
Abstract
Sponges are an ancient group of animals that diverged from other metazoans over 600 million years ago. Here we present the draft genome sequence of Amphimedon queenslandica, a demosponge from the Great Barrier Reef, and show that it is remarkably similar to other animal genomes in content, structure and organization. Comparative analysis enabled by the sequencing of the sponge genome reveals genomic events linked to the origin and early evolution of animals, including the appearance, expansion and diversification of pan-metazoan transcription factor, signalling pathway and structural genes. This diverse ‘toolkit’ of genes correlates with critical aspects of all metazoan body plans, and comprises cell cycle control and growth, development, somatic- and germ-cell specification, cell adhesion, innate immunity and allorecognition. Notably, many of the genes associated with the emergence of animals are also implicated in cancer, which arises from defects in basic processes associated with metazoan multicellularity.
Similar content being viewed by others

Main
The emergence of multicellular animals from single-celled ancestors over 600 million years ago required the evolution of mechanisms for coordinating cell division, growth, specialization, adhesion and death. Dysfunction of these mechanisms drives diseases such as cancers, in which social controls on multicellularity fail, and autoimmune disorders, in which distinctions between self and non-self are disrupted. The hallmarks of metazoan multicellularity are therefore intimately related to those of cancer1 and immunity2.
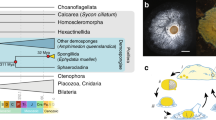
Sponges have a critical role in the search for the origins of metazoan multicellular processes3, as they are generally recognized as the oldest surviving metazoan phyletic lineage. Although the kinship of sponges to other animals was recognized by the nineteenth century4, the absence of a gut and nervous system had relegated sponges to the ‘Parazoa’5, a grade below the ‘Eumetazoa’ or ‘true animals’ (that is, cnidarians, ctenophores and bilaterians)6. Nevertheless, sponges share key adhesion and signalling genes7,8,9,10,11 with eumetazoans, as well as other genes important in body plan patterning such as developmental transcription factors12,13,14,15; sponge embryos and larvae (Fig. 1) are readily comparable to those of other animals12,16. Sponges are diverse and their phylogeny is poorly resolved17,18,19, allowing for the possibility that sponges are paraphyletic20, which implies that other animals evolved from sponge-like ancestors.

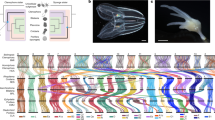
a, Amphimedon queenslandica adult. Scale bar, 5 cm. b, Embryos in a brood chamber. Scale bar, 1 mm. c, Larva. Scale bar, 100 μm. d, Animal phylogeny based on whole-genome data. This unrooted tree is inferred from 229 concatenated nuclear protein-coding genes with 44,616 amino acids using Bayesian inference. All clades are supported with a posterior probability of 1. Coloured boxes mark the nodes for which origins of genes are inferred in Figs 3 and 4. The same topology is supported by the nuclear gene data sets generated by alternative methods as well as by other inference methods (Supplementary Note 7). The metazoan stem leading to the animal radiation is shown in bold. Contrary to the current consensus of eukaryotic relationships, Amoebozoa are not a sister-group to Opisthokonta in this tree (Supplementary Note 7).
Here we report on the genome of Amphimedon queenslandica, a haplosclerid demosponge, the adult organization and lifestyle of which is typical for sponges, feeding on microbes and particulate organic matter filtered by flagellated collar cells that resemble choanoflagellates. Although the diversity of sponges and their uncertain phylogeny make it doubtful that any single species can reveal the intricacies of early animal evolution, comparison of the A. queenslandica draft genome with sequences from other species can provide a conservative estimate of the genome of the common ancestor of all animals and the timing and nature of the genomic events that led to the origin and early evolution of animal lineages.
The A. queenslandica genome harbours an extensive repertoire of developmental signalling and transcription factor genes, indicating that the metazoan ancestor had a developmental ‘toolkit’ similar to that of modern complex bilaterians. The origins of many of these and other genes specific to animal processes such as cell adhesion, and social control of cell proliferation, death and differentiation can be traced to genomic events (gene birth, subfamily expansions, intron gain/loss, and so on) that occurred in the lineage that led to the metazoan ancestor, after animals diverged from their unicellular ‘cousins’. In addition to possessing a wide range of metazoan-specific genes, the Amphimedon draft genome is missing some genes that are conserved in other animals, indicative of gene origin and expansion in eumetazoans after their divergence from the demosponge lineage and/or gene loss in Amphimedon.
Genome sequencing and annotation
Amphimedon queenslandica is a hermaphroditic spermcast spawner, and cannot be readily inbred in the laboratory (Fig. 1a–c and Supplementary Note 1)21. Adult sponges also harbour many commensal microbes. To minimize allelic variation and microbial contamination we sequenced genomic DNA from multiple embryos and larvae from a single mother. This DNA contains four dominant parental haplotypes (∼3% polymorphism), although a single brood may have multiple fathers (Supplementary Notes 2.1 and 3). We used ∼9-fold whole-genome Sanger shotgun coverage to produce a ∼167-megabase-pair assembly that typically represents each locus once rather than splitting alleles (Supplementary Notes 2 and 3) and captures ∼97% of the protein-coding gene content (Supplementary Note 2.5). We also recovered an alpha-proteobacterial genome that is probably a vertically transmitted commensal microbe of Amphimedon embryos (Supplementary Note 2.7).
