
Abstract
Potato (Solanum tuberosum L.) is the world’s most important non-grain food crop and is central to global food security. It is clonally propagated, highly heterozygous, autotetraploid, and suffers acute inbreeding depression. Here we use a homozygous doubled-monoploid potato clone to sequence and assemble 86% of the 844-megabase genome. We predict 39,031 protein-coding genes and present evidence for at least two genome duplication events indicative of a palaeopolyploid origin. As the first genome sequence of an asterid, the potato genome reveals 2,642 genes specific to this large angiosperm clade. We also sequenced a heterozygous diploid clone and show that gene presence/absence variants and other potentially deleterious mutations occur frequently and are a likely cause of inbreeding depression. Gene family expansion, tissue-specific expression and recruitment of genes to new pathways contributed to the evolution of tuber development. The potato genome sequence provides a platform for genetic improvement of this vital crop.
Similar content being viewed by others

Main
Potato (Solanum tuberosum L.) is a member of the Solanaceae, an economically important family that includes tomato, pepper, aubergine (eggplant), petunia and tobacco. Potato belongs to the asterid clade of eudicot plants that represents ∼25% of flowering plant species and from which a complete genome sequence has not yet, to our knowledge, been published. Potato occupies a wide eco-geographical range1 and is unique among the major world food crops in producing stolons (underground stems) that under suitable environmental conditions swell to form tubers. Its worldwide importance, especially within the developing world, is growing rapidly, with production in 2009 reaching 330 million tons (http://www.fao.org). The tubers are a globally important dietary source of starch, protein, antioxidants and vitamins2, serving the plant as both a storage organ and a vegetative propagation system. Despite the importance of tubers, the evolutionary and developmental mechanisms of their initiation and growth remain elusive.
Outside of its natural range in South America, the cultivated potato is considered to have a narrow genetic base resulting originally from limited germplasm introductions to Europe. Most potato cultivars are autotetraploid (2n = 4x = 48), highly heterozygous, suffer acute inbreeding depression, and are susceptible to many devastating pests and pathogens, as exemplified by the Irish potato famine in the mid-nineteenth century. Together, these attributes present a significant barrier to potato improvement using classical breeding approaches. A challenge to the scientific community is to obtain a genome sequence that will ultimately facilitate advances in breeding.
To overcome the key issue of heterozygosity and allow us to generate a high-quality draft potato genome sequence, we used a unique homozygous form of potato called a doubled monoploid, derived using classical tissue culture techniques3. The draft genome sequence from this genotype, S. tuberosum group Phureja DM1-3 516 R44 (hereafter referred to as DM), was used to integrate sequence data from a heterozygous diploid breeding line, S. tuberosum group Tuberosum RH89-039-16 (hereafter referred to as RH). These two genotypes represent a sample of potato genomic diversity; DM with its fingerling (elongated) tubers was derived from a primitive South American cultivar whereas RH more closely resembles commercially cultivated tetraploid potato. The combined data resources, allied to deep transcriptome sequence from both genotypes, allowed us to explore potato genome structure and organization, as well as key aspects of the biology and evolution of this important crop.
Genome assembly and annotation
We sequenced the nuclear and organellar genomes of DM using a whole-genome shotgun sequencing (WGS) approach. We generated 96.6 Gb of raw sequence from two next-generation sequencing (NGS) platforms, Illumina Genome Analyser and Roche Pyrosequencing, as well as conventional Sanger sequencing technologies. The genome was assembled using SOAPdenovo4, resulting in a final assembly of 727 Mb, of which 93.9% is non-gapped sequence. Ninety per cent of the assembly falls into 443 superscaffolds larger than 349 kb. The 17-nucleotide depth distribution (Supplementary Fig. 1) suggests a genome size of 844 Mb, consistent with estimates from flow cytometry5. Our assembly of 727 Mb is 117 Mb less than the estimated genome size. Analysis of the DM scaffolds indicates 62.2% repetitive content in the assembled section of the DM genome, less than the 74.8% estimated from bacterial artificial chromosome (BAC) and fosmid end sequences (Supplementary Table 1), indicating that much of the unassembled genome is composed of repetitive sequences.
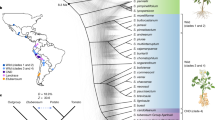
We assessed the quality of the WGS assembly through alignment to Sanger-derived phase 2 BAC sequences. In an alignment length of ∼1 Mb (99.4% coverage), no gross assembly errors were detected (Supplementary Table 2 and Supplementary Fig. 2). Alignment of fosmid and BAC paired-end sequences to the WGS scaffolds revealed limited (≤0.12%) potential misassemblies (Supplementary Table 3). Extensive coverage of the potato genome in this assembly was confirmed using available expressed sequence tag (EST) data; 97.1% of 181,558 available Sanger-sequenced S. tuberosum ESTs (>200 bp) were detected. Repetitive sequences account for at least 62.2% of the assembled genome (452.5 Mb) (Supplementary Table 1) with long terminal repeat retrotransposons comprising the majority of the transposable element classes, representing 29.4% of the genome. In addition, subtelomeric repeats were identified at or near chromosomal ends (Fig. 1). Using a newly constructed genetic map based on 2,603 polymorphic markers in conjunction with other available genetic and physical maps, we genetically anchored 623 Mb (86%) of the assembled genome (Supplementary Fig. 3), and constructed pseudomolecules for each of the 12 chromosomes (Fig. 1), which harbour 90.3% of the predicted genes.

a, Ideograms of the 12 pseudochromosomes of potato (in Mb scales). Each of the 12 pachytene chromosomes from DM was digitally aligned with the ideogram (the amount of DNA in each unit of the pachytene chromosomes is not in proportion to the scales of the pseudochromosomes). b, Gene density represented as number of genes per Mb (non-overlapping, window size = 1 Mb). c, Percentage of coverage of repetitive sequences (non-overlapping windows, window size = 1 Mb). d, Transcription state. The transcription level for each gene was estimated by averaging the fragments per kb exon model per million mapped reads (FPKM) from different tissues in non-overlapping 1-Mb windows. e, GC content was estimated by the per cent G+C in 1-Mb non-overlapping windows. f, Distribution of the subtelomeric repeat sequence CL14_cons.
To aid annotation and address a series of biological questions, we generated 31.5 Gb of RNA-Seq data from 32 DM and 16 RH libraries representing all major tissue types, developmental stages and responses to abiotic and biotic stresses (Supplementary Table 4). For annotation, reads were mapped against the DM genome sequence (90.2% of 824,621,408 DM reads and 88.6% of 140,375,647 RH reads) and in combination with ab initio gene prediction, protein and EST alignments, we annotated 39,031 protein-coding genes. RNA-Seq data revealed alternative splicing; 9,875 genes (25.3%) encoded two or more isoforms, indicative of more functional variation than represented by the gene set alone. Overall, 87.9% of the gene models were supported by transcript and/or protein similarity with only 12.1% derived solely from ab initio gene predictions (Supplementary Table 5).
