
Abstract
Monozygotic or ‘identical’ twins have been widely studied to dissect the relative contributions of genetics and environment in human diseases. In multiple sclerosis (MS), an autoimmune demyelinating disease and common cause of neurodegeneration and disability in young adults, disease discordance in monozygotic twins has been interpreted to indicate environmental importance in its pathogenesis1,2,3,4,5,6,7,8. However, genetic and epigenetic differences between monozygotic twins have been described, challenging the accepted experimental model in disambiguating the effects of nature and nurture9,10,11,12. Here we report the genome sequences of one MS-discordant monozygotic twin pair, and messenger RNA transcriptome and epigenome sequences of CD4+ lymphocytes from three MS-discordant, monozygotic twin pairs. No reproducible differences were detected between co-twins among ∼3.6 million single nucleotide polymorphisms (SNPs) or ∼0.2 million insertion-deletion polymorphisms. Nor were any reproducible differences observed between siblings of the three twin pairs in HLA haplotypes, confirmed MS-susceptibility SNPs, copy number variations, mRNA and genomic SNP and insertion-deletion genotypes, or the expression of ∼19,000 genes in CD4+ T cells. Only 2 to 176 differences in the methylation of ∼2 million CpG dinucleotides were detected between siblings of the three twin pairs, in contrast to ∼800 methylation differences between T cells of unrelated individuals and several thousand differences between tissues or between normal and cancerous tissues. In the first systematic effort to estimate sequence variation among monozygotic co-twins, we did not find evidence for genetic, epigenetic or transcriptome differences that explained disease discordance. These are the first, to our knowledge, female, twin and autoimmune disease individual genome sequences reported.
Similar content being viewed by others


Main
We sought to assess the magnitude of genetic, epigenetic and transcriptomic differences in CD4+ lymphocytes from MS-affected and unaffected monozygotic twin sibships (Supplementary Fig. 1). CD4+ T cells are involved in the pathophysiology of MS (Online Mendelian Inheritance in Man (OMIM) accession 126200)1. mRNA, genomic DNA (gDNA) and reduced-representation, bisulphite-treated gDNA were prepared from negatively isolated, CD4+ T lymphocytes from three pairs of adult, monozygotic twins who were discordant for MS (-001, affected; -101, unaffected). Affected individuals fulfilled McDonald criteria for MS diagnosis13. A lack of sibling affectation was assessed by clinical evaluation, and, for twin 041896-101, confirmed by magnetic resonance brain imaging and cerebrospinal studies. Monozygotic twin pair 041896 was female, of Ashkenazi Jewish origin and beyond the susceptibility age-range for MS at the time of study (Supplementary Table 1). Twin pair 230178 was female and African-American, whereas twins 041907 were white males. Individual 041896-001 had an onset of MS at age 30 years, and is at present in the secondary progressive phase; individuals 230178-001 and 041907-001 had MS onset at ages 38 and 13, respectively, and have relapsing-remitting disease. Molecular typing of HLA loci showed identical genotypes within the three twin pairs (Supplementary Table 1). Only co-twins 041907 had DRB1*1501, the strongest genetic susceptibility factor for MS14.
Nucleic acid samples were sequenced by sequencing-by-synthesis with reversible-terminator chemistry15,16,17,18. mRNA was prepared from blood samples drawn on different days from twin pair 041896 to ascertain sampling variance. A total of 50–68-million, high-quality, 36–44-nucleotide, singleton sequences from each of eight mRNA samples were aligned to the NCBI human genome reference, and read-counts per gene were calculated18,19,20 (Supplementary Table 2). Sequencing to this depth (median relative transcript coverage of 5.0-fold and 6.4-fold for 041896-001 and 041896-101, respectively) allowed the determination of the diversity of the polyadenylated transcriptome in CD4+ lymphocytes: ∼92% of 20,601 genes with exon annotations were expressed, as assessed by aligned reads and the upper asymptote of the best-fit sigmoid curve (Supplementary Table 2 and Supplementary Fig. 2). The distribution of transcript abundance was a left-skewed, bell-shaped curve with >7 log10 dynamic range (Supplementary Fig. 2), in agreement with a previous study17. Digital gene expression values correlated well with exon-resolution array hybridization results (Supplementary Fig. 3), in agreement with another report21. Surprisingly, diagnosis or treatment of MS accounted for only 9.4% of variance in transcript abundance in T cells of monozygotic twins, compared with 57.3% being attributable to twin-pair-to-twin-pair differences, 23.6% to day-to-day variation (as assessed in twin pair 041896 alone), and 3.5% to lane-to-lane sequencing variation (Supplementary Figs 4–7). The variance in transcript abundance attributable to MS was within the range of variances obtained by random permutation of MS diagnosis labels (Supplementary Fig. 8 and Supplementary Table 3). Thus, robust gene expression differences were not observed between MS-affected and unaffected twins in CD4+ lymphocytes that were inexplicable by other variables.
One-billion, high-quality, shotgun, whole-genome sequences were generated from twins 041896-001 and -101, corresponding to 21.7- and 22.5-fold aligned coverage, and representing 99.6% and 99.5% of the NCBI human reference genome, respectively (Supplementary Table 4). Comparisons of genome coverage of the twins with the AK1 genome, which was determined using identical procedures, showed no individual coverage bias15 (Supplementary Figs 9 and 10).