The assembled A. queenslandica genome encodes ∼30,000 predicted protein-coding loci (Supplementary Note 4). This is an overestimate of the true gene number due to overprediction, unrecognized transposable elements and gene fragmentation at contig or scaffold boundaries. Nevertheless, 18,693 (63%) have identifiable homologues in other organisms in the Swiss-Prot database; there are no doubt novel or rapidly evolving sponge genes unknown in other species. CpG dinucleotides are depleted, and TpG and CpA dinucleotides augmented, relative to overall G+C composition, which is indicative of germline cytosine methylation in the Amphimedon genome. This is consistent with the presence of a DNMT3-related putative de novo methytransferase as well as proteins with predicted methyl CpG binding domains.
Analysis of the Amphimedon gene set reveals marked conservation of gene structure (intron phase and position) and genome organization (synteny) relative to other animals (Supplementary Notes 5 and 6). In Amphimedon, intragenic position and phase are retained for 84% of the introns inferred for the metazoan ancestor, comparable to the 76% and 88% retention in human and sea anemone, respectively22,23. The organization of genes shows conserved synteny (that is, conserved linkage without necessarily requiring colinearity) relative to other animals. In particular, 83 of the 153 longest Amphimedon scaffolds (those that contain genes from more than ten distinct metazoan gene families, sufficient for synteny to be assessed) show segments of conserved synteny with other animals (Supplementary Note 6). This indicates that portions of the 15 ancestral linkage groups inferred for the cnidarian–bilaterian ancestor22,24 were already in place in the demosponge–eumetazoan ancestor. No such conserved synteny was detected between animals and the choanoflagellate Monosiga brevicollis.
Animal relationships
We addressed the controversial phyletic branching of early animal lineages by comparing sets of orthologous genes in A. queenslandica and a diverse sampling of 18 complete genomes (Supplementary Note 7). Our analyses support the grouping of placozoans, cnidarians and bilaterians into a eumetazoan clade, with demosponges as an earlier-branching lineage25, and reject the diploblast–triploblast phylogeny17 in favour of a more conventional ‘sponges first’ tree19,20 (Fig. 1d). In our discussion below we therefore refer to descendants of the placozoan–cnidarian–bilaterian last common ancestor as Eumetazoa, and reserve ‘Eumetazoa sensu stricto’ for the more limited clade defined by descendants of the cnidarian–bilaterian ancestor.
Our analysis emphasizes the quantitative divergence between metazoans and their closest living unicellular relatives. For example, 28% of the amino acid substitutions between humans and their last common ancestor with choanoflagellates occurred on the metazoan stem lineage (bold line in Fig. 1d), before the divergence of sponges from other animals. This pre-metazoan period can be crudely estimated to be ∼150–200 million years (Supplementary Note 7.6).
The zootype and origin of metazoan genes
With multiple animal genomes now in hand, we can extend the ‘zootype’ concept26 to include other shared derived genomic characteristics of animals. Out of 4,670 pan-metazoan gene families defined by clustering sponge and eumetazoan peptides, 1,286 (27%) seem to be metazoan-specific (see Supplementary Note 9.2). Similarly, there are eumetazoan, eumetazoan sensu stricto and bilaterian genomic synapomorphies, as well as sponge-specific gene families (for example, kinases, see Supplementary Note 8). Owing to residual incompleteness of the sponge genome draft, and possible gene losses in the Amphimedon lineage, this analysis provides a conservative estimate.
Nearly three-quarters of the 1,286 animal-specific gene families arose by gene duplication on the metazoan stem (Supplementary Note 9). These include the early duplication of transcription factor families such as homeodomains and basic helix–loop–helix domains13,14,27. Additional gene duplication and divergence in eumetazoans further increased transcription factor gene family number, which in general are 2 to 34 times larger in eumetazoans than in Amphimedon. In contrast, substantial diversification of kinase gene families occurred before the divergence of the sponge and eumetazoan lineages (see below)28. We can assess the role of tandem duplication in the creation of these families by seeking evidence for linkages among anciently diverged paralogues (Supplementary Note 10). A significant fraction remain linked (up to 30%, as found in Trichoplax, P < 0.0001, with lower levels in other contemporary metazoan genomes), indicating that many gene family expansions originally occurred as tandem or proximal duplications, and that these genomically local duplications have remained linked over time. This is consistent with the overall preservation of relict linkages observed here and in other basal metazoan genomes22,24,25.
We find 235 animal-specific protein domains and 769 animal-specific domain combinations that evolved along the metazoan stem (Supplementary Note 9). Additionally, lineage-specific changes to these animal domain architectures occurred in early metazoan evolution16,29,30. For example, new combinations of domains in death-fold domain proteins and laminins possibly allow for the modification of protein interactions and pathways involved in programmed cell death and cell adhesion, respectively (Supplementary Note 9.3), and the co-option of sponge-, eumetazoan- or bilaterian-specific architectures into novel functions.
The 705 Amphimedon kinases represent the largest reported metazoan kinome, and include members of >70% of human kinase classes (compared with 59% in choanoflagellate, 83% in sea anemone, 70% in Caenorhabditis elegans and 77% in fruitfly; see Supplementary Note 8.7). Amphimedon has single copies of most metazoan kinase classes, but has several expansions of over 50 genes per class. The largest expansions are in the tyrosine kinase and tyrosine-kinase-like groups, and include over 150 likely receptor tyrosine kinases (RTKs). Unlike Monosiga, where RTKs could not be classified into metazoan families28, Amphimedon has kinase domains from six known animal families (epidermal growth factor receptor (EGFR), Met, discoidin domain receptor (DDR), regeneron orphan receptor (ROR), Eph and Sevenless). The EGFR and some Eph extracellular domain architectures are as in their eumetazoan counterparts, but many other RTKs have unique extracellular domains. For instance, DDRs have immunoglobulin repeats, and sushi domains are found in some members of the expanded Eph and Met families. This indicates that the activating ligands, presumably found largely in the external environment, may be distinct from those of eumetazoans.