Karyotypes of RH and DM suggested similar heterochromatin content6 (Supplementary Table 6 and Supplementary Fig. 4) with large blocks of heterochromatin located at the pericentromeric regions (Fig. 1). As observed in other plant genomes, there was an inverse relationship between gene density and repetitive sequences (Fig. 1). However, many predicted genes in heterochromatic regions are expressed, consistent with observations in tomato7 that genic ‘islands’ are present in the heterochromatic ‘ocean’.
Genome evolution
Potato is the first sequenced genome of an asterid, a clade within eudicots that encompasses nearly 70,000 species characterized by unique morphological, developmental and compositional features8. Orthologous clustering of the predicted potato proteome with 11 other green plant genomes revealed 4,479 potato genes in 3,181 families in common (Fig. 2a); 24,051 potato genes clustered with at least one of the 11 genomes. Filtering against transposable elements and 153 nonasterid and 57 asterid publicly available transcript-sequence data sets yielded 2,642 high-confidence asterid-specific and 3,372 potato-lineage-specific genes (Supplementary Fig. 5); both sets were enriched for genes of unknown function that had less expression support than the core Viridiplantae genes. Genes encoding transcription factors, self-incompatibility, and defence-related proteins were evident in the asterid-specific gene set (Supplementary Table 7) and presumably contribute to the unique characteristics of asterids.

a, Clusters of orthologous and paralogous gene families in 12 plant species as identified by OrthoMCL33. Gene family number is listed in each of the components; the number of genes within the families for all of the species within the component is noted within parentheses. b, Genome duplication in dicot genomes as revealed through 4DTv analyses. c, Syntenic blocks between A. thaliana, potato, and V. vinifera (grape) demonstrating a high degree of conserved gene order between these taxa.
Structurally, we identified 1,811 syntenic gene blocks involving 10,046 genes in the potato genome (Supplementary Table 8). On the basis of these pairwise paralogous segments, we calculated an age distribution based on the number of transversions at fourfold degenerate sites (4DTv) for all duplicate pairs. In general, two significant groups of blocks are seen in the potato genome (4DTv ∼0.36 and ∼1.0; Fig. 2b), suggesting two whole-genome duplication (WGD) events. We also identified collinear blocks between potato and three rosid genomes (Vitis vinifera, Arabidopsis thaliana and Populus trichocarpa) that also suggest both events (Fig. 2c and Supplementary Fig. 6). The ancient WGD corresponds to the ancestral hexaploidization (γ) event in grape (Fig. 2b), consistent with a previous report based on EST analysis that the two main branches of eudicots, the asterids and rosids, may share the same palaeo-hexaploid duplication event9. The γ event probably occurred after the divergence between dicots and monocots about 185 ± 55 million years ago10. The recent duplication can therefore be placed at ∼67 million years ago, consistent with the WGD that occurred near the Cretaceous–Tertiary boundary (∼65 million years ago)11. The divergence of potato and grape occurred at ∼89 million years ago (4DTv ∼0.48), which is likely to represent the split between the rosids and asterids.
Haplotype diversity
High heterozygosity and inbreeding depression are inherent to potato, a species that predominantly outcrosses and propagates by means of vegetative organs. Indeed, the phenotypes of DM and RH differ, with RH more vigorous than DM (Fig. 3a). To explore the extent of haplotype diversity and possible causes of inbreeding depression, we sequenced and assembled 1,644 RH BAC clones generating 178 Mb of non-redundant sequence from both haplotypes (∼10% of the RH genome with uneven coverage) (Supplementary Tables 9–11). After filtering to remove repetitive sequences, we aligned 99 Mb of RH sequence (55%) to the DM genome. These regions were largely collinear with an overall sequence identity of 97.5%, corresponding to one single-nucleotide polymorphism (SNP) every 40 bp and one insertion/deletion (indel) every 394 bp (average length 12.8 bp). Between the two RH haplotypes, 6.6 Mb of sequence could be aligned with 96.5% identity, corresponding to 1 SNP per 29 bp and 1 indel per 253 bp (average length 10.4 bp).

a, Plants and tubers of DM and RH showing that RH has greater vigour. b, Illumina K-mer volume histograms of DM and RH. The volume of K-mers (y-axis) is plotted against the frequency at which they occur (x-axis). The leftmost truncated peaks at low frequency and high volume represent K-mers containing essentially random sequencing errors, whereas the distribution to the right represents proper (putatively error-free) data. In contrast to the single modality of DM, RH exhibits clear bi-modality caused by heterozygosity. c, Genomic distribution of premature stop, frameshift and presence/absence variation mutations contributing to inbreeding depression. The hypothetical RH pseudomolecules were solely inferred from the corresponding DM ones. Owing to the inability to assign heterozygous PS and FS of RH to a definite haplotype, all heterozygous PS and FS were arbitrarily mapped to the left haplotype of RH. d, A zoom-in comparative view of the DM and RH genomes. The left and right alignments are derived from the euchromatic and heterochromatic regions of chromosome 5, respectively. Most of the gene annotations, including PS and RH-specific genes, are supported by transcript data.
Current algorithms are of limited use in de novo whole-genome assembly or haplotype reconstruction of highly heterozygous genomes such as RH, as shown by K-mer frequency count histograms (Fig. 3b and Supplementary Table 12). To complement the BAC-level comparative analysis and provide a genome-wide perspective of heterozygosity in RH, we mapped 1,118 million whole-genome NGS reads from RH (84× coverage) onto the DM assembly. A total of 457.3 million reads uniquely aligned providing 90.6% (659.1 Mb) coverage. We identified 3.67 million SNPs between DM and one or both haplotypes of RH, with an error rate of 0.91% based on evaluation of RH BAC sequences. We used this data set to explore the possible causes of inbreeding depression by quantifying the occurrence of premature stop, frameshift and presence/absence variants12, as these disable gene function and contribute to genetic load (Supplementary Tables 13–16). We identified 3,018 SNPs predicted to induce premature stop codons in RH, with 606 homozygous (in both haplotypes) and 2,412 heterozygous. In DM, 940 premature stop codons were identified. In the 2,412 heterozygous RH premature stop codons, 652 were shared with DM and the remaining 1,760 were found in RH only (Fig. 3c and Supplementary Table 13). Frameshift mutations were identified in 80 loci within RH, 49 homozygous and 31 heterozygous, concentrated in seven genomic regions (Fig. 3c and Supplementary Table 14). Finally, we identified presence/absence variations for 275 genes; 246 were RH specific (absent in DM) and 29 were DM specific, with 125 and 9 supported by RNA-Seq and/or Gene Ontology13 annotation for RH and DM, respectively (Supplementary Tables 15 and 16). Collectively, these data indicate that the complement of homozygous deleterious alleles in DM may be responsible for its reduced level of vigour (Fig. 3a).
The divergence between potato haplotypes is similar to that reported between out-crossing maize accessions14 and, coupled with our inability to successfully align 45% of the BAC sequences, intra- and inter-genome diversity seem to be a significant feature of the potato genome. A detailed comparison of the three haplotypes (DM and the two haplotypes of RH) at two genomic regions (334 kb in length) using the RH BAC sequence (Fig. 3d and Supplementary Tables 17 and 18) revealed considerable sequence and structural variation. In one region (‘euchromatic’; Fig. 3d) we observed one instance of copy number variation, five genes with premature stop codons, and seven RH-specific genes. These observations indicate that the plasticity of the potato genome is greater than revealed from the unassembled RH NGS. Improved assembly algorithms, increased read lengths, and de novo sequences of additional haplotypes will reveal the full catalogue of genes critical to inbreeding depression.