Viral infection has been suggested to contribute to the aetiology of MS. After re-alignment of unaligned sequences to 2,864 viral genomes, ∼0.02% of DNA reads from twins 041896 and 0.2% of RNA reads from the three twin pairs matched 310 viral genomes. A large majority of these alignments reflected simple sequence repeats or endogenous retroviral sequences. After reverse-transcription and PCR, no reproducible differences were found between sequences aligning to viral genomes in T cells from MS-affected and unaffected individuals.
Approximately 3.6 million SNPs and ∼0.2 million insertions and deletions (indels) were detected in the genomes of subjects 041896-001 and -101, using optimized criteria, which are similar to values reported for male genomes (ref. 15 and Supplementary Table 5). Indels varied in size from -31 to +8 nucleotides, with an approximately normal frequency distribution. Of 13 common risk variants previously associated with MS susceptibility14, co-twins 041896 were homozygous for five, heterozygous for five, and three were absent. This genetic load is predicted to increase the risk for development of MS ∼8-fold under an additive model (Supplementary Table 6). Co-twins 230178 were homozygous for seven susceptibility loci and heterozygous for two, and co-twins 041907 were homozygous for eight risk alleles and heterozygous for two, conferring a 14-fold and 43-fold increased risk, respectively (Supplementary Table 6). These data should be interpreted cautiously because translation of genetic burden into risk for complex disorders is rudimentary. Clustering of 9.9 million SNPs in eight individual genome sequences showed close similarity of the twins 041896, female genomes and their separation from six male genomes (Supplementary Fig. 11).
SNP genotype differences were sought between affected and unaffected twin siblings in genomic DNA and mRNA (Supplementary Fig. 1). First, stringent bioinformatic filters were trained both to call SNPs in aligned genome and mRNA sequences and to infer SNP genotypes, by comparing genotypes obtained from duplicate Affymetrix 6.0 SNP array hybridizations with those derived from genome and mRNA sequencing (Supplementary Tables 7 and 8 and Supplementary Fig. 12)15. These filters excluded low coverage or repetitive genomic sequences (<11-fold or >44-fold coverage, respectively), yielding high positive predictive values (PPVs) to enable meaningful co-twin comparisons. Second, these filters were used to determine SNP genotypes in aligned genomic sequences of twin pair 041896 and in aligned mRNA sequences of the three twin pairs. Third, identities and differences in inferred SNP genotypes were sought between affected and unaffected twin siblings. Co-twin genotype differences were categorized as changes from homozygous reference allele to heterozygote, or from heterozygote to homozygous variant (Table 1). Of 1,089,550 SNP genotypes inferred in genomes 041896 using these filters, 3,241 (0.3%) differed between twins (Table 1). Of more than 730,000 genomic SNP genotypes determined by duplicate array hybridizations, 126 (0.02%), 153 (0.02%), and 120 (0.02%) differed between siblings in the three twin pairs, respectively, which was considerably less than ∼8,500 SNPs that were discordant between repeated hybridizations of individual DNA samples (Supplementary Table 9). mRNA sequencing covered ∼65.6 megabases (Mb) of annotated exons to a depth of ∼5-fold. Three-hundred-and-twenty-two (0.6%), 1,017 and 380 SNP genotypes inferred in mRNA sequences differed between siblings of twin pairs 041896, 230178 and 041907, respectively (Table 1). Finally, replication of co-twin SNP genotype identities and differences was sought. No differences in SNP genotypes inferred by one approach were recapitulated by a second method. In contrast, >98% of SNPs that were identical in twin siblings and genotyped by two methods (array hybridization, mRNA sequencing or genomic DNA sequencing) were replicated (Table 1). Furthermore, Sanger resequencing showed identical genotypes in twin pair 041896 for a set of 15 SNP differences well supported by at least one method.
The SNP genotyping filters were also used to infer indel genotypes in genome and mRNA sequences of the twins: 91.9% of indels detected in both genome and mRNA sequences had identical genotypes (Table 1). Of 26,908 indel genotypes inferred in the genomes of twins 041896, 213 (0.8%) differed between siblings. Of 1,322, 1,073 and 407 indel genotypes inferred in mRNA sequences from twins 041896, 230178 and 041907, 8, 39 and 10 differed between twin siblings, respectively (Table 1). No indel genotype differences identified by one approach were recapitulated by a second method. In summary, siblings in three monozygotic twin pairs exhibited no replicable nucleotide variation differences in non-repetitive sequences, as assessed by genome and mRNA sequencing and SNP array hybridization. Much longer reads and lower error rates will be required to evaluate variation differences in repetitive sequences comprehensively. Detection of no replicable SNP genotype differences between siblings of any of the three twin pairs in peripheral CD4+ T cells accords with estimated rates of somatic mutation of 8.4 × 10-9 to 4.6 × 10-10 per nucleotide per generation in human tumours, Saccharomyces cerevisiae and Drosophila melanogaster22,23,24.