Six hallmarks of animal multicellularity
The A. queenslandica genome allows us to assess systematically the origin of the six hallmarks of metazoan multicellularity: (1) regulated cell cycling and growth; (2) programmed cell death; (3) cell–cell and cell–matrix adhesion; (4) developmental signalling and gene regulation; (5) allorecognition and innate immunity; and (6) specialization of cell types. These cardinal features of metazoan multicellularity have their origins on the metazoan stem and often are the result of metazoan gene novelties combining with more ancient factors. A recurring theme is the overlap of these core ‘multicellularity’ genes with genes perturbed in cancer, a disease of aberrant multicellularity (see oncogenes and tumour suppressors in Figs 2 and 3).

Reference pathways from human and other vertebrates are depicted here for comparative purposes. Gene products are coloured by their node of origin as per Fig. 1. White text denotes known oncogenes or tumour suppressor genes. Genes with eumetazoan origin are found in either Nematostella or Trichoplax or both. a, Cell cycle; b, growth signalling; c, apoptosis. Dashed outlines indicate cases where proteins could not be affiliated to a subtype (see Supplementary Note 8.3).

Reference cellular structures from human and other vertebrates are depicted here for comparative purposes. Gene products are coloured by their node of origin as per Fig. 1. White text denotes known oncogenes or tumour suppressor genes. Genes with eumetazoan origin are found in either Nematostella or Trichoplax or both. a, Cell adhesion and polarity in epithelia. The asterisk indicates that collagen IV genes have not been found in the Amphimedon genome but have been reported as present in the homoscleromorph sponge Pseudocorticium jarrei48. The ancient origins of integrins reflect the recent findings of ref. 47. b, Synaptic and signalling elements in neurons.
Regulated cell cycling and growth
Although the core machinery of the animal cell cycle traces back to early eukaryotes (Fig. 2a and Supplementary Note 8.2), some critical metazoan regulatory mechanisms emerged more recently. For example, whereas the p53/p63/p73 tumour suppressor family is holozoan-specific31, the HIPK kinase that phosphorylates p53 in the presence of DNA breaks is metazoan-specific, and the MDM2 ubiquitin ligase that regulates p53 appears as a eumetazoan feature. Thus, the p53-mediated response to DNA damage may have emerged before the divergence of eumetazoans. The Myc oncogene illustrates how intramolecular regulation has also evolved. Although Amphimedon shares the four-amino-acid N-terminal DCMW motif present in other animal Myc proteins, this motif is missing in the Myc orthologue found in the unicellular Monosiga31. Because mutation of this motif disrupts Myc function in vertebrates, it may have an important role in all animals.
Tumour suppressors encoded by two classes of cyclin-dependent kinase (CDK) inhibitors mediate growth-factor-dependent regulation of the cell cycle. Although the INK4/CDKN2 class (p15/p16/p18/p19) regulates the eumetazoan-specific CDK4/6-cyclin D kinase and is chordate-specific, the Cip/Kip/CDKN1 class (p21/p27/p57) is more general, regulating many CDKs, and seems to have arisen on the eumetazoan stem. In bilaterians, Cip/Kip genes integrate external growth signals, and are regulated transcriptionally and post-transcriptionally by the major growth pathways (see below). The emergence of this class of CDK inhibitors on the eumetazoan stem suggests a central regulatory role even in early animals.
Although cell growth and cell division are tightly coupled in unicellular species, they can be separately regulated in multicellular organisms. In bilaterians, growth is regulated by six major signalling pathways (RTK signalling via Ras, insulin signalling via the phosphatidylinositol-3-OH kinase (PI(3)K) pathway, Rheb/Tor, cytokine-JAK/STAT, Warts/Hippo, and the Myc oncogene) that also modulate the cell cycle (Supplementary Note 8.2). Whereas the Rheb/Tor pathway dates back to early eukaryotes, the other pathways contain several genes that are holozoan and metazoan innovations. For example, the insulin receptor substrate and phosphotyrosine binding proteins GAB1/GAB2 emerged on the metazoan stem after the divergence of choanoflagellates, indicating that an insulin-signalling-like pathway may have been a key regulator of growth in early animals by tying into the ancient PDK1 and Akt kinases (Fig. 2b). However, because p21, p27 and MDM2 are all eumetazoan novelties, this pathway may not have acquired the ability to regulate cell proliferation until after the divergence of sponges from eumetazoans.
Programmed cell death
In contrast to the cell cycle machinery, most of the apoptotic circuitry is unique to animals, increasing in complexity along metazoan, eumetazoan and bilaterian stems (Fig. 2c and Supplementary Note 8.3). Both intrinsic and extrinsic programmed cell death pathways require caspases, a metazoan-specific family of cysteine aspartyl proteases. Amphimedon encodes initiator caspases with the characteristic caspase recruitment and death effector domains, as well as an expanded repertoire of effector capases.
The intrinsic pathway drives cell death by permeabilization of the outer mitochondrial membrane and is regulated by the Bcl-2 oncogene family of pro- and antiapoptotic factors. The pro-apoptotic protein Bak arose in the metazoan lineage, whereas Bax and Bok seem to be eumetazoan-specific. Bcl-2/Bcl-X are antiapoptotic and metazoan-specific. Mitochondrial permeabilization releases proteins of varying evolutionary origin, including the ancient apoptosis-inducing factor (AIF) that contributes to caspase-independent apoptosis, metazoan-specific apoptotic protease activating factor 1 (Apaf-1), and eumetazoan sensu stricto-specific caspase-activated DNase (CAD) and its regulator ICAD.