Tuber biology
In developing DM and RH tubers, 15,235 genes were expressed in the transition from stolons to tubers, with 1,217 transcripts exhibiting >5-fold expression in stolons versus five RH tuber tissues (young tuber, mature tuber, tuber peel, cortex and pith; Supplementary Table 19). Of these, 333 transcripts were upregulated during the transition from stolon to tuber, with the most highly upregulated transcripts encoding storage proteins. Foremost among these were the genes encoding proteinase inhibitors and patatin (15 genes), in which the phospholipase A function has been largely replaced by a protein storage function in the tuber15. In particular, a large family of 28 Kunitz protease inhibitor genes (KTIs) was identified with twice the number of genes in potato compared to tomato. The KTI genes are distributed across the genome with individual members exhibiting specific expression patterns (Fig. 4a, b). KTIs are frequently induced after pest and pathogen attack and act primarily as inhibitors of exogenous proteinases16; therefore the expansion of the KTI family may provide resistance to biotic stress for the newly evolved vulnerable underground organ.

a, KTI gene organization across the potato genome. Black arrows indicate the location of individual genes on six scaffolds located on four chromosomes. b, Phylogenetic tree and KTI gene expression heat map. The KTI genes were clustered using all potato and tomato genes available with the Populus KTI gene as an out-group. The tissue specificity of individual members of the highly expanded potato gene family is shown in the heat map. Expression levels are indicated by shades of red, where white indicates no expression or lack of data for tomato and poplar. c, A model of starch synthesis showing enzyme activities is shown on the left. AGPase, ADP-glucose pyrophosphorylase; F16BP, fructose-1,6-biphosphatase; HexK, hexokinase; INV, invertase; PFK, phosphofructokinase; PFPP, pyrophosphate-fructose-6-phosphate-1-phosphotransferase; PGI, phosphoglucose isomerase; PGM, phosphoglucomutase; SBE, starch branching enzyme; SP, starch phosphorylase; SPP, sucrose phosphate phosphatase; SS, starch synthase; SuSy, sucrose synthase; SUPS, sucrose phosphate synthase; UDP-GPP, UDP-glucose pyrophosphorylase. The grey background denotes substrate (sucrose) and product (starch) and the red background indicates genes that are specifically upregulated in RH versus DM. On the right, a heat map of the genes involved in carbohydrate metabolism is shown. ADP-glucose pyrophosphorylase large subunit, AGPase (l); ADP-glucose pyrophosphorylase small subunit, AGPase (s); ADP-glucose pyrophosphorylase small subunit 3, AGPase 3 (s); cytosolic fructose-1,6-biphosphatase, F16BP (c); granule bound starch synthase, GBSS; leaf type L starch phosphorylase, Leaf type SP; plastidic phosphoglucomutase, pPGM; starch branching enzyme II, SBE II; soluble starch synthase, SSS; starch synthase V, SSV; three variants of plastidic aldolase, PA.
The stolon to tuber transition also coincides with strong upregulation of genes associated with starch biosynthesis (Fig. 4c). We observed several starch biosynthetic genes that were 3–8-fold more highly expressed in tuber tissues of RH compared to DM (Fig. 4c). Together this suggests a stronger shift from the relatively low sink strength of the ATP-generating general carbon metabolism reactions towards the plastidic starch synthesis pathway in tubers of RH, thereby causing a flux of carbon into the amyloplast. This contrasts with the cereal endosperm where carbon is transported into the amyloplast in the form of ADP-glucose via a specific transporter (brittle 1 protein17). Carbon transport into the amyloplasts of potato tubers is primarily in the form of glucose-6-phosphate18, although recent evidence indicates that glucose-1-phosphate is quantitatively important under certain conditions19. The transport mechanism for glucose-1-phosphate is unknown and the genome sequence contains six genes for hexose-phosphate transporters with two highly and specifically expressed in stolons and tubers. Furthermore, an additional 23 genes encode proteins homologous to other carbohydrate derivative transporters, such as triose phosphate, phosphoenolpyruvate, or UDP-glucuronic acid transporters and two loci with homologues for the brittle 1 protein. By contrast, in leaves, carbon-fixation-specific genes such as plastidic aldolase, fructose-1,6-biphosphatase and distinct leaf isoforms of starch synthase, starch branching enzyme, starch phosphorylase and ADP-glucose pyrophosphorylase were upregulated. Of particular interest is the difference in tuber expression of enzymes involved in the hydrolytic and phosphorolytic starch degradation pathways. Considerably greater levels of α-amylase (10–25-fold) and β-amylase (5–10-fold) mRNAs were found in DM tubers compared to RH, whereas α-1,4 glucan phosphorylase mRNA was equivalent in DM and RH tubers. These gene expression differences between the breeding line RH and the more primitive DM are consistent with the concept that increasing tuber yield may be partially attained by selection for decreased activity of the hydrolytic starch degradation pathway.
Recent studies using a potato genotype strictly dependent on short days for tuber induction (S. tuberosum group Andigena) identified a potato homologue (SP6A) of A. thaliana FLOWERING LOCUS T (FT) as the long-distance tuberization inductive signal. SP6A is produced in the leaves, consistent with its role as the mobile signal (S. Prat, personal communication). SP/FT is a multi-gene family (Supplementary Text and Supplementary Fig. 7) and expression of a second FT homologue, SP5G, in mature tubers suggests a possible function in the control of tuber sprouting, a photoperiod-dependent phenomenon20. Likewise, expression of a homologue of the A. thaliana flowering time MADS box gene SOC1, acting downstream of FT21, is restricted to tuber sprouts (Supplementary Fig. 8). Expression of a third FT homologue, SP3D, does not correlate with tuberization induction but instead with transition to flowering, which is regulated independently of day length (S. Prat, personal communication). These data indicate that neofunctionalization of the day-length-dependent flowering control pathway has occurred in potato to control formation and possibly sprouting of a novel storage organ, the tuber (Supplementary Fig. 9).
Disease resistance
Potato is susceptible to a wide range of pests and pathogens and the identification of genes conferring disease resistance has been a major focus of the research community. Most cloned disease resistance genes in the Solanaceae encode nucleotide-binding site (NBS) and leucine-rich-repeat (LRR) domains. The DM assembly contains 408 NBS-LRR-encoding genes, 57 Toll/interleukin-1 receptor/plant R gene homology (TIR) domains and 351 non-TIR types (Supplementary Table 20), similar to the 402 resistance (R) gene candidates in Populus22. Highly related homologues of the cloned potato late blight resistance genes R1, RB, R2, R3a, Rpi-blb2 and Rpi-vnt1.1 were present in the assembly. In RH, the chromosome 5 R1 cluster contains two distinct haplotypes; one is collinear with the R1 region in DM (Supplementary Fig. 10), yet neither the DM nor the RH R1 regions are collinear with other potato R1 regions23,24. Comparison of the DM potato R gene sequences with well-established gene models (functional R genes) indicates that many NBS-LRR genes (39.4%) are pseudogenes owing to indels, frameshift mutations, or premature stop codons including the R1, R3a and Rpi-vnt1.1 clusters that contain extensive chimaeras and exhibit evolutionary patterns of type I R genes25. This high rate of pseudogenization parallels the rapid evolution of effector genes observed in the potato late blight pathogen, Phytophthora infestans26. Coupled with abundant haplotype diversity, tetraploid potato may therefore contain thousands of R-gene analogues.