Expression quantitative trait loci (eQTL) are emerging as a molecular mechanism for common SNPs that are significant in genome-wide association studies of disease25. In light of an absence of significant MS-associated genotypic or mRNA expression differences between twins, we sought allele-specific differences in mRNA expression. For heterozygous coding SNPs (cSNPs), the expression of both alleles in CD4+ lymphocytes was measured to address deviation from the 1:1 expected ratio (allelic imbalance). A total of 268 heterozygous cSNPs exhibited allelic imbalance in cis at 188 loci in twin 041896 transcriptomes, as determined by significant deviation of aligned genomic and mRNA read counts (Supplementary Table 10). Single base mismatches do not cause systematic bias in GSNAP alignments. Two imprinted genes showed altered allelic expression in both co-twins (ZNF331 and GNAS), as did three genes that exhibit altered allelic expression in human cerebellar cortex (ABLIM1, UBE2I and KIAA1267, S.F.K. et al., unpublished data), and two that have previously shown altered allelic expression in CD4+ lymphocytes (the MS-associated gene CD6 and acid trehalase-like 1 (ATHL1))14,26. We used quantitative PCR to validate each of the three possible outcomes: (1) where both twins showed an expected 1:1 ratio of allelic expression; (2) where both twins show skewed expression of an allele in the same direction and magnitude, indicative of a cis-acting eQTL or imprinting; and (3) where the direction or magnitude of the imbalance differed between the twins (Supplementary Fig. 13). Notably, 115 (43%) cSNPs differed between twins (that is, differential allelic expression; Fig. 1 and Supplementary Table 10). These results indicate that some gene expression differences between twins represent chromatid-specific alterations in transcription. Variance in allelic expression between samples mirrored that observed in overall mRNA levels, with twin-pair-to-twin-pair accounting for 51.2%, day-to-day variation for 27.7% and MS diagnosis for 8.0% of variance. No cSNPs showing allelic imbalance were shared among the three twin pairs. Notably, however, cSNPs that show allelic imbalance were significantly closer to transcription-factor-binding sites than random SNPs, providing a new, potential mechanism of action.

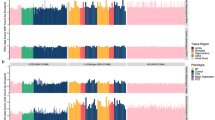
a, b, Allelic imbalance for 041896-001 (a) and 041896-101 (b) was detected in cSNPs called by ≥10 gDNA reads with Q ≥ 20 and where 20–80% of uniquely aligning gDNA reads called the SNP, together with detection in ≥10 mRNA reads with Q ≥ 20. Out of 14,461 heterozygous cSNPs, 268 (1.9%) showed significant allelic imbalance in expression (P < 10-7), of which 153 (57%) were of the same magnitude and direction in both subjects. TCRVB is the T cell receptor beta locus, V (variable) segment, locus symbol TRB@. WDR40B is also known as DCAF12L1.
Structural variants were identified in the six genomes by hybridization of duplicate arrays. In contrast to a recent report, we found no copy number variants or allelic gains/losses that differed between siblings in any twin pair12. Twins 041896 displayed 143 structural variants comprising 89 Mb, twins 230178 exhibited 13 variants comprising 3 Mb, and twins 041907 had 58 variants encompassing 33 Mb (Supplementary Figs 1, 14, 15 and Supplementary Table 11). Of note, seven structural variants were common to all three twin pairs, and changed the copy number of two genes (late cornified envelope-3B (LCE3B) and T-cell receptor gamma chain alternate reading frame protein (TARP)) and one pseudogene (ADAM metallopeptidase domain 6 (ADAM6)) (Supplementary Table 12). LCE3B was not expressed in T-cell mRNA samples from these patients. TARP was expressed at a level of 12.9 ± 6.1 reads per million (mean ± s.d.) and did not show altered expression in MS. These genes have not been previously associated with MS.
A further axis of heritable genetic information in human genomic DNA is cytosine methylation, which serves several functions including regulation of gene expression, silencing of retrotransposons, genomic imprinting and X-chromosome inactivation, and has been implicated in several diseases27,28. We sought to compare genome-scale DNA methylation profiles between twin siblings at nucleotide resolution. We aligned 50–90 million, high-quality, 50-nucleotide, reduced representation bisulphite sequences (RRBS) from ten samples—the three pairs of twin T lymphocytes, normal lung and lung cancer, and normal breast and breast cancer16 (Supplementary Table 13). For twins 041896, these corresponded to 45.5- and 32.7-fold coverage of 1.4 million uniquely aligning, non-repetitive MspI fragments, and 2,146,620 and 2,033,078 CpG dinucleotides from the -001 and -101 genomes, respectively (Table 2). Bisulphite conversion of non-CpG cytosines was >99%. Almost identical numbers of CpG sites were identified in the forward and reverse strands, as expected (Supplementary Table 14). As reported for mouse, methylation levels of CpG dinucleotides in human T cells showed a bimodal distribution, with most being unmethylated or extensively methylated (>95% of reads in either state) (Fig. 2a and Supplementary Fig. 16)16. Approximately one-quarter of CpGs were methylated. More than 90% of CpG sites were common to siblings within each twin pair (Table 2). CpGs aggregated into clusters (corresponding to CpG islands16) at a ratio of 1.58–1.74 CpGs per cluster. More than 92% of CpG clusters were common to siblings within each twin pair (Table 2 and Supplementary Table 14). Highly congruent results were obtained with two alignment algorithms (Supplementary Table 14 and Supplementary Figs 17 and 18) and two reference genome data sets. Of ∼2 million CpGs represented by ≥10 high-quality reads in twins 041896, only two showed a switch between siblings from ≤20% methylated to ≥80% by ELAND and four by GSNAP, none of which was supported by both methods (Fig. 2b and Table 2). Likewise, 10 