools for molecular population genomics and genome-wide association analysis are publicly available (Table 1). The DGRP lines contain at least 4,672,297 SNPs, 105,799 polymorphic microsatellites and 36,810 transposable elements, as well as insertion/deletion events and copy number variants and are a valuable resource for understanding the genetic architecture of quantitative traits of ecological and evolutionary relevance as well as Drosophila models of human quantitative traits. These novel mutations have survived the sieve of natural selection and will enhance the functional annotation of the Drosophila genome, complementing the Drosophila Gene Disruption Project42 and the Drosophila modENCODE project43.
Genome-wide molecular population genetic analyses show that patterns of polymorphism, but not divergence, differ by autosomal chromosome region, and between the X chromosome and autosomes. Polymorphism is lower in autosomal centromeric than non-centromeric regions, but not for the X chromosome. We propose that the correlation of polymorphism with recombination in regions where recombination is < 2 cM Mb−1 is due to the reduced effective population size in regions of low recombination8. Selection is less efficient in regions of low recombination32, consistent with our observation that the fraction of strongly deleterious mutations and positively selected sites are reduced in these regions.
All molecular population genomic analyses support the ‘faster X’ hypothesis44. Relative to the autosomes, the X chromosome shows lower polymorphism, faster rates of molecular evolution, a higher percentage of gene regions undergoing adaptive evolution, a higher fraction of strongly deleterious sites, and a lower level of weak negative selection and relaxation of selection. New X-linked mutations are directly exposed to selection each generation in hemizygous males, and the X chromosome has greater recombination than autosomes44; both of these factors could contribute to this observation.
Genome-wide association analyses of three fitness-related quantitative traits reveal hundreds of novel candidate genes, highlighting our ignorance of the genetic basis of complex traits. Most variants associated with the traits are at low frequency, and there is an inverse relationship between frequency and effect. Given that low-frequency alleles are likely to be deleterious for traits under directional or stabilizing selection, these results are consistent with the mutation–selection balance hypothesis1 for the maintenance of quantitative genetic variation. Regression models incorporating significant SNPs explain most of the phenotypic variance of the traits, in contrast with human association studies, where significant SNPs have tiny effects and together explain a small fraction of the total phenotypic variance7. If the genetic architecture of human complex traits is also dominated by low-frequency causal alleles, we expect estimates of effect size based on linkage disequilibrium with common variants to be strongly biased downwards.
In the future, the full power of Drosophila genetics can be applied to validating marker-trait associations: mutations, RNA interference constructs and quantitative trait loci mapping populations. The DGRP is an ideal resource for systems genetics analyses of the relationship between molecular variation, causal molecular networks and genetic variation for complex traits4,38,45, and will anchor evolutionary studies in comparison with sequenced Drosophila species to assess to what extent variation within a species corresponds to variation among species.
Methods Summary
The full Methods are in the Supplementary Information. Information on sequencing and bioinformatics includes methods for DNA isolation; library construction and genomic sequencing; sequence read alignment; SNP, microsatellite and transposable element identification; genotypes for assurance of sample identity; and Wolbachia detection. Methods for molecular population genomics analysis include details of recombination estimates; diversity measures, linkage disequilibrium and neutrality tests; software used for population genomic analysis; data visualization (popDrowser); standard and generalized McDonald–Kreitman tests, statistical analysis methods; quality assessment and data filtering; and gene ontology analyses. Methods for quantitative genetic analyses include phenotype measures, quantitative genetic analyses of phenotypes, statistical analyses of genotype–phenotype associations and predictive models, and a web-based association analysis pipeline.