Conclusions and future directions
We sequenced a unique doubled-monoploid potato clone to overcome the problems associated with genome assembly due to high levels of heterozygosity and were able to generate a high-quality draft potato genome sequence that provides new insights into eudicot genome evolution. Using a combination of data from the vigorous, heterozygous diploid RH and relatively weak, doubled-monoploid DM, we could directly address the form and extent of heterozygosity in potato and provide the first view into the complexities that underlie inbreeding depression. Combined with other recent studies, the potato genome sequence may elucidate the evolution of tuberization. This evolutionary innovation evolved exclusively in the Solanum section Petota that encompasses ∼200 species distributed from the southwestern United States to central Argentina and Chile. Neighbouring Solanum species, including the Lycopersicon section, which comprises wild and cultivated tomatoes, did not acquire this trait. Both gene family expansion and recruitment of existing genes for new pathways contributed to the evolution of tuber development in potato.
Given the pivotal role of potato in world food production and security, the potato genome provides a new resource for use in breeding. Many traits of interest to plant breeders are quantitative in nature and the genome sequence will simplify both their characterization and deployment in cultivars. Whereas much genetic research is conducted at the diploid level in potato, almost all potato cultivars are tetraploid and most breeding is conducted in tetraploid material. Hence, the development of experimental and computational methods for routine and informative high-resolution genetic characterization of polyploids remains an important goal for the realization of many of the potential benefits of the potato genome sequence.
Methods Summary
DM1-3 516 R44 (DM) resulted from chromosome doubling of a monoploid (1n = 1x = 12) derived by anther culture of a heterozygous diploid (2n = 2x = 24) S. tuberosum group Phureja clone (PI 225669)27. RH89-039-16 (RH) is a diploid clone derived from a cross between a S. tuberosum ‘dihaploid’ (SUH2293) and a diploid clone (BC1034) generated from a cross between two S. tuberosum × S. tuberosum group Phureja hybrids28 (Supplementary Fig. 11). Sequence data from three platforms, Sanger, Roche 454 Pyrosequencing, and Illumina Sequencing-by-Synthesis, were used to assemble the DM genome using the SOAPdenovo assembly algorithm4. The RH genotype was sequenced using shotgun sequencing of BACs and WGS in which reads were mapped to the DM reference assembly. Superscaffolds were anchored to the 12 linkage groups using a combination of in silico and genetic mapping data. Repeat sequences were identified through sequence similarity at the nucleotide and protein level29. Genes were annotated using a combined approach30 on the repeat masked genome with ab initio gene predictions, protein similarity and transcripts to build optimal gene models. Illumina RNA-Seq reads were mapped to the DM draft sequence using Tophat31 and expression levels from the representative transcript were determined using Cufflinks32.
Online Methods
DM whole-genome shotgun sequencing and assembly
Libraries were constructed from DM genomic DNA and sequenced on the Sanger, Illumina Genome Analyser 2 (GA2) and Roche 454 platforms using standard protocols (see Supplementary Text). A BAC library and three fosmid libraries were end sequenced using the Sanger platform. For the Illumina GA2 platform, we generated 70.6 Gb of 37–73 bp paired-end reads from 16 libraries with insert lengths of 200–811 bp (Supplementary Tables 21 and 22). We also generated 18.7 Gb of Illumina mate-pair libraries (2, 5 and 10 kb insert size). In total, 7.2 Gb of 454 single-end data were generated and applied for gap filling to improve the assembly, of which 4.7 Gb (12,594,513 reads) were incorporated into the final assembly. For the 8 and 20 kb 454 paired-end reads, representing 0.7 and 1.0 Gb of raw data respectively, 90.7 Mb (511,254 reads) and 211 Mb (1,525,992 reads), respectively, were incorporated into the final assembly.
We generated a high-quality potato genome using the short read assembly software SOAPdenovo4 (Version 1014). We first assembled 69.4 Gb of GA2 paired-end short reads into contigs, which are sequence assemblies without gaps composed of overlapping reads. To increase the assembly accuracy, only 78.3% of the reads with high quality were considered. Then contigs were further linked into scaffolds by paired-end relationships (∼300 to ∼550 bp insert size), mate-pair reads (2 to approximately 10 kb), fosmid ends (∼40 kb, 90,407 pairs of end sequences) and BAC ends (∼100 kb, 71,375 pairs of end sequences). We then filled gaps with the entire short-read data generated using Illumina GA2 reads. The primary contig N50 size (the contig length such that using equal or longer contigs produces half of the bases of the assembled genome) was 697 bp and increased to 1,318 kb after gap-filling (Supplementary Tables 23 and 24). When only the paired-end relationships were used in the assembly process, the N50 scaffold size was 22.4 kb. Adding mate-pair reads with 2, 5 and 10 kb insert sizes, the N50 scaffold size increased to 67, 173 and 389 kb, respectively. When integrated with additional libraries of larger insert size, such as fosmid and BAC end sequences, the N50 reached 1,318 kb. The final assembly size was 727 Mb, 93.87% of which is non-gapped sequence. We further filled the gaps with 6.74 fold coverage of 454 data, which increased the N50 contig size to 31,429 bp with 15.4% of the gaps filled.
The single-base accuracy of the assembly was estimated by the depth and proportion of disconcordant reads. For the DM v3.0 assembly, 95.45% of 880 million usable reads could be mapped back to the assembled genome by SOAP 2.20 (ref. 34) using optimal parameters. The read depth was calculated for each genomic location and peak depth for whole genome and the CDS regions are 100 and 105, respectively. Approximately 96% of the assembled sequences had more than 20-fold coverage (Supplementary Fig. 1). The overall GC content of the potato genome is about 34.8% with a positive correlation between GC content and sequencing depth (data not shown). The DM potato should have few heterozygous sites and 93.04% of the sites can be supported by at least 90% reads, suggesting high base quality and accuracy.
RH genome sequencing
Whole-genome sequencing of genotype RH was performed on the Illumina GA2 platform using a variety of fragment sizes and reads lengths resulting in a total of 144 Gb of raw data (Supplementary Table 25). These data were filtered using a custom C program and assembled using SOAPdenovo 1.03 (ref. 4). Additionally, four 20-kb mate-pair libraries were sequenced on a Roche 454 Titanium sequencer, amounting to 581 Mb of raw data (Supplementary Table 26). The resulting sequences were filtered for duplicates using custom Python scripts.