out of 1.7 million CpG sites in twins 230178 and 176 out of 1.7 million CpG sites in twins 041907 showed a switch in methylation by ELAND (Fig. 2c, d and Supplementary Table 15). Two CpG methylation switches between affected and unaffected siblings were common to twin pairs 230178 and 041907, albeit with opposite directions of change (>80% → <20% methylated CpG sites (mCpG) in 041907-001 and -101, respectively, whereas <20% → >80% mCpG in 230178-001 and -101, at a CpG site 9,912 nucleotides 5′ of TMEM1 and 8,536 and 10,659 nucleotides 5′ of PEX14). To put these findings in context, we evaluated the magnitude of methylation changes in CD4+ T cells from unrelated individuals, between tissues and between normal and cancerous tissue. We observed 586–827 inter-individual <20% → >80% CpG methylation differences (Fig. 2e, f), and 4,255–7,180 CpG methylation shifts between T lymphocytes, lung and breast tissues (Table 2 and Fig. 2i, j). Breast and lung cancers showed 1,557 and 16,509 CpG methylation shifts, when compared with normal breast and lung tissue, respectively (Fig. 2g, h and Table 2). A second pattern of change in CpG methylation was observed in comparison of male and female samples: 394 CpGs were <5% methylated in 041907-001 T lymphocytes (male) but 20–50% methylated in 041896-001 (female). Likewise, 406 CpG sites were <5% methylated in 041907-101 (male) and 20–50% methylated in 041896-101 (female). Of these, 385 and 389, respectively, mapped to chromosome X, consistent with female X inactivation (Fig. 2e). Similarly, a very large number of CpG sites that were <10% methylated in normal lung were 20–70% methylated in lung cancer (Fig. 2h). A previous study has shown epigenetic differences between dizygotic twins to be qualitatively greater than between monozygotic twins29. Here we show the magnitude of epigenetic differences between monozygotic twin sibling CD4+ lymphocytes to be at least an order of magnitude less than those between individuals, and ∼three orders less than those observed between tissues and in malignant transformation.

a, Frequency distribution of CpG site methylation in 041896-001 (blue) and -101 (red) using ELAND-extended. b–j, Pairwise comparisons of CpG site methylation using ELAND-extended in CD4+ lymphocytes from monozygotic twin siblings 041896-001 and -101 (b), 230178-001 and -101 (c) and 041907-001 and -101 (d); inter-individual differences between CD4+ lymphocytes from 041896-001 and 041907-001 (e) and 041896-001 and 230178-101 (f); neoplastic differences between breast tissue and breast cancer (g) and between normal lung tissue and lung cancer (h); and between-tissue differences between CD4+ lymphocytes and breast tissue (i) and lung tissue (j).
In summary, the recent genome-wide association study (GWAS)-identification of novel risk loci is opening a broad window into genetic intricacies underpinning complex diseases. Although genetic knowledge remains incomplete, a new generation of sequencing and analytical tools may prove to hold great potential, as shown here. Likewise, a discordant monozygotic twins study controls for many genetic and non-genetic confounders, enhancing the tractability of mechanisms in complex disorders. We sought genetic, epigenetic or transcriptomic differences between CD4+ T cells of twin siblings that might explain MS-discordance. Although MS is a neurological disease, T cells are fundamentally involved in its pathophysiology1. However, no reproducible differences in SNPs, indels, copy number variants (CNVs), gene expression levels or sequences aligning to viral genomes were detected between CD4+ T cells of co-twins. To provide analytical rigor, SNP and indel differences were sought using at least two different approaches and CNV experiments were performed in duplicate. However, analysis of nucleotide variants was limited in scope by exclusion of low coverage regions and repetitive sequences (because the latter cannot be reliably interrogated by alignment of short reads or array hybridization), by moderate sensitivity for detection of structural variants of size 50–1,500 nucleotides (which fall between the resolution of sequencing and array hybridization), and by limited feasibility to detect possible somatic mosaicism. A previous study has shown differences in selection of T-cell receptors after antigen stimulation between monozygotic twins discordant for MS30. Quantitative analysis of T-cell repertoire or immunoglobulin locus recombination was not possible at ∼22× depth of aligned coverage. Progress in single molecule sequencing technologies with longer reads and deeper coverage should overcome many of these limitations in the future, as would examination of further cellular compartments of innate and adaptive immunity. Furthermore, deep RRBS showed very few changes in CpG methylation between CD4+ T cells of twin siblings and no differences common to two or more twin pairs. It should be noted, however, that RRBS was limited to the investigation of marked shifts in CpG methylation in a relatively broad population of T cells. Other epigenetic mechanisms, differences within lymphocyte subsets, mono-allelic differences or other tissues were not examined. These caveats aside, however, monozygotic twins lacked genetic, epigenetic or transcriptomic differences in T cells to explain MS-discordance. Several tantalizing, new, differences were detected that will require replication and further studies: 43% of eQTLs had a different direction or magnitude of imbalance in twin siblings. In summary, a singular genetic, epigenetic or transcriptomic mechanism underpinning MS-discordance in monozygotic twins was not detected in a study of unprecedented resolution. Although disease-discordant monozygotic twins seem to provide a framework for analysis of complex disorders that has fewer variables, further stratification and/or concomitant measurement of several data types may be necessary to yield molecular mechanisms underpinning disease.