The extrinsic apoptotic pathway is activated by external signals through transmembrane tumour necrosis factor receptors (TNFRs) whose intracellular death domain interacts with downstream adaptors. Amphimedon encodes a nerve growth factor receptor (NGFR) p75-like protein, although it lacks the crucial death domain that is seen in Nematostella and bilaterians (see ref. 32); other death TNFRs (that is, Fas, DR4, DR5 and TNFR1) are vertebrate-specific32,33. Because the intrinsic cascade is composed of components that pre-date metazoans, it is likely to be the original mechanism for inducing apoptosis.
Cell–cell and cell–matrix adhesion
The diagnostic domains of two major cell–cell adhesion superfamilies, the cadherins and the immunoglobulins, are present in Monosiga within the extracellular region of putative transmembrane proteins31,34 (Supplementary Note 8.8). Amphimedon cadherins differ from those of Monosiga in having proteins with domain architectures diagnostic for the metazoan-specific classical cadherin and seven pass transmembrane cadherin subfamilies31,35. A considerable expansion of immunoglobulin-like domain-containing proteins occurred on the metazoan stem, with 218 predicted in Amphimedon versus 5 in Monosiga31. The combination of N-terminal immunoglobulin domains with C-terminal FN3 repeats is found only in metazoans.
Similarly, metazoan extracellular matrix (ECM) proteins use domains that evolved on the holozoan stem. For example, Monosiga encodes proteins with collagen triple helix repeats and other genes with fibrillar collagen C-terminal domains, but these domains only appear together in metazoans30,31. Thrombospondin domain architectures are found in Amphimedon; however, agrin, netrin and perlecan seem to be eumetazoan innovations. The extracellular matrix receptors, α and β integrin (Int), are present in Amphimedon and other metazoans, but absent from the Monosiga and the other non-metazoan eukaryotic genomes we considered (Fig. 3a; see note added in proof).
Developmental signalling and transcription
Components of the major metazoan developmental signalling pathways, as well as classes of developmental transcription factors, are mostly present in Amphimedon and absent from Monosiga and other non-metazoan genomes13,14,16,27,29, suggesting that ontogenetic development, including primary germ cell formation (Supplementary Note 8.4), originated on the metazoan stem3,11,12. Although Amphimedon possesses a characteristically metazoan repertoire of transcription factor families (Supplementary Note 8.6)13,14,27,31, in general these families are further expanded in eumetazoans13. Some differences between sponges and eumetazoans correlate with morphological complexity. For example, sponges do not seem to have a mesoderm and accordingly Amphimedon lacks transcription factors involved in mesoderm development (Fkh, Gsc, Twist, Snail). In contrast, sponges possess several transcription factors involved in determination or differentiation of muscles and nerves despite lacking a neuromuscular system (PaxB, Lhx genes, SoxB, Msx, Mef2, Irx and bHLH neurogenic factors)13,14,27. Amphimedon lacks Hox genes and some other transcription factor subfamilies that are involved in specifying and patterning bilaterian nervous systems and body plans13,14,27,36,37.
Signalling cascades, such as the Wnt, TGF-β, Notch and Hedgehog pathways, pattern embryos by specifying cellular identity and coordinating morphogenetic events. The ligands and receptors of all of these cascades are metazoan innovations at the cell surface (Supplementary Note 8.5), except the eumetazoan sensu stricto-specific Hedgehog ligand29. The transcription factors specific to these pathways are also metazoan-specific (Tcf/Lef, Smads, CSL, Gli), whereas the cytosolic signal transducers generally have more ancient origins. This pattern suggests that these pathways arose by the engagement of novel ligands and receptors with already active signalling mechanisms, enabling multicellular communication.
Amphimedon also has fewer ligands and receptors in each pathway compared to eumetazoans (three Wnt and two Fzd, eight TGF-β ligands and five TGF-β receptors, one Notch and five Deltas) (Supplementary Note 8.5), as observed for many transcription factor families. In contrast to transcription factors13,14,27, however, these proteins generally can not be assigned to eumetazoan subfamilies or are obvious recent sponge-specific duplications. This lack of phylogenetic resolution may reflect a period of rapid evolution and diversification of ligand/receptor molecules in sponge and eumetazoan lineages. Perhaps as a consequence, the inhibitors that interact with ligands and receptors to modulate pathway activity also appear to be lineage-specific. In particular, inhibitors described from bilaterians were not found in Amphimedon (for example, Chordin, Numb, I-Smads, Wif).
Allorecognition and innate immunity
The transition to multicellularity was accompanied by mechanisms to defend against invading pathogens and to prevent the fusion of genetically distinct conspecifics2. Although some metazoan immunity genes originated early in eukaryotic evolution, many are restricted to animals, as illustrated by the signalling cascades shared by the Toll-like receptor (TLR) and the interleukin1 receptor (IL-1R) (Supplementary Note 8.10). An ancestral form belonging to this receptor superfamily was probably present in the last common metazoan ancestor and independently diversified in poriferan and cnidarian lineages. Nuclear factor κB (NF-κB), Tollip and ECSIT genes are present in holozoans; however, most TLR/IL-1R pathway proteins are either composed of metazoan-specific domains (for example, Pellino) or architectures (for example, the death domain with TIR and protein kinase domains in MyD88 and IRAKs, respectively). Immune effector systems also consist largely of metazoan innovations, such as the macrophage-expressed gene 1 (MPEG1) that participates directly in pathogen elimination38. Likewise all animals share specific antiviral defence factors such as MDA5-like RNA helicases, and interferon regulatory factor-like proteins, although other systems (for example, RNAi) have more ancient origins39. A primordial complement pathway appears to have evolved exclusively on the eumetazoan sensu stricto stem and further diversified in bilaterians40.
Amphimedon and other demosponges encode unique extracellular Calx-β domain-containing proteoglycans called aggregation factors, which promote cell adhesion and may also be involved in allorecognition41. The presence of a cluster of aggregation-factor-related genes in the Amphimedon genome indicates that allorecognition could be under the control of a multigene family.