The RH BACs were sequenced using a combination of Sanger and 454 sequencing at various levels of coverage (Supplementary Tables 9–11). Consensus base calling errors in the BAC sequences were corrected using custom Python and C scripts using a similar approach to that described previously35 (Supplementary Text). Sequence overlaps between BACs within the same physical tiling path were identified using megablast from BLAST 2.2.21 (ref. 36) and merged with megamerger from the EMBOSS 6.1.0 package37. Using the same pipeline, several kilobase-sized gaps were closed through alignment of a preliminary RH whole-genome assembly. The resulting non-redundant contigs were scaffolded by mapping the RH whole-genome Illumina and 454 mated sequences against these contigs using SOAPalign 2.20 (ref. 34) and subsequently processing these mapping results with a custom Python script. The scaffolds were then ordered into superscaffolds based on the BAC order in the tiling paths of the FPC map. This procedure removed 25 Mb of redundant sequence, reduced the number of sequence fragments from 17,228 to 3,768, and increased the N50 sequence length from 24 to 144 kb (Supplementary Tables 9 and 10).
Construction of the DM genetic map and anchoring of the genome
To anchor and fully orientate physical contigs along the chromosome, a genetic map was developed de novo using sequence-tagged-site (STS) markers comprising simple sequence repeats (SSR), SNPs, and diversity array technology (DArT). SSR and SNP markers were designed directly from assembled sequence scaffolds, whereas polymorphic DArT marker sequences were searched against the scaffolds for high-quality unique matches. A total of 4,836 STS markers including 2,174 DArTs, 2,304 SNPs and 358 SSRs were analysed on 180 progeny clones from a backcross population ((DM × DI) × DI) developed at CIP between DM and DI (CIP no. 703825), a heterozygous diploid S. tuberosum group Stenotomum (formerly S. stenotomum ssp. goniocalyx) landrace clone. The data from 2,603 polymorphic STS markers comprising 1,881 DArTs, 393 SNPs and 329 SSR alleles were analysed using JoinMap 4 (ref. 38) and yielded the expected 12 potato linkage groups. Supplementary Fig. 3 represents the mapping and anchoring of the potato genome, using chromosome 7 as an example.
Anchoring the DM genome was accomplished using direct and indirect approaches. The direct approach employed the ((DM × DI) × DI) linkage map whereby 2,037 of the 2,603 STS markers comprised of 1,402 DArTs, 376 SNPs and 259 SSRs could be uniquely anchored on the DM superscaffolds. This approach anchored ∼52% (394 Mb) of the assembly arranged into 334 superscaffolds (Supplementary Table 27 and Supplementary Fig. 3).
RH is the male parent of the mapping population of the ultra-high-density (UHD) linkage map28 used for construction and genetic anchoring of the physical map using the RHPOTKEY BAC library39. The indirect mapping approach exploited in silico anchoring using the RH genetic and physical map28,40, as well as tomato genetic map data from SGN (http://solgenomics.net/). Amplified fragment length polymorphism markers from the RH genetic map were linked to DM sequence scaffolds via BLAST alignment36 of whole-genome-profiling sequence tags41 obtained from anchored seed BACs in the RH physical map, or by direct alignment of fully sequenced RH seed BACs to the DM sequence. The combined marker alignments were processed into robust anchor points. The tomato sequence markers from the genetic maps were aligned to the DM assembly using SSAHA2 (ref. 42). Positions of ambiguously anchored superscaffolds were manually checked and corrected. This approach anchored an additional ∼32% of the assembly (229 Mb). In 294 cases, the two independent approaches provided direct support for each other, anchoring the same scaffold to the same position on the two maps.
Overall, the two strategies anchored 649 superscaffolds to approximate positions on the genetic map of potato covering a length of 623 Mb. The 623 Mb (∼86%) anchored genome includes ∼90% of the 39,031 predicted genes. Of the unanchored superscaffolds, 84 were found in the N90 (622 scaffolds greater than 0.25 Mb), constituting 17 Mb of the overall assembly or 2% of the assembled genome. The longest anchored superscaffold is 7 Mb (from chromosome 1) and the longest unanchored superscaffold is 2.5 Mb.
Identification of repetitive sequences
Transposable elements (TEs) in the potato genome assembly were identified at the DNA and protein level. RepeatMasker29 was applied using Repbase43 for TE identification at the DNA level. At the protein level, RepeatProteinMask29,44 was used in a WuBlastX36 search against the TE protein database to further identify TEs. Overlapping TEs belonging to the same repeat class were collated, and sequences were removed if they overlapped >80% and belonged to different repeat classes.
Gene prediction
To predict genes, we performed ab initio predictions on the repeat-masked genome and then integrated the results with spliced alignments of proteins and transcripts to genome sequences using GLEAN30. The potato genome was masked by identified repeat sequences longer than 500 bp, except for miniature inverted repeat transposable elements which are usually found near genes or inside introns45. The software Augustus46 and Genscan47 was used for ab initio predictions with parameters trained for A. thaliana. For similarity-based gene prediction, we aligned the protein sequences of four sequenced plants (A. thaliana, Carica papaya, V. vinifera and Oryza sativa) onto the potato genome using TBLASTN with an E-value cut-off of 1 × 10−5, and then similar genome sequences were aligned against the matching proteins using Genewise48 for accurately spliced alignments. In EST-based predictions, EST sequences of 11 Solanum species were aligned against the potato genome using BLAT (identity ≥0.95, coverage ≥0.90) to generate spliced alignments. All these resources and prediction approaches were combined by GLEAN30 to build the consensus gene set. To finalize the gene set, we aligned the RNA-Seq from 32 libraries, of which eight were sequenced with both single- and paired-end reads, to the genome using Tophat31 and the alignments were then used as input for Cufflinks32 using the default parameters. Gene, transcript and peptide sets were filtered to remove small genes, genes modelled across sequencing gaps, TE-encoding genes, and other incorrect annotations. The final gene set contains 39,031 genes with 56,218 protein-coding transcripts, of which 52,925 nonidentical proteins were retained for analysis.
Transcriptome sequencing
RNA was isolated from many tissues of DM and RH that represent developmental, abiotic stress and biotic stress conditions (Supplementary Table 4 and Supplementary Text). cDNA libraries were constructed (Illumina) and sequenced on an Illumina GA2 in the single- and/or paired-end mode. To represent the expression of each gene, we selected a representative transcript from each gene model by selecting the longest CDS from each gene. The aligned read data were generated by Tophat31 and the selected transcripts used as input into Cufflinks32, a short-read transcript assembler that calculates the fragments per kb per million mapped reads (FPKM) as expression values for each transcript. Cufflinks was run with default settings, with a maximum intron length of 15,000. FPKM values were reported and tabulated for each transcript (Supplementary Table 19).
Comparative genome analyses
Paralogous and orthologous clusters were identified using OrthoMCL49 using the predicted proteomes of 11 plant species (Supplementary Table 28). After removing 1,602 TE-related genes that were not filtered in earlier annotation steps, asterid-specific and potato-lineage-specific genes were identified using the initial OrthoMCL clustering followed by BLAST searches (E-value cut-off of 1 × 10−5) against assemblies of ESTs available from the PlantGDB project (http://plantgdb.org; 153 nonasterid species and 57 asterid species; Supplementary Fig. 5 and Supplementary Table 29). Analysis of protein domains was performed using the Pfam hmm models identified by InterProScan searches against InterPro (http://www.ebi.ac.uk/interpro). We compared the Pfam domains of the asterid-specific and potato-lineage-specific sets with those that are shared with at least one other nonasterid genome or transcriptome. A Fisher’s exact test was used to detect significant differences in Pfam representation between protein sets.