Methods Summary
The study was approved by the University of California, San Francisco (UCSF) Institutional Review Board. Informed, written consent was obtained from all individuals. CD4+ lymphocytes were isolated from peripheral blood and nucleic acids extracted with standard methods. Two samples were obtained on different days from twins 041896 and single samples from the others. HLA typing was by AlleleSEQR (Atria Genetics) and Assign SBT software (Conexio Genomics). Genome-wide genotypes and CNVs were detected with Affymetrix 6.0 arrays in duplicate. Log-R ratios were generated with Affymetrix Genotyping Console 3.0.2 and analysed with Nexus software (BioDiscovery Inc.). Short- and long-insert, paired-end libraries were generated from gDNA, mRNA and reduced-representation, bisulphite-treated gDNA as described15,16,17,18. Paired-end and singleton, 36–130-nucleotide reads were generated using Illumina GAIIx instruments. Sequences were aligned principally to the NCBI reference genome build 36.3, with GSNAP and tolerance of 5% mismatches15. SNPs, indels and gene expression were analysed with Alpheus using filters trained with array results15,18,19,20: genomic SNP calling filters were >20% and >4 uniquely aligning reads with average quality score (Q) ≥ 20 (Supplementary Table 7). mRNA SNP calling filters were Q ≥ 20, presence in ≥20% and ≥2 reads and ≥1 uniquely aligning read. Nucleotides with coverage 11–44× and Q ≥ 20 were genotyped according to frequency cutoffs in Supplementary Table 8. Genotype differences were called where frequencies differed by >50%. eQTLs were detected by allelic mRNA read counts differing from equality with χ2 P-values of <10-7. Gene expression was assessed by log2-transformed aligned read counts. Putative SNP differences were validated by Sanger sequencing and putative gene expression differences using Affymetrix Human Exon 1.0 ST arrays. Putative eQTLs and virus alignments were validated by quantitative PCR (with allele specificity for the former). Statistical analysis used JMP-Genomics (SAS Institute) or R (http://www.R-project.org).
Online Methods
The study was approved by the University of California, San Francisco (UCSF) Institutional Review Board. Informed, written consent was obtained from all individuals. CD4+ lymphocytes were isolated from peripheral blood and nucleic acids extracted with standard methods. Two samples were obtained on different days from twins 041896 and single samples from the others. HLA typing was by AlleleSEQR (Atria Genetics) and Assign SBT software (Conexio Genomics). Putative SNP differences were validated by Sanger sequencing and putative gene expression differences using Affymetrix Human Exon 1.0 ST arrays. Putative eQTLs and virus alignments were validated by quantitative PCR (with allele specificity for the former). Statistical analysis used JMP-Genomics (SAS Institute) or R (http://www.R-project.org).
Array-based genotyping and CNV detection
Genome-wide genotypes (>900,000 SNPs) and CNVs (∼1.8 million probes) were detected with Affymetrix 6.0 arrays. Genomic DNA from each individual was tested on duplicate arrays. Log-R ratios (normalized probe intensities) were generated with Affymetrix Genotyping Console 3.0.2 and analysed with Nexus software (BioDiscovery Inc.), which identifies CNVs with a circular binary algorithm using intensity data from all probes, and allele ratios from SNP probes.
Alignment of mRNA and gDNA sequences to reference databases
Short- and long-insert, paired-end libraries were generated from gDNA, mRNA and reduced-representation, bisulphite-treated gDNA as described15,16,17,18. Paired-end and singleton, 36–130-nucleotide reads were generated using Illumina GAIIx instruments. mRNA-Seq and whole-genome shotgun sequences were aligned to the NCBI reference genome (build 36.3) with GSNAP and tolerance of 5% mismatches15,20 (Supplementary Tables 2 and 4). For definition of exon boundaries, annotations from RefSeq Transcript (downloaded 2 September 2008) and from 5,224 non-redundant UCSC transcripts (downloaded 13 April 2009) were appended to Build 36.3 of the reference human genome. Long (75–130 nucleotides) genomic reads were found to align poorly using these criteria, owing to low terminal quality scores and higher rates of mismatch. Therefore, unaligned long, genomic, paired reads were further aligned to the NCBI reference genome with GSNAP by trimming to paired 75 nucleotides and tolerance of ≤10 mismatches.
mRNA-Seq and whole-genome shotgun reads not mapping to the human genome were aligned to 2,864 NCBI viral genome sequences (release 35) with GSNAP and tolerance of 5% mismatches. Alignments were visualized using Alpheus20 and CMTV31. High likelihood true alignments were identified on the basis of: (1) significant read coverage of the viral genome; (2) elimination of reads composed primarily of simple sequence repeats; (3) unique read alignments; (4) paired read alignments with correct orientation and distance separating read pairs; and (5) alignments of non-clonal reads to contiguous stretches of viral genome sequence.