Specialized cell types
Polarized epithelia
Sponge cells adhere to form tissue-like layers, but a true epithelial cell layer, characterized by aligned cell polarity, belt-form junctions and underlying basal lamina, is thought to be a eumetazoan innovation. Amphimedon possesses all the main components of the Par, Crumbs and Discs Large (Dlg) complexes, a set of interacting proteins that are largely metazoan-specific and determine polarity in epithelial cells (Fig. 3a and Supplementary Note 8.8). The main proteins comprising bilaterian spot-form and zonula adherens junctions are also present in Amphimedon and appear to be metazoan-specific34,42. By contrast, septate junction and basal lamina proteins appear to be largely eumetazoan innovations (Fig. 3a); Amphimedon does possess several genes with laminin-like domain architectures (Supplementary Note 9.3).
Sensory systems and the neuron
Sponges can sense and respond to their environment, although nerve cells seem to be restricted to eumetazoans sensu stricto43,44. However, the expression of orthologues of post-synaptic structural and proneural regulatory proteins in Amphimedon larval globular cells suggests an evolutionary connection with an ancestral protoneuron36,42. Amphimedon possesses homologues of bilaterian proteins involved in nervous system development (for example, elav- and musashi-like RNA-binding proteins, neural transcription factors), pre- and post-synaptic organization (for example, Discs large)42, endogenous and exogenous signalling (for example, G-protein-coupled receptors (GPCRs)), and neuroendocrine secretion, although bilaterian peptide hormones are not detected (Supplementary Note 8.9). Some key synaptic genes are conspicuously missing from Amphimedon (Fig. 3b and Supplementary Note 8.9), including the ionotropic glutamate receptor family42, whereas neuronal-type metabotropic glutamate, dopamine and serotonin receptors are present. Amphimedon has a homologue of the ephrin receptor, an axon guidance protein, although the ephrin ligand and developmental genes involved in axon guidance (for example, slit, netrin, unc-5 and robo) are not present. Amphimedon also possesses over 200 GPCRs, which includes a large lineage-specific expansion of rhodopsin-related GPCRs (Rh-GPCRs) that are encoded largely by clusters of single exon genes as observed in other metazoans (Supplementary Note 8.9). From these observations we infer that the metazoan ancestor possessed a complex sensory system, and many of the molecular requirements for neural development and nerve cell function. This suggests that exaptation was critical for the genesis of the first nerve cell, with eumetazoan-specific gene innovations providing the regulatory and structural requirements to connect these protoneural components into a functional neuron (Fig. 3b).
Molecular correlates of morphological complexity
With a diverse sample of genomes in hand, we sought differences in gene repertoire that are associated with gross morphological complexity. Figure 4 shows molecular function categories that are significantly enriched (P < 1×10−10) in one or more metazoan complexity group, with the relative frequencies of genes with these functions in each species shown by colour code. Here we have defined broad groupings representing three grades of morphological complexity, guided by the number of described cell types45, including non-bilaterian (or ‘basal’) metazoans (Nematostella, Trichoplax, Amphimedon; ∼5–15 cell types), invertebrate bilaterians (Drosophila, C. elegans, sea urchin; ∼50–100 cell types), and vertebrates (∼225 cell types, represented by the human genome), with a selection of non-animals as an outgroup (Supplementary Note 11). Similarly, using a principal component analysis, we also identified suites of molecular functions that are associated with complexity (Supplementary Figure 11.2). The first component differentiates between metazoans and non-metazoans; the second component partly differentiates between metazoan complexity groups.

Molecular function categories that show significant enrichment (P<1×10−10) in Fisher’s exact tests were selected (Supplementary Note 11). Significance of enrichments (grey background) and depletions (white background) for the three ‘metazoan complexity groups’ (non-bilaterian (basal) metazoans; invertebrate bilaterians; vertebrates) are indicated in the columns to the left of the heatmap. The heatmap shows normalized gene counts of PANTHER molecular function categories for the species in the analysis. Aqu, Amphimedon queenslandica; Ath, Arabidopsis thaliana; Cel, Caenorhabditis elegans; Ddi, Dictyostelium discoideum; Dme, Drosophila melanogaster; Hma, Hydra magnipapillata; Hsa, Homo sapiens; Mbr, Monosiga brevicollis; Ncr, Neurospora crassa; Nve, Nematostella vectensis; Pte, Paramecium tetraurelia; Spu, Strongylocentrotus purpuratus; Tad, Trichoplax adhaerens.
Included among the functional categories that correlate with increase in metazoan morphological complexity are (Fig. 4 and Supplementary Table 11.1.1): GPCRs, ion channels, cell adhesion proteins, and defence and immunity proteins, which are enriched in basal metazoans relative to non-animals; homeobox transcription factors and gap junction proteins, which are enriched in bilaterians relative to non bilaterian animals; and immunoglobulin receptor family members, immunoglobulins, MHC antigens, and cytokine receptors, which are enriched in vertebrates relative to invertebrate bilaterians. These broad associations with complexity are evidently superimposed on notable lineage-specific variation as seen in Fig. 4 (for example, serine protease gene loss in C. elegans, and voltage-gated ion channel expansion in Paramecium). Similar functional categories contribute to principal components (Supplementary Table 11.2.1).
Conclusions
The Amphimedon genome, combined with recently sequenced genomes of diverse invertebrates and a choanoflagellate, identifies innovations that underlie the emergence and early diversification of the Metazoa. These genomic comparisons reconstruct a common animal ancestor of remarkable complexity. Metazoans can now be defined by a long list of genomic synapomorphies—gene content, intron–exon structure and syntenies—as well as characteristics common to all animal life such as sex, development, controlled cellular proliferation, differentiation and growth, and immunity. To what extent the ancestral functioning of this gene set is reflected in modern poriferans is unclear, although studies of both sponge development, which yields a highly patterned larva with axial polarity12, and sponge immunity provide points of direct comparison with the eumetazoan condition.