After removing the self and multiple matches, the syntenic blocks (≥5 genes per block) were identified using MCscan9 and i-adhore 3.0 (ref. 50) based on the aligned protein gene pairs (Supplementary Table 8). For the self-aligned results, each aligned block represents the paralogous segments pair that arose from the genome duplication whereas, for the inter-species alignment results, each aligned block represents the orthologous pair derived from the shared ancestor. We calculated the 4DTv (fourfold degenerate synonymous sites of the third codons) for each gene pair from the aligned block and give a distribution for the 4DTv value to estimate the speciation or WGD event that occurred in evolutionary history.
Identification of disease resistance genes
Predicted open reading frames (ORFs) from the annotation of S. tuberosum group Phureja assembly V3 were screened using HMMER V.3 (http://hmmer.janelia.org/software) against the raw hidden Markov model (HMM) corresponding to the Pfam NBS (NB-ARC) family (PF00931). The HMM was downloaded from the Pfam home page (http://pfam.sanger.ac.uk/). The analysis using the raw HMM of the NBS domain resulted in 351 candidates. From these, a high quality protein set (<1 × 10−60) was aligned and used to construct a potato-specific NBS HMM using the module ‘hmmbuild’. Using this new potato-specific model, we identified 500 NBS-candidate proteins that were individually analysed. To detect TIR and LRR domains, Pfam HMM searches were used. The raw TIR HMM (PF01582) and LRR 1 HMM (PF00560) were downloaded and compared against the two sets of NBS-encoding amino acid sequences using HMMER V3. Both TIR and LRR domains were validated using NCBI conserved domains and multiple expectation maximization for motif elicitation (MEME)51. In the case of LRRs, MEME was also useful to detect the number of repeats of this particular domain in the protein. As previously reported52, Pfam analysis could not identify the CC motif in the N-terminal region. CC domains were thus analysed using the MARCOIL53 program with a threshold probability of 90 (ref. 52) and double-checked using paircoil2 (ref. 54) with a P-score cut-off of 0.025 (ref. 55). Selected genes (±1.5 kb) were searched using BLASTX against a reference R-gene set56 to find a well-characterized homologue. The reference set was used to select and annotate as pseudogenes those peptides that had large deletions, insertions, frameshift mutations, or premature stop codons. DNA and protein comparisons were used.
Haplotype diversity analysis
RH reads generated by the Illumina GA2 were mapped onto the DM genome assembly using SOAP2.20 (ref. 34) allowing at most four mismatches and SNPs were called using SOAPsnp. Q20 was used to filter the SNPs owing to sequencing errors. To exclude SNP calling errors caused by incorrect alignments, we excluded adjacent SNPs separated by <5 bp. SOAPindel was used to detect the indels between DM and RH. Only indels supported by more than three uniquely mapped reads were retained. Owing to the heterozygosity of RH, the SNPs and indels were classified into heterozygous and homozygous SNPs or indels.
On the basis of the annotated genes in the DM genome assembly, we extracted the SNPs located at coding regions and stop codons. If a homozygous SNP in RH within a coding region induced a premature stop codon, we defined the gene harbouring this SNP as a homozygous premature stop gene in RH. If the SNP inducing a premature stop codon was heterozygous, the gene harbouring this SNP was considered a heterozygous premature stop codon gene in RH. In addition, both categories can be further divided into premature stop codons shared with DM or not shared with DM. As a result, the numbers of premature stop codons are 606 homozygous PS genes in RH, 1,760 heterozygous PS genes in RH but not shared with DM, 288 PS in DM only, and 652 heterozygous premature stop codons in RH and shared by DM.
To identify genes with frameshift mutations in RH, we identified all the genes containing indels of which the length could not be divided by 3. We found 80 genes with frameshift mutations, of which 31 were heterozygous and 49 were homozygous.
To identify DM-specific genes, we mapped all the RH Illumina GA2 reads to the DM genome assembly. If the gene was not mapped to any RH read, it was considered a DM-specific gene. We identified 35 DM-specific genes, 11 of which are supported by similarity to entries in the KEGG database57. To identify RH-specific genes, we assembled the RH Illumina GA2 reads that did not map to the DM genome into RH-specific scaffolds. Then, these scaffolds were annotated using the same strategy as for DM. To exclude contamination, we aligned the CDS sequences against the protein set of bacteria with the E-value cut-off of 1 × 10−5 using Blastx. CDS sequences with >90% identity and >90% coverage were considered contaminants and were excluded. In addition, all DM RNA-seq reads were mapped onto the CDS sequences, and CDS sequences with homologous reads were excluded because these genes may be due to incorrect assembly. In total, we predicted 246 RH specific genes, 34 of which are supported by Gene Ontology annotation17.
Accession codes
Primary accessions
ArrayExpress
GenBank/EMBL/DDBJ
Sequence Read Archive
Data deposits
BAC and fosmid end sequences have been deposited in the GSS division of GenBank (BAC: GS025503–GS026177, GS262924–GS365942, GS504213–GS557003; fosmid: FI900795–FI901529, FI907952–FI927051, GS557234–GS594339, GS635316–GS765761). DM Illumina GA2 WGS and Roche 454 sequences have been deposited in the NCBI Sequence Read Archive (SRA029323) and EBI Short Read Archive (ERP000411) respectively. RH NGS sequences have been deposited in the EBI Short Read Archive (ERP000627). DM and RH RNA-Seq reads have been deposited in the NCBI Sequence Read Archive (SRA030516; study SRP005965) and the European Nucleotide Database ArrayExpress Database (E-MTAB-552; study ERP000527), respectively. The DM Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AEWC00000000. The version described in this paper is the First Version, AEWC01000000. Genome sequence and annotation can be obtained and viewed at http://potatogenome.net.