Putative, new viral sequences with average quality scores (Q) ≥ 20 were assembled by ABySS32 or by reference-guided assembly with AMOScmp-shortReads-alignmentTrimmed33. Default parameters were used. Contigs were aligned to the NCBI nr database using BLASTN 2.2.21.
mRNA-Seq-based measurement of gene expression changes
After alignment of mRNA-Seq reads, read counts were calculated per gene for each lane of sequence and log2 transformed. Distribution analysis (Supplementary Fig. 4) and Mahalanobis differences (Supplementary Fig. 6) were assessed for log-transformed read counts from each lane of mRNA-Seq and outlier lanes were removed. Principal component analysis (Supplementary Fig. 6) and variance decomposition of principal components were undertaken for log-transformed read counts from each lane to assess sources of variability in gene expression (Supplementary Fig. 7). Because diagnosis (MS-affected versus non-affected) accounted for 9.4% of variance, all possible permutations of lanes of sequence were examined to determine whether diagnosis-associated variance was greater than a random permutation (experimental design file in Supplementary Table 3). Principal component analysis and variance decomposition of principal components were repeated with log-transformed read counts from each lane for each permutation to assess permuted diagnosis-associated variance in gene expression (Supplementary Fig. 8). Because true diagnosis-associated variance was not greater than permuted variance, genes differing between MS-affected and unaffected individuals were not assessed by weighted ANOVA. eQTLs were detected by allelic mRNA read counts differing from equality with χ2 P-values of <10-7.
ELAND alignment of RRBS
Treatment of DNA with bisulphite converts cytosine residues to uracil, but leaves 5-methylcytosine residues unaffected. Thus, alignments of 50-bp, singleton, RRBS to the human genome are complicated by the simplification of the genetic code from four to three bases, except at methylcytosine (mC) locations. ELAND-extended performs alignments of the first 32 nucleotides of a read with up to two substitutions, and then extends the alignment with unlimited mismatches. Alignment of 3-base reads (after conversion of residual cytosines to thymidines in the RRBS reads) to a 3-base genome (after conversion of all cytosines to thymidines) with ELAND-extended resulted in many non-unique alignments. To circumvent this problem, we made use of the fact that all RRBS start at an MspI site (which comprise most CpG residues and the large majority of CpG islands16). Thus, 3-base reads were aligned to a 3-base version of ∼3.7% of the human genome, comprising 2.3 million MspI fragments of up to 50 nucleotides in length, derived from the NCBI human genome sequence, version 36.3, totalling 113 Mb in length (Supplementary Table 16). The fragments were of two types: 133,609 fragments of 30–50 bp that were flanked by MspI sites on both ends, and 2.2 million 50-bp fragments with a 5′ flanking MspI site (representing genomic MspI fragments of greater than 50 bp in length). Only unique alignments with Phred-like scores >4 (greater than 50% likelihood of being correct alignments) and only those starting with a 5′ thymidine (base 1 of a converted MspI fragment) were retained (Supplementary Table 13). Alignments to fragments of less than 50 nucleotides terminated at the end of the fragment. ELAND does not align to MspI fragments of less than 30 nucleotides in length. After alignment of converted reads, thymidine residues were corrected to their original sequence in the RRBS and reference, and C-to-T transitions were identified. The percentage methylation for CpG sites was scored by the ratio of C/(C+T) calls for each C that was followed by a G. The percentage conversion of C to T when followed by another base was used for estimation of the bisulphite conversion rate, and was >99.8%.
RRBS alignment with GSNAP
RRBS were also aligned with GSNAP to the NCBI human genome reference sequence, version 36.3, allowing 5% mismatches and without penalizing C-to-T transitions (Supplementary Table 13). Because GSNAP reports only the best alignments (those with the fewest mismatches) using the entire 50-nucleotide alignment, unique alignments were possible using the entire genome without penalizing C-to-T transitions. The percentage methylation was assessed for CpG sites with at least tenfold coverage, based on all alignments (that is, not restricted to unique). Only CpG sites within MspI fragments were considered. For identification of differences between subjects from ‘largely methylated’ to ‘largely unmethylated’, we sought positions where there was at least 80% cytosine in one subject and less than 20% cytosine in the other.
GSNAP is a short-read alignment program based on GMAP that uses a hash table and a compressed version of the reference genome, which is constructed once for that genome34. The reference may include arbitrary contigs (up to 4 billion), so that one may also align to a reference transcriptome, with redundancy allowed among the contigs. The hash table contains the locations of a given 12-nucleotide sequence in the genome, subject to sampling. The sampling step occurs during pre-processing of the genome, so that genomic locations are stored only for every third 12-nucleotide sequence in the genome. Sampling is needed to reduce the memory footprint of the program below 4 gigabytes for a human-sized genome. GSNAP can handle short reads of >24 or more nucleotides, with each read in the input potentially having a varying length. There is theoretically no upper bound on the length of the query sequence, except that this bound is compiled into GSNAP by default at 200 nucleotides; longer sequences can be handled simply by changing this constant at compile time.
GSNAP has specialized algorithms for identifying exact mappings, one-mismatch mappings, multiple-mismatch mappings, and indel mappings (including a user-specified number of mismatches). Exact mappings are identified by taking the intersection of genomic positions over a spanning set of 12-nucleotide sequences in the query sequence. The spanning set must contain 12-nucleotide sequences in the same phase modulo 3, to account for the sampling used in pre-processing the genome, so the program must test each of the three possible phases. For spanning set members that overhang the ends of the query sequence by 1 or 2 nucleotides, the relevant genomic positions can be obtained by substituting 1 or 2 wildcard nucleotides, respectively, and taking the union of genomic locations in the hash table.
Candidates for one-mismatch mappings are similarly identified by computing an incomplete intersection, in which one 12-nucleotide sequence in the spanning set does not contain the given genomic location. These candidate genomic mappings are then compared against a compressed version of the genome to verify that only one mismatch was present.