References
Falconer, D. S. & Mackay, T. F. C. Introduction to Quantitative Genetics 4th edn (Longman, 1996)
Lynch, M. & Walsh, B. Genetics and Analysis of Quantitative Traits (Sinauer Associates, 1998)
Flint, J. & Mackay, T. F. C. Genetic architecture of quantitative traits in flies, mice and humans. Genome Res. 19, 723–733 (2009)
Mackay, T. F. C., Stone, E. A. & Ayroles, J. F. The genetics of quantitative traits: challenges and prospects. Nature Rev. Genet. 10, 565–577 (2009)
Altshuler, D., Daly, M. J. & Lander, E. S. Genetic mapping in human disease. Science 322, 881–888 (2008)
Donnelly, P. Progress and challenges in genome-wide association studies in humans. Nature 456, 728–731 (2008)
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009)
Hill, W. G. & Robertson, A. The effect of linkage on limits to artificial selection. Genet. Res. 8, 269–294 (1966)
Werren, J. H. Biology of Wolbachia. Annu. Rev. Entomol. 42, 587–609 (1997)
Clark, A. G. et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450, 203–218 (2007)
Smith, N. G. & Eyre-Walker, A. Adaptive protein evolution in Drosophila. Nature 415, 1022–1024 (2002)
Andolfatto, P. Adaptive evolution of non-coding DNA in Drosophila. Nature 437, 1149–1152 (2005)
Presgraves, D. C. Recombination enhances protein adaptation in Drosophila melanogaster. Curr. Biol. 15, 1651–1656 (2005)
Casillas, S., Barbadilla, A. & Bergman, C. Purifying selection maintains highly conserved noncoding sequences in Drosophila. Mol. Biol. Evol. 24, 2222–2234 (2007)
Sella, G. et al. Pervasive natural selection in the Drosophila genome? PLoS Genet. 5, e1000495 (2009)
Sackton, T. B. et al. Population genomic inferences from sparse high-throughput sequencing of two populations of Drosophila melanogaster. Genome Biol. Evol. 1, 449–465 (2009)
Nei, M. Molecular Evolutionary Genetics (Columbia Univ. Press, 1987)
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276 (1975)
Jukes, T. H. & Cantor, C. R. in Mammalian Protein Metabolism vol. 3 (eds Munro, H. N. & Allison, J. B.) 21–132 (Academic Press, 1969)
Andolfatto, P. & Przeworski, M. Regions of lower crossing over harbor more rare variants in African Drosophila melanogaster. Genetics 158, 657–665 (2001)
Begun, D. J. & Aquadro, C. F. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356, 519–520 (1992)
Charlesworth, B., Morgan, M. T. & Charlesworth, D. The effect of deleterious mutations on neutral molecular variation. Genetics 134, 1289–1303 (1993)
McDonald, J. H. & Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (1991)
Jenkins, D. L., Ortori, C. A. & Brookfield, J. F. A test for adaptive change in DNA sequences controlling transcription. Proc. R. Soc. Lond. B 261, 203–207 (1995)
Egea, R., Casillas, S. & Barbadilla, A. Standard and generalized McDonald–Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 36, W157–W162 (2008)
Sawyer, S. A. & Hartl, D. L. Population genetics of polymorphism and divergence. Genetics 132, 1161–1176 (1992)
Nielsen, R. Statistical tests of selective neutrality in the age of genomics. Heredity 86, 641–647 (2001)
Eyre-Walker, A. Changing effective population size and the McDonald-Kreitman test. Genetics 162, 2017–2024 (2002)
Charlesworth, J. & Eyre-Walker, A. The McDonald-Kreitman test and slightly deleterious mutations. Mol. Biol. Evol. 25, 1007–1015 (2008)
Eyre-Walker, A. & Keightley, P. D. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol. Biol. Evol. 26, 2097–2108 (2009)
Fay, J. C., Wyckoff, G. J. & Wu, C. I. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature 415, 1024–1026 (2002)
Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 246, 96–98 (1973)
David, J. R. & Capy, P. Genetic variation of Drosophila melanogaster natural populations. Trends Genet. 4, 106–111 (1988)
Begun, D. J. & Aquadro, C. F. African and North American populations of Drosophila melanogaster are very different at the DNA level. Nature 365, 548–550 (1993)
Tajima, F. Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989)
Smith, N. G. & Eyre-Walker, A. Adaptive protein evolution in Drosophila. Nature 415, 1022–1024 (2002)
Stoletzki, N. & Eyre-Walker, A. Estimation of the neutrality index. Mol. Biol. Evol. 28, 63–70 (2011)
Ayroles, J. F. et al. Systems genetics of complex traits in Drosophila melanogaster. Nature Genet. 41, 299–307 (2009)
Yamamoto, A. et al. Neurogenetic networks for startle-induced locomotion in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 105, 12393–12398 (2008)
Harbison, S. T., Yamamoto, A. H., Fanara, J. J., Norga, K. K. & Mackay, T. F. C. Quantitative trait loci affecting starvation resistance in Drosophila melanogaster. Genetics 166, 1807–1823 (2004)
Pospisilik, J. A. et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 140, 148–160 (2010)
Bellen, H. J. et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167, 761–781 (2004)
The ModENCODE Consortium. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797 (2010)
Charlesworth, B., Coyne, J. A. & Barton, N. H. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130, 113–146 (1987)
Sieberts, S. K. & Schadt, E. E. Moving toward a system genetics view of disease. Mamm. Genome 18, 389–401 (2007)
Acknowledgements
This work was supported by National Institutes of Health grant GM 45146 to T.F.C.M., E.A.S. and R.R.H.A.; R01 GM 059469 to R.R.H.A., MCI BFU 2009-09504 to A.B., R01 GM 085183 to K.R.T., NHGRI U54 HG003273 to R.A.G.; and an award through the NVIDIA Foundation’s “Compute the Cure” programme to D.M.