References
Hijmans, R. J. Global distribution of the potato crop. Am. J. Potato Res. 78, 403–412 (2001)
Burlingame, B., Mouillé, B. & Charrondiére, R. Nutrients, bioactive non-nutrients and anti-nutrients in potatoes. J. Food Compost. Anal. 22, 494–502 (2009)
Paz, M. M. & Veilleux, R. E. Influence of culture medium and in vitro conditions on shoot regeneration in Solanum phureja monoploids and fertility of regenerated doubled monoploids. Plant Breed. 118, 53–57 (1999)
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010)
Arumuganathan, K. & Earle, E. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218 (1991)
Tang, X. et al. Assignment of genetic linkage maps to diploid Solanum tuberosum pachytene chromosomes by BAC-FISH technology. Chromosome Res. 17, 899–915 (2009)
Peters, S. A. et al. Solanum lycopersicum cv. Heinz 1706 chromosome 6: distribution and abundance of genes and retrotransposable elements. Plant J. 58, 857–869 (2009)
Albach, D. C., Soltis, P. S. & Soltis, D. E. Patterns of embryological and biochemical evolution in the Asterids. Syst. Bot. 26, 242–262 (2001)
Tang, H. et al. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 18, 1944–1954 (2008)
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007)
Fawcett, J. A., Maere, S. & Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous–Tertiary extinction event. Proc. Natl Acad. Sci. USA 106, 5737–5742 (2009)
Lai, J. et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nature Genet. 42, 1027–1030 (2010)
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nature Genet. 25, 25–29 (2000)
Gore, M. A. et al. A first-generation haplotype map of maize. Science 326, 1115–1117 (2009)
Prat, S. et al. Gene expression during tuber development in potato plants. FEBS Lett. 268, 334–338 (1990)
Glaczinski, H., Heibges, A., Salamini, R. & Gebhardt, C. Members of the Kunitz-type protease inhibitor gene family of potato inhibit soluble tuber invertase in vitro . Potato Res. 45, 163–176 (2002)
Shannon, J. C., Pien, F. M. & Liu, K. C. Nucleotides and nucleotide sugars in developing maize endosperms: synthesis of ADP-glucose in brittle-1 . Plant Physiol. 110, 835–843 (1996)
Tauberger, E. et al. Antisense inhibition of plastidial phosphoglucomutase provides compelling evidence that potato tuber amyloplasts import carbon from the cytosol in the form of glucose-6-phosphate. Plant J. 23, 43–53 (2000)
Fettke, J. et al. Glucose 1-phosphate is efficiently taken up by potato (Solanum tuberosum) tuber parenchyma cells and converted to reserve starch granules. New Phytol. 185, 663–675 (2010)
Sonnewald, U. Control of potato tuber sprouting. Trends Plant Sci. 6, 333–335 (2001)
Yoo, S. K. et al. CONSTANS activates SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 through FLOWERING LOCUS T to promote flowering in Arabidopsis . Plant Physiol. 139, 770–778 (2005)
Kohler, A. et al. Genome-wide identi?cation of NBS resistance genes in Populus trichocarpa . Plant Mol. Biol. 66, 619–636 (2008)
Ballvora, A. et al. Comparative sequence analysis of Solanum and Arabidopsis in a hot spot for pathogen resistance on potato chromosome V reveals a patchwork of conserved and rapidly evolving genome segments. BMC Genomics 8, 112 (2007)
Kuang, H. et al. The R1 resistance gene cluster contains three groups of independently evolving, type I R1 homologues and shows substantial structural variation among haplotypes of Solanum demissum . Plant J. 44, 37–51 (2005)
Kuang, H., Woo, S. S., Meyers, B. C., Nevo, E. & Michelmore, R. W. Multiple genetic processes result in heterogeneous rates of evolution within the major cluster disease resistance genes in lettuce. Plant Cell 16, 2870–2894 (2004)
Haas, B. J. et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans . Nature 461, 393–398 (2009)
Haynes, F. L. in Prospects for the Potato in the Developing World: an International Symposium on Key Problems and Potentials for Greater Use of the Potato in the Developing World (ed. French, E. R. ) 100–110 (International Potato Center (CIP), 1972)
van Os, H. et al. Construction of a 10,000-marker ultradense genetic recombination map of potato: providing a framework for accelerated gene isolation and a genomewide physical map. Genetics 173, 1075–1087 (2006)
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14 (2004)
Elsik, C. G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009)
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 28, 511–515 (2010)
Li, L., Stoeckert, C. J., Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003)
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009)
Chaisson, M., Pevzner, P. & Tang, H. Fragment assembly with short reads. Bioinformatics 20, 2067–2074 (2004)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000)
Van Ooijen, J. W. in JoinMap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations (ed. Kyazma, B. V. ) (Wageningen, 2006)
Borm, T. J. Construction and Use of a Physical Map of Potato. PhD thesis, Wageningen Univ. (2008)
Visser, R. G. F. et al. Sequencing the potato genome: outline and first results to come from the elucidation of the sequence of the world's third most important crop. Am. J. Potato Res. 86, 417–429 (2009)
Van der Vossen, E. et al. in Whole Genome Profiling of the Diploid Potato Clone RH89-039-16 (Plant & Animal Genomes XVIII Conference, 2010)
Ning, Z., Cox, A. J. & Mullikin, J. C. SSAHA: a fast search method for large DNA databases. Genome Res. 11, 1725–1729 (2001)
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005)
Jiang, Z., Hubley, R., Smit, A. & Eichler, E. E. DupMasker: a tool for annotating primate segmental duplications. Genome Res. 18, 1362–1368 (2008)
Kuang, H. et al. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res. 19, 42–56 (2009)
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004)
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997)
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004)
Chen, F., Mackey, A. J., Vermunt, J. K. & Roos, D. S. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS ONE 2, e383 (2007)
Simillion, C., Janssens, K., Sterck, L. & Van de Peer, Y. i-ADHoRe 2.0: an improved tool to detect degenerated genomic homology using genomic profiles. Bioinformatics 24, 127–128 (2008)
Bailey, T. L. & Elkan, C. The value of prior knowledge in discovering motifs with MEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 3, 21–29 (1995)
Mun, J. H., Yu, H. J., Park, S. & Park, B. S. Genome-wide identification of NBS-encoding resistance genes in Brassica rapa . Mol. Genet. Genomics 282, 617–631 (2009)
Delorenzi, M. & Speed, T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625 (2002)
McDonnell, A. V., Jiang, T., Keating, A. E. & Berger, B. Paircoil2: improved predictions of coiled coils from sequence. Bioinformatics 22, 356–358 (2006)
Porter, B. W. et al. Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Mol. Genet. Genomics 281, 609–626 (2009)
Sanseverino, W. et al. PRGdb: a bioinformatics platform for plant resistance gene analysis. Nucleic Acids Res. 38, D814–D821 (2010)
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 (2004)
Acknowledgements