Whereas the eumetazoan lineage produced a wide diversity of body forms, the sponge body plan has been stable for over 600 million years. What can explain this disparity in evolved morphological complexity? Although we have seen that sponges and eumetazoans share many common pathways related to morphogenesis and cell-type specification, there are notable genomic differences, including different microRNA assemblages46, lineage-specific domains and domain architectures, and the differential expansions of gene families. Although there has been minimal characterization of cis-regulatory architectures in non-bilaterians, we note that as most classes of bilaterian transcription factors are also present in sponges, cnidarians and placozoans, it may be that quantitative rather than qualitative differences in cis-regulatory mechanisms were needed to produce more diverse body plans.
The sexually-reproducing, heterotrophic metazoan ancestor had the capacity to sense, respond to, and exploit the surrounding environment while maintaining multicellular homeostasis. Although sponges lack some of the cell types found in eumetazoans, including neurons and muscles, they share with all other animals genes that are essential for the form and function of integrated multicellular organisms. With these genomic innovations enabling the regulation of cellular proliferation, death, differentiation and cohesion, metazoans transcended their microbial ancestry.
Note added in proof: After completing our analysis, integrins and other cell-adhesion-related genes were discovered outside metazoa47. The presumed earlier origin of integrins has been incorporated in Fig 3a.
Methods Summary
Detailed methods are described in Supplementary Information. The genome assembly, gene model sequences, predicted proteins, EST clusters and sequences have been deposited with DDBJ/EMBL/GenBank as project accession ACUQ00000000 and can be accessed from http://www.metazome.net/amphimedon.
Online Methods
A detailed description of methods used in this study can be found in the Supplementary Information.
Genome sequencing
Genomic DNA was sheared and cloned into plasmid and fosmid vectors for whole genome shotgun sequencing as described49. The data were assembled using a custom approach described in the Supplementary Information. The Amphimedon 9X assembly and the preliminary data analysis has been deposited at DDBJ/EMBL/GenBank as project accession ACUQ00000000.
Gene prediction and annotation
Protein-coding genes were annotated using homology-based methods (Augustus50, Genomescan51) and one ab initio method (SNAP52). Protein-coding gene predictions can be accessed from http://www.metazome.net/amphimedon.
Phylogenetic methods
Three data sets of orthologous genes from eighteen genomes were aligned using default parameters using CLUSTALW53 and poorly aligned regions were excluded using Gblocks54.
Phylogenetic analyses were conducted using Bayesian inference and maximum likelihood with bootstrap using MrBayes55,56, and PHYML57 respectively. Alternative likelihood topologies were tested using TREEPUZZLE58 and CONSEL59. Bayesian analysis using site-heterogeneous models were done using aamodel (J. Huelsenbeck, unpublished) and PhyloBayes60,61.
Identification of Amphimedon orthologues of specific bilaterian genes
Putative orthologues of genes involved in various processes in bilaterians were identified by reciprocal BLAST of human, mouse, or Drosophila genes against the Amphimedon gene models (blastp) or the assembly (tblastn). PFAM62 domain composition, assignment of PANTHER HMMs63,64 and phylogenetic trees were used to determine orthology. Trees were built using the neighbour-joining method in Phylip65 with one-hundred bootstrap replicates.
Molecular function enrichments and correlation of complexity
Metazoan gene families were assigned molecular functions using PANTHER63 annotations. Fisher’s exact test as implemented in R66 was run to test for enrichment or depletion of numbers of gene families for each molecular function category in the novel versus ancestral gene sets. Numbers of genes (not gene families) for various molecular function categories were tested for enrichment between different pairs of four eukaryotic complexity groups (vertebrate, non-vertebrate bilaterian, basal metazoan, non-animal) to identify molecular function families that correlate with the differences in complexity. Principal components analysis was used to identify the contribution of each molecular function category to a eukaryotic complexity group.
Accession codes
Primary accessions
DDBJ/GenBank/EMBL
Data deposits
The genome sequence data can be accessed from DDBJ/EMBL/GenBank as project accession ACUQ00000000.