Candidates for multiple-mismatch mappings are determined by processing a sorted list of genomic locations from all 12-nucleotide sequences in the query sequence. This sorted list is computed efficiently using a heap-based priority queue. For each candidate genomic location, a floor on the number of mismatches can be computed from the pattern of query positions of the 12-nucleotide sequences that match the genomic location. Candidates with a sufficiently low floor (based either on a user-specified limit or on the best mapping determined so far) are then compared against the compressed genome to determine the actual number of mismatches.
For identifying indel mappings, GSNAP accumulates partial genomic alignments during the multiple-mismatch algorithm, where a partial alignment can be supported by a single 12-nucleotide sequence in the query sequence. These partial alignments are then scanned in genomic order to identify pairs that are sufficiently close to constitute a candidate indel, where the default distances are 30 nucleotides for an insertion and 12 nucleotides for a deletion. These candidate pairs are then compared against the compressed genome to determine the number of mismatches. To identify indels occurring at either end of the query sequence, the program computes floors that exclude the 12-nucleotide sequences on either end. Candidates with a sufficiently low floor are then compared against the compressed genome to identify a possible indel at the end and to count the actual number of mismatches.
Although GSNAP allows repetitive regions of the genome to be masked before building the genomic data structure, in typical usage (as described here) the genome is not pre-masked. Therefore, GSNAP is able to align sequences to redundant regions in the genome, including repetitive regions, and report all such alignments. In default mode (as described here), the program reports only the best alignments, those with the fewest mismatches, although the program also can be run to identify and report suboptimal alignments. GSNAP differs from ELAND in that it processes the reference genome first, constructs a hash table of the genome, and then aligns the short reads to the genome. In contrast, ELAND processes the short reads first, constructs a hash table of the short reads, and then scans the genome to find matches.
Identification of optimal bioinformatic filters for SNP detection and genotyping
SNP detection in Illumina GAII sequences is complicated by relatively high sequencing error rates, particularly at nucleotides 50–130 using the chemistry and base calling software available during the first half of 2009. SNP genotyping in Illumina GAII sequences is complicated by a continuous, albeit trimodal, distribution of frequencies of SNP- and reference-sequence-containing reads at a given location (Supplementary Fig. 12). To translate SNP- and reference-sequence-containing read frequencies into genotypes and to understand the sensitivity and specificity of SNP detection and genotyping, comparisons between array-based SNP genotypes and sequencing results were performed extensively. Unambiguous SNP genotypes from duplicate array hybridizations (with SNP calls and concordant genotypes in both replicates) were assessed to be true. Subsets of SNPs common to Affymetrix 6.0 arrays and sequence data sets were identified. Optimal SNP genotyping filters (those with maximal PPVs and near-optimal sensitivity) for each sequence data set were identified by determining the number of true positives, false positives and false negatives, and determining the PPV and sensitivity of all combinations of the following criteria: number of reads calling the SNP, number of uniquely aligning reads calling the SNP, percentage reads calling the SNP, average quality score (Q), and minimum quality score. To detect changes in SNP genotype, each possible genotype in a diploid genome was modelled (homozygous reference allele, heterozygote, and homozygous variant allele) and the optimal change in allele frequency was determined. Genomic SNP calling filters were >20% and >4 uniquely aligning reads with Q ≥ 20 (Supplementary Table 7). mRNA SNP calling filters were Q ≥ 20, presence in ≥20% and ≥2 reads and ≥1 uniquely aligning read. Nucleotides with coverage 11–44× and Q ≥ 20 were genotyped according to frequency cutoffs in Supplementary Table 8. Genotype differences were called where frequencies differed by >50%. These methods represent a refinement of those used previously15, and which were extensively validated by Sanger resequencing and genotyping arrays.
Identification of allele-specific expression
Allele-specific expression in mRNA sequences was identified by methods similar to those described25. Frequencies of frequencies of SNP- and reference-sequence-containing reads at a given heterozygous location in mRNA sequences are continuous, albeit unimodal (Supplementary Fig. 12), reflecting both random reference and variant-containing read sequencing, effects of clonal reads and allele-specific expression. Unambiguous heterozygous SNP locations in each individual were determined based on duplicate array hybridizations (with SNP calls and concordant genotypes in both replicates) and by the SNP calling criteria developed above. Allele-specific expression effects were assessed by application of genome-wide P values to significance testing of deviation from 50:50 read frequencies. Artefactual allele-specific expression associated with enrichment of clonal reads was evaluated for many, putative allele-specific expression SNPs by visualization of start and stop sites of reads using Alpheus. Artefactual allele-specific expression associated with bias in GSNAP alignment of reads containing or lacking specific SNPs was evaluated as discussed above.
Accession codes
Data deposits
Data is deposited at dbGaP under accession phs000239.v1.p1.