We acknowledge the assistance of W. Amoros, B. Babinska, R. V. Baslerov, B. K. Bumazhkin, M. F. Carboni, T. Conner, J. Coombs, L. Daddiego, J. M. D’Ambrosio, G. Diretto, S. B. Divito, D. Douches, M. Filipiak, G. Gianese, R. Hutten, E. Jacobsen, E. Kalinska, S. Kamoun, D. Kells, H. Kossowska, L. Lopez, M. Magallanes-Lundback, T. Miranda, P. S. Naik, A. N. Panteleeva, D. Pattanayak, E. O. Patutina, M. Portantier, S. Rawat, R. Simon, B. P. Singh, B. Singh, W. Stiekema, M. V. Sukhacheva and C. Town in providing plant material, generating data, annotation, analyses, and discussions. We are indebted to additional faculty and staff of the BGI-Shenzhen, J Craig Venter Institute, and MSU Research Technology Support Facility who contributed to this project. Background and preliminary data were provided by the Centre for BioSystems Genomics (CBSG), EU-project (APOPHYS EU-QLRT-2001-01849) and US Department of Agriculture National Institute of Food and Agriculture SolCAP project (2008-55300-04757 and 2009-85606-05673). We acknowledge the funding made available by the “863” National High Tech Research Development Program in China (2006AA100107), “973” National Key Basic Research Program in China (2006CB101904, 2007CB815703, 2007CB815705, 2009CB119000), Board of Wageningen University and Research Centre, CAPES - Brazilian Ministry of Education, Chinese Academy of Agricultural Sciences (seed grant to S.H.), Chinese Ministry of Agriculture (The “948” Program), Chinese Ministry of Finance (1251610601001), Chinese Ministry of Science and Technology (2007DFB30080), China Postdoctoral Science Foundation (20070420446 to Z.Z.), CONICET (Argentina), DAFF Research Stimulus Fund (07-567), CONICYT-Chile (PBCT-PSD-03), Danish Council for Strategic Research Programme Commission on Health, Food and Welfare (2101-07-0116), Danish Council for Strategic Research Programme Commission on Strategic Growth Technologies (Grant 2106-07-0021), FINCyT ((099-FINCyT-EQUIP-2009)/(076-FINCyT-PIN-2008), Préstamo BID no. 1663/OC-PE, FONDAP and BASAL-CMM), Fund for Economic Structural Support (FES), HarvestPlus Challenge Program, Indian Council of Agricultural Research, INIA-Ministry of Agriculture of Chile, Instituto Nacional de Innovacion Agraria-Ministry of Agriculture of Peru, Instituto Nacional de Tecnología Agropecuaria (INTA), Italian Ministry of Research (Special Fund for Basic Research), International Potato Center (CIP-CGIAR core funds), LBMG of Center for Genome Regulation and Center for Mathematical Modeling, Universidad de Chile (UMI 2807 CNRS), Ministry of Education and Science of Russia (contract 02.552.11.7073), National Nature Science Foundation of China (30671319, 30725008, 30890032, 30971995), Natural Science Foundation of Shandong Province in China (Y2006D21), Netherlands Technology Foundation (STW), Netherlands Genomics Initiative (NGI), Netherlands Ministries of Economic Affairs (EZ) and Agriculture (LNV), New Zealand Institute for Crop & Food Research Ltd Strategic Science Initiative, Perez Guerrero Fund, Peruvian Ministry of Agriculture-Technical Secretariat of coordination with the CGIAR, Peruvian National Council of Science and Technology (CONCYTEC), Polish Ministry of Science and Higher Education (47/PGS/2006/01), Programa Cooperativo para el Desarrollo Tecnológico Agroalimentario y Agroindustrial del Cono Sur (PROCISUR), Project Programa Bicentenario de Ciencia y Tecnología - Conicyt, PBCT - Conicyt PSD-03, Russian Foundation for Basic Research (09-04-12275), Secretaría de Ciencia y Tecnología (SECyT) actual Ministerio de Ciencia y Tecnología (MINCyT), Argentina, Shenzhen Municipal Government of China (CXB200903110066A, ZYC200903240077A, ZYC200903240076A), Solexa project (272-07-0196), Special Multilateral Fund of the Inter-American Council for Integral Development (FEMCIDI), Teagasc, Teagasc Walsh Fellowship Scheme, The New Zealand Institute for Plant & Food Research Ltd Capability Fund, UK Potato Genome Sequencing grant (Scottish Government Rural and Environment Research and Analysis Directorate (RERAD), Department for Environment, Food and Rural Affairs (DEFRA), Agriculture and Horticulture Development Board - Potato Council), UK Biotechnology and Biological Sciences Research Council (Grant BB/F012640), US National Science Foundation Plant Genome Research Program (DBI-0604907 /DBI-0834044), Virginia Agricultural Experiment Station USDA Hatch Funds (135853), and Wellcome Trust Strategic award (WT 083481).
Author information
Authors and Affiliations
Consortia
Contributions
A.D.G., A.G., A.N.M., A.V.B., A.V.M., B.B.K., B.K., B.R.W., B.S., B.t.L.H., B.V., B.X., B.Z., C.L., C.R.B., C.W.B.B., D.F.M., D. Martinez, D. Milbourne, D.M.A.M., D.M.B., D.D., D.M., E.D., F.G., G.A.M., G.A.T., G.D.l.C., G.G., G.J. Bishop, G.J. Bryan, G.L., G.O., G.P., G.Z., H.K., H.L., H.v.E., I.N., J.d.B., J.G., J.H., J.J., J.M.E.J., J.W., J.X., K.L.N., K.O’B., L.D., L.E.B., M.B., M.D., M.d.R.H., M.F., M. Geoffroy, M.Ghislain, M.I., M.P., M.S., M.T., N.M., N.V.R., O.P., P.F., P.N., P.S., Q.H., R.C.H.J.v.H., R.E.V., R.G., R.G.F.V., R. Lozano, R. Li, S.C., S.E.F., S.H., S.J.T., S.K.C., S.K.S., S.L., S.P., S.Y., T.B., T.V.K., V.U.P., X. Xiong, X. Xu, Y.D., Y.H., Y.L., Y.Y., Y.Z. and Z.Z. were involved in experimental design, data generation and/or data analysis. A.N.M., B.K., C.R.B., C.W.B.B., D.D., D. Milbourne, D.M.A.M., D.M.B., E.D., G.G., G.J. Bishop, G.J. Bryan, G.O., H.L., I.N., J.d.B., J.J., J.M.E.J., K.L.N., M.B., M.F., M.D., M.S., O.P., R.C.H.J.v.H., R.E.V., R.G.F.V., R. Lozano, R.W., S.E.F., S.H., S.J.T., S.K.S., T.B. and X. Xu wrote the manuscript. B.S., C.R.B., C.W.B.B., D.F.M., D. Milbourne, D.M.A.M., D.Q., G.G., G.J. Bishop, G.J. Bryan, G.O., G.P., J.M.E.J., J.W., K.G.S., R.G.F.V., R. Li, R.W., S.E.F., S.H., S.K.C., S.Y., W.Z. and Y.D. supervised data generation/analysis and managed the project. C.R.B., C.W.B.B., G.J. Bryan, G.O., J.M.E.J. and S.H. are members of The Potato Genome Sequencing Consortium Steering Committee.
Corresponding authors
Ethics declarations
Competing interests
The author declare no competing financial interests.
Supplementary information
Supplementary Information
The file contains Supplementary Text, Supplementary Figures 1-15 with legends and Supplementary Tables 1-18 and 20-34. This is a 500-page document and may take a short while to download. This file was replaced on 9 May 2014 as Supplementary Figure 12 was corrupted. (PDF 14371 kb)
Supplementary Tables
The zipped file contains two Supplementary Tables: Supplementary Table 19A displaying FPKM levels in DM tissues. Supplementary Table 19B displaying FPKM levels in RH tissues. (ZIP 12701 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
The Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011). https://doi.org/10.1038/nature10158
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10158
This article is cited by
-
Whole-genome sequencing of tetraploid potato varieties reveals different strategies for drought tolerance
Scientific Reports (2024)
-
Transcriptome Analysis Reveals Molecular Signatures Associated with Apical Rooted Cutting (ARC) Technology in Seed Potato Production
Journal of Plant Growth Regulation (2024)
-
Potato and sweetpotato breeding at the International Potato Center: approaches, outcomes and the way forward
Theoretical and Applied Genetics (2024)
-
Diploid Interspecific Recombinant Inbred Lines for Genetic Mapping in Potato
American Journal of Potato Research (2024)
-
CRISPR/Cas system for the traits enhancement in potato (Solanum tuberosum L.): present status and future prospectives
Journal of Plant Biochemistry and Biotechnology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