References
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000)
Muller, W. E. & Muller, I. M. Origin of the metazoan immune system: identification of the molecules and their functions in sponges. Integr. Comp. Biol. 43, 281–292 (2003)
Muller, W. E. et al. Bauplan of Urmetazoa: basis for genetic complexity of metazoa. Int. Rev. Cytol. 235, 53–92 (2004)
Grant, R. E. Animal Kingdom. in The Cyclopaedia of Anatomy and Physiology, Vol. 1 (ed. Todd, R. B.) (Sherwood-Gilbert-Piper, 1836)
Sollas, W. J. Report on the Tetractinellida collected by H.M.S. Challenger, during the years 1873–1876. in Report on the Scientific Results of the Voyage of H.M.S. Challenger During the Years 1873–76 (Neill and Company, 1888)
Hyman, L. H. The Invertebrates, Vol. 1 Protozoa through Ctenophora (McGraw-Hill, 1940)
Müller, W. E. Origin of metazoan adhesion molecules and adhesion receptors as deduced from cDNA analyses in the marine sponge Geodia cydonium: a review. Cell Tissue Res. 289, 383–395 (1997)
Muller, W. E. & Schacke, H. Characterization of the receptor protein-tyrosine kinase gene from the marine sponge Geodia cydonium . Prog. Mol. Subcell. Biol. 17, 183–208 (1996)
Suga, H., Katoh, K. & Miyata, T. Sponge homologs of vertebrate protein tyrosine kinases and frequent domain shufflings in the early evolution of animals before the parazoan-eumetazoan split. Gene 280, 195–201 (2001)
Skorokhod, A. et al. Origin of insulin receptor-like tyrosine kinases in marine sponges. Biol. Bull. 197, 198–206 (1999)
Nichols, S. A., Dirks, W., Pearse, J. S. & King, N. Early evolution of animal cell signaling and adhesion genes. Proc. Natl Acad. Sci. USA 103, 12451–12456 (2006)
Larroux, C. et al. Developmental expression of transcription factor genes in a demosponge: insights into the origin of metazoan multicellularity. Evol. Dev. 8, 150–173 (2006)
Larroux, C. et al. Genesis and expansion of metazoan transcription factor gene classes. Mol. Biol. Evol. 25, 980–996 (2008)
Simionato, E. et al. Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evol. Biol. 7, 33 (2007)
Gazave, E. et al. NK homeobox genes with choanocyte-specific expression in homoscleromorph sponges. Dev. Genes Evol. 218, 479–489 (2008)
Adamska, M. et al. Wnt and TGF-β expression in the sponge Amphimedon queenslandica and the origin of metazoan embryonic patterning. PLoS ONE 2, e1031 (2007)
Schierwater, B. et al. Concatenated analysis sheds light on early metazoan evolution and fuels a modern “urmetazoon” hypothesis. PLoS Biol. 7, e20 (2009)
Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008)
Pick, K. S. et al. Improved phylogenomic taxon sampling noticeably affects non-bilaterian relationships. Mol. Biol. Evol. 10.1093/molbev/msq089 (2010)
Sperling, E. A., Peterson, K. J. & Pisani, D. Phylogenetic-signal dissection of nuclear housekeeping genes supports the paraphyly of sponges and the monophyly of Eumetazoa. Mol. Biol. Evol. 26, 2261–2274 (2009)
Degnan, B. et al. The demosponge Amphimedon queenslandica: Reconstructing the ancestral metazoan genome and deciphering the origin of animal multicellularity. in Emerging Model Organisms: A Laboratory Manual, Vol. 1 (Cold Spring Harbor Laboratory Press, 2009)
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007)
Sullivan, J. C., Reitzel, A. M. & Finnerty, J. R. A high percentage of introns in human genes were present early in animal evolution: evidence from the basal metazoan Nematostella vectensis . Genome Inform 17, 219–229 (2006)
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071 (2008)
Srivastava, M. et al. The Trichoplax genome and the nature of placozoans. Nature 454, 955–960 (2008)
Slack, J. M., Holland, P. W. & Graham, C. F. The zootype and the phylotypic stage. Nature 361, 490–492 (1993)
Larroux, C. et al. The NK homeobox gene cluster predates the origin of Hox genes. Curr. Biol. 17, 706–710 (2007)
Manning, G., Young, S. L., Miller, W. T. & Zhai, Y. The protist, Monosiga brevicollis, has a tyrosine kinase signaling network more elaborate and diverse than found in any known metazoan. Proc. Natl Acad. Sci. USA 105, 9674–9679 (2008)
Adamska, M. et al. The evolutionary origin of hedgehog proteins. Curr. Biol. 17, R836–R837 (2007)
Exposito, J. Y. et al. Demosponge and sea anemone fibrillar collagen diversity reveals the early emergence of A/C clades and the maintenance of the modular structure of type V/XI collagens from sponge to human. J. Biol. Chem. 283, 28226–28235 (2008)
King, N. et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451, 783–788 (2008)
Robertson, A. J. et al. The genomic underpinnings of apoptosis in Strongylocentrotus purpuratus . Dev. Biol. 300, 321–334 (2006)
Huang, S. et al. Genomic analysis of the immune gene repertoire of amphioxus reveals extraordinary innate complexity and diversity. Genome Res. 18, 1112–1126 (2008)
Abedin, M. & King, N. The premetazoan ancestry of cadherins. Science 319, 946–948 (2008)
Tepass, U., Truong, K., Godt, D., Ikura, M. & Peifer, M. Cadherins in embryonic and neural morphogenesis. Nature Rev. Mol. Cell Biol. 1, 91–100 (2000)
Richards, G. S. et al. Sponge genes provide new insight into the evolutionary origin of the neurogenic circuit. Curr. Biol. 18, 1156–1161 (2008)
Srivastava, M. et al. Evolution of the LIM homeobox gene family in basal metazoans. BMC Biol. 8, 4 (2010)
Wiens, M. et al. Innate immune defense of the sponge Suberites domuncula against bacteria involves a MyD88-dependent signaling pathway. Induction of a perforin-like molecule. J. Biol. Chem. 280, 27949–27959 (2005)
de Jong, D. et al. Multiple dicer genes in the early-diverging metazoa. Mol. Biol. Evol. 26, 1333–1340 (2009)
Kimura, A., Sakaguchi, E. & Nonaka, M. Multi-component complement system of Cnidaria: C3, Bf, and MASP genes expressed in the endodermal tissues of a sea anemone, Nematostella vectensis . Immunobiology 214, 165–178 (2009)
Fernandez-Busquets, X. & Burger, M. M. Circular proteoglycans from sponges: first members of the spongican family. Cell. Mol. Life Sci. 60, 88–112 (2003)
Sakarya, O. et al. A post-synaptic scaffold at the origin of the animal kingdom. PLoS ONE 2, e506 (2007)