Author information
Authors and Affiliations
Contributions
T.F.C.M., S.R. and R.A.G. conceived the project. T.F.C.M., S.R., A.B. and E.A.S. wrote the main manuscript. T.F.C.M., S.R., A.B., E.A.S., J.F.A., K.R.T., J.M.C., C.M.B. and D.M. wrote the Supplementary methods. M.M.M., C.B., K.P.B., M.A.C., L.C., L.D., Y.H., M.J., J.C.J., S.N.J., K.W.J., F. Lara, F. Lawrence, S.L.L., R.F.L., M.M., D.M.M., L.N., I.M., L.P., L.L.P., C.Q., J.G.R., S.M.R., L.T., K.C.W., Y.-Q.W., A.Y. and Y.Z. performed experiments. T.F.C.M., A.B., J.F.A., D.Z., S.C., M.M.M., J.M.C., M.F.R., M.B., D.C., R.S.L., A.M., C.M.B., K.R.T., D.M. and E.A.S. did the bioinformatics and data analysis. J.F.A., S.C., M.M.M., Z.H., P.L., M.R., J.R. and E.A.S. wrote the Methods and did the web site development. R.R.H.A. contributed resources.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Sequences have been deposited at the National Center for Biotechnology Information Short Read Archives (http://www.ncbi.nlm.nih.gov/sra?term=DGRP).
Supplementary information
Supplementary Information
This file contains Supplementary Methods and Data (see Contents for more details) and Supplementary References. (PDF 0 kb)
Supplementary Figures
This file contains Supplementary Figures 1-16 with legends. (PDF 2118 kb)
Supplementary Tables
This file contains Supplementary Tables 1-17, 19-22 and 25-28 – see separate files for Supplementary Tables 18, 23 and 24. (PDF 6582 kb)
Supplementary Table 18
This file contains GO categories, selective constraint and positive selection. (XLS 72 kb)
Supplementary Table 23
This file contains GWA analysis results. (XLS 392 kb)
Supplementary Table 24
This file contains Microsatellite analysis results. (XLS 64 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
Mackay, T., Richards, S., Stone, E. et al. The Drosophila melanogaster Genetic Reference Panel. Nature 482, 173–178 (2012). https://doi.org/10.1038/nature10811
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10811
This article is cited by
-
Genome-wide association in Drosophila identifies a role for Piezo and Proc-R in sleep latency
Scientific Reports (2024)
-
Genome Wide Association Studies of Early Fitness Traits in Drosophila melanogaster Unveil Plasticity and Decoupling of Different Aspects of Phenotype
Evolutionary Biology (2024)
-
Genetic basis and repeatability for desiccation resistance in Drosophila melanogaster (Diptera: Drosophilidae)
Genetica (2024)
-
The composition of piRNA clusters in Drosophila melanogaster deviates from expectations under the trap model
BMC Biology (2023)
-
Rapid seasonal changes in phenotypes in a wild Drosophila population
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