References
Chitnis, T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int. Rev. Neurobiol. 79, 43–72 (2007)
Oksenberg, J. R., Baranzini, S. E., Sawcer, S. & Hauser, S. L. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nature Rev. Genet. 9, 516–526 (2008)
Sadovnick, A. D. et al. Evidence for genetic basis of multiple sclerosis. Lancet 347, 1728–1730 (1996)
Nielsen, N. M. et al. Familial risk of multiple sclerosis: a nationwide cohort study. Am. J. Epidemiol. 162, 774–778 (2005)
Mumford, C. J. et al. The British Isles survey of multiple sclerosis in twins. Neurology 44, 11–15 (1994)
Willer, C. J. et al. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc. Natl Acad. Sci. USA 100, 12877–12882 (2003)
Islam, T. et al. Differential twin concordance for multiple sclerosis by latitude of birthplace. Ann. Neurol. 60, 56–64 (2006)
French Research Group on Multiple Sclerosis. Multiple sclerosis in 54 twinships: concordance rate is independent of zygosity. Ann. Neurol. 32, 724–727 (1992)
Machin, G. A. Some causes of genotypic and phenotypic discordance in monozygotic twin pairs. Am. J. Med. Genet. 61, 216–228 (1996)
Gringras, P. & Chen, W. Mechanisms for differences in monozygous twins. Early Hum. Dev. 64, 105–117 (2001)
Fraga, M. F. et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl Acad. Sci. USA 102, 10604–10609 (2005)
Bruder, C. E. et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 82, 763–771 (2008)
Kim, J.-I. et al. A highly annotated whole genome sequence of a Korean individual. Nature 460, 1011–1015 (2009)
McDonald, W. I. et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann. Neurol. 50, 121–127 (2001)
De Jager, P. L. et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nature Genet. 41, 776–782 (2009)
Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008)
Wang, E. T. et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008)
Mudge, J. et al. Genomic convergence analysis of schizophrenia: mRNA sequencing reveals altered synaptic vesicular transport in post-mortem cerebellum. PLoS One 3, e3625 (2008)
Sugarbaker, D. J. et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl Acad. Sci. USA 105, 3521–3526 (2008)
Miller, N. A. et al. Management of high-throughput DNA sequencing projects: Alpheus . J. Comput. Sci. Syst. Biol. 1, 132–148 (2008)
Mane, S. P. et al. Transcriptome sequencing of the Microarray Quality Control (MAQC) RNA reference samples using next generation sequencing. BMC Genomics 10, 264 (2009)
Jones, S. et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl Acad. Sci. USA 105, 4283–4288 (2008)
Lynch, M. et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl Acad. Sci. USA 105, 9272–9277 (2008)
Haag-Liautard, C. et al. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila . Nature 445, 82–85 (2007)
Moffatt, M. F. et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448, 470–473 (2007)
Heap, G. A. et al. Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum. Mol. Genet. 19, 122–134 (2010)
Jones, P. A. & Baylin, S. B. The epigenomics of cancer. Cell 128, 683–692 (2007)
Cedar, H. & Bergman, Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature Rev. Genet. 10, 295–304 (2009)
Kaminsky, Z. A. et al. DNA methylation profiles in monozygotic and dizygotic twins. Nature Genet. 41, 240–245 (2009)
Utz, U. et al. Skewed T-cell receptor repertoire in genetically identical twins correlates with multiple sclerosis. Nature 364, 243–247 (1993)
Sawkins, M. C. et al. Comparative map and trait viewer (CMTV): an integrated bioinformatic tool to construct consensus maps and compare QTL and functional genomics data across genomes and experiments. Plant Mol. Biol. 56, 465–480 (2004)
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009)
Pop, M., Phillippy, A., Delcher, A. L. & Salzberg, S. L. Comparative genome assembly. Brief. Bioinform. 5, 237–248 (2004)
Wu, T. D. &. N. a. c. u. S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 10.1093/bioinformatics/btq057 (10 February 2010)
Acknowledgements
We thank the study subjects. This work was supported by grants from Small Ventures USA Inc., A. J. Brass Foundation, Nancy Davis Foundation, NIH grants RR016480 to F.D.S., RO1NS26799 to S.L.H., RO1NS46297 to J.R.O. and National MS Society (NMSS) grants RG3060C8 and RG2901D9 to J.R.O. S.E.B is a Harry Weaver Neuroscience Scholar of NMSS.
Author information
Authors and Affiliations
Contributions
S.E.B., G.P.S., J.R.O., S.L.H. and S.F.K. designed the project. S.F.K., S.E.B., J.M. and J.R.O. wrote the paper with input from the other authors. S.E.B., J.M., J.C.v.V., L.Z., R.W.K., G.D.M., J.E.W., S.J.C., J.P.M., R.G., M.J.P., L.E.C., E.E.G., F.D.S., J.J.H. and S.L. performed the experiments. S.E.B., J.M., J.C.v.V., P.K., I.K., N.A.M., L.Z., A.D.F., C.J.B., T.R., S.L., P.K., T.D.W., G.P.S., J.R.O., S.L.H. and S.F.K. analysed the data. S.L.H., J.R.O. and O.A.K. supervised patient recruitment.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1-16 and Supplementary Figures 1-18 with legends. (PDF 22526 kb)
PowerPoint slides
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Baranzini, S., Mudge, J., van Velkinburgh, J. et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464, 1351–1356 (2010). https://doi.org/10.1038/nature08990
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature08990
This article is cited by
-
Comprehensive pan-cancer analysis reveals that C5orf34 regulates the proliferation and mortality of lung cancer
Functional & Integrative Genomics (2024)
-
Stochastic gene expression and environmental stressors trigger variable somite segmentation phenotypes
Nature Communications (2023)
-
Association between vaccination and the risk of central demyelination: results from a case-referent study
Journal of Neurology (2023)
-
Quantitative proteomics reveals protein dysregulation during T cell activation in multiple sclerosis patients compared to healthy controls
Clinical Proteomics (2022)
-
Differences between germline genomes of monozygotic twins
Nature Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