Pavans de Ceccatty, M. Coordination in sponges. The foundations of integration. Am. Zool. 14, 895–903 (1974)
Leys, S. P. & Degnan, B. M. The cytological basis of photoresponsive behavior in a sponge larva. Biol. Bull. 201, 323–338 (2001)
Valentine, J. W. Late Precambrian bilaterians: grades and clades. Proc. Natl Acad. Sci. USA 91, 6751–6757 (1994)
Grimson, A. et al. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455, 1193–1197 (2008)
Sebe-Pedros, A., Roger, A. J., Lang, F. B., King, N. & Ruiz-Trillo, I. Ancient origin of the integrin-mediated adhesion and signaling machinery. Proc. Natl Acad. Sci. USA 107, 10142–10147 (2010)
Boute, N. et al. Type IV collagen in sponges, the missing link in basement membrane ubiquity. Biol. Cell 88, 37–44 (1996)
Aparicio, S. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes . Science 297, 1301–1310 (2002)
Stanke, M., Tzvetkova, A. & Morgenstern, B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7 (Suppl. 1). S11.1–S11.8 (2006)
Yeh, R. F., Lim, L. P. & Burge, C. B. Computational inference of homologous gene structures in the human genome. Genome Res. 11, 803–816 (2001)
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004)
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994)
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000)
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003)
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001)
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003)
Schmidt, H. A., Strimmer, K., Vingron, M. & von Haeseler, A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18, 502–504 (2002)
Shimodaira, H. & Hasegawa, M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247 (2001)
Lartillot, N., Brinkmann, H. & Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 7 (Suppl. 1). S4 (2007)
Lartillot, N. & Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109 (2004)
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 32, D138–D141 (2004)
Thomas, P. D. et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 (2003)
Thomas, P. D. et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31, 334–341 (2003)
Felsenstein, J. PHYLIP–Phylogeny Inference Package (Version 3.2). Cladistics 5, 164–166 (1989)
Team, R. D. C. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2009)
Acknowledgements
This study was supported by funds from the Australian Research Council (B.M.D., Maj.A), US Department of Energy Joint Genome Institute (B.M.D., D.S.R., S.P.L.) Harvey Karp (K.S.K.), NSF (T.H.O.), NIH/NHGRI (G.M.), University of Queensland Postdocotral Fellowship (Maj.A., S.F.C), Sars International Centre for Marine Molecular Biology (Maj.A.), DFG (M.St.), ANR (M.V.), CNRS (M.V.), Gordon and Betty Moore Foundation (D.S.R.) and Richard Melmon (D.S.R.). We thank J. Huelsenbeck and I. Hariharan for help with phylogenetic analyses and growth pathways, respectively. The work conducted by the US Department of Energy Joint Genome Institute was supported by the Office of Science of the US Department of Energy under contract no. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
Genome and EST sequencing, assembly, annotation and analysis: J.C., T.M., U.H., N.H.P., M.St., A.D., Y.Z., Mar.A., A.C., D.M.G., D.J.J., S.S., B.J.W. and D.S.R. Phylogenetics: M.Sr. and D.S.R. Gene family and biological process analyses: M.Sr., B.F., M.E.A.G., G.S.R., C.C., M.D., C.L., Maj.A., S.M.D., T.H.O., D.C.P., S.F.C., C.H., M.V., K.S.K., G.M., B.M.D. and D.S.R. Clustering, novelty, domain content and complexity analyses: O.S. and D.S.R. Gene family expansion analyses: M.Sr., O.S., D.S.R. Writing: M.Sr., B.M.D., D.S.R., O.S., J.C., B.F., M.G., G.S.R., G.M., K.S.K., M.V., C.L., S.M.D., N.H.P., A.D., C.C., M.A., T.H.O. and S.P.L. Project design and coordination: B.M.D and D.S.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Information (see pages 1-2 for Table of Contents), Supplementary Tables and Figures (see page 47) and References (see page 154). See separate excel files for tables listed below. (PDF 7809 kb)
Supplementary Table 8.7.1
List of Amphimedon queenslandica kinases classified into known kinase families. (XLS 112 kb)
Supplementary Table 8.9.5
List of Amphimedon queenslandica GPCR genes classified into adhesion/secretin, glutamate, and rhodopsin-like families. (XLS 90 kb)
Supplementary Table 9.2.1
List of gene clusters that appear to be novel to metazoans, eumetazoans, eumetazoans sensu stricto, and bilaterians. (XLS 844 kb)
Supplementary Table 9.3.1
List of PFAM domains and domain architectures that appear to be novel to metazoans, eumetazoans, eumetazoans sensu stricto, and bilaterians. (XLS 399 kb)
Supplementary Table 9.4.1
List of molecular function categories that are enriched or depleted in novel metazoan genes. (XLS 48 kb)
Supplementary Table 11.1.1
List of molecular function categories that are enriched in animal complexity group comparisons. (XLS 133 kb)
Supplementary Table 11.1.2
List of molecular function categories that are depleted in animal complexity group comparisons. (XLS 125 kb)
Supplementary Table 11.2.1
Molecular function principle components that explain differences between animal complexity groups. (XLS 36 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Srivastava, M., Simakov, O., Chapman, J. et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466, 720–726 (2010). https://doi.org/10.1038/nature09201
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature09201
This article is cited by
-
Origins of cancer: ain’t it just mature cells misbehaving?
The EMBO Journal (2024)
-
Highly conserved and extremely evolvable: BMP signalling in secondary axis patterning of Cnidaria and Bilateria
Development Genes and Evolution (2024)
-
Respiration kinetics and allometric scaling in the demosponge Halichondria panicea
BMC Ecology and Evolution (2023)
-
The compact genome of the sponge Oopsacas minuta (Hexactinellida) is lacking key metazoan core genes
BMC Biology (2023)
-
Antioxidant enzymes that target hydrogen peroxide are conserved across the animal kingdom, from sponges to mammals
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
