
Abstract
Domestic animals are excellent models for genetic studies of phenotypic evolution1,2,3. They have evolved genetic adaptations to a new environment, the farm, and have been subjected to strong human-driven selection leading to remarkable phenotypic changes in morphology, physiology and behaviour. Identifying the genetic changes underlying these developments provides new insight into general mechanisms by which genetic variation shapes phenotypic diversity. Here we describe the use of massively parallel sequencing to identify selective sweeps of favourable alleles and candidate mutations that have had a prominent role in the domestication of chickens (Gallus gallus domesticus) and their subsequent specialization into broiler (meat-producing) and layer (egg-producing) chickens. We have generated 44.5-fold coverage of the chicken genome using pools of genomic DNA representing eight different populations of domestic chickens as well as red jungle fowl (Gallus gallus), the major wild ancestor4. We report more than 7,000,000 single nucleotide polymorphisms, almost 1,300 deletions and a number of putative selective sweeps. One of the most striking selective sweeps found in all domestic chickens occurred at the locus for thyroid stimulating hormone receptor (TSHR), which has a pivotal role in metabolic regulation and photoperiod control of reproduction in vertebrates. Several of the selective sweeps detected in broilers overlapped genes associated with growth, appetite and metabolic regulation. We found little evidence that selection for loss-of-function mutations had a prominent role in chicken domestication, but we detected two deletions in coding sequences that we suggest are functionally important. This study has direct application to animal breeding and enhances the importance of the domestic chicken as a model organism for biomedical research.
Similar content being viewed by others

Main
For most of their history, domestic chicken populations have been bred for two purposes, egg laying and meat production5. The effective chicken population size must have been huge in the past, before specialized commercial populations were established during the twentieth century, as a large proportion of farms kept a group of chickens interconnected with other groups by trade between regions, countries and continents. This is consistent with the extensive sequence diversity present in domestic chicken (∼5 single nucleotide polymorphisms (SNPs) per kilobase (kb) in pairwise comparisons)6. During the twentieth century, specialized layer and broiler breeds were established to circumvent the inherent conflict in selecting for both growth traits (meat production) and reproductive traits (egg production) in the same bird. This approach, combined with the implementation of modern breeding methods based on quantitative genetics theory, has been extremely successful in improving productivity7.
Our experimental design involved the resequencing of birds representing eight populations of domestic chickens as well as populations of red jungle fowl (Fig. 1). The aim was to identify the most common allele at the majority of polymorphic sites in the genome and to identify selective sweeps shared by populations selected for the same trait. We used the Applied Biosystems SOLiD technology to generate short reads (35 base pairs (bp)) to a sequence depth of four- to fivefold coverage from pools of chickens, each representing different populations, which is an effective approach for finding common alleles favoured by positive selection. The sequencing of DNA pools from individual lines is also effective for detecting selective sweeps, because heterozygosity can be calculated in sliding windows from sequences drawn from a pool of haplotypes. We sampled four different layer populations, four broiler populations and a pool of red jungle fowl representing two different zoo populations (Supplementary Table 1). Finally, we resequenced the particular red jungle fowl female (from the partly inbred UCD 001 line) previously used to generate the classical Sanger-sequenced reference chicken genome8. The inclusion of the reference bird provided an excellent quality check of the SOLiD data.

Schematic tree based on breeding history (branch lengths not to scale). The minimum time of divergence, when known, is indicated. Pools of genomic DNA from the following populations were resequenced: red jungle fowl (n = 8), commercial broiler 1 (CB-1; n = 10), commercial broiler 2 (CB-2; n = 10), high growth line (n = 11), low growth line (n = 11), Rhode Island Red (RIR; n = 8), obese strain (n = 10), White Leghorn line 13 (WL-A; n = 11) and a commercial White Leghorn line (WL-B; n = 8). The single red jungle fowl female from the partly inbred UCD 001 line used to generate the reference genome sequence8 was also included.
We aligned the 35-bp reads to the chicken reference sequence. The uniquely placed reads covered 92% of the 1,043 megabases (Mb) in the current genome assembly (Supplementary Table 1), and these were used for all further analyses of sequence variation. The 90 Mb not covered by any read from any line were mostly repetitive sequences but to some extent probably represent sequences under-represented after the emulsion-PCR step. We identified SNPs for each population individually and also for several pools of populations. Three independent reads of the same non-reference nucleotide were required to declare a position polymorphic. This criterion was chosen to minimize the number of false-positives. Bioinformatic analysis showed that the great majority of SNPs (>95%) reported in this study represent true SNPs (Supplementary Information), and this conclusion was supported by experimental verification of more than 300 SNPs (Supplementary Table 2). Furthermore, we eliminated ∼40,000 putative SNPs because we did not find any support for the reference allele, and assume that these represent rare sequence errors in the current assembly (Supplementary Fig. 1). In total, 7,453,845 SNP loci of which we are confident remained after this filtering and were used in the subsequent analyses. Supplementary Table 1 shows the sequence coverage and the number of non-reference alleles found in each population. We used the allele frequencies of these SNPs to construct a genetic distance tree summarizing the genetic relationships among populations (Supplementary Information and Supplementary Fig. 2).
To detect putative selective sweeps, we searched the genome for regions with high degrees of fixation. The major challenge in such an analysis is to distinguish true sweeps from fixation due to genetic drift. In general, there is a high degree of genetic diversity in the chicken genome. However, some of the populations included were maintained with a limited effective population size, which could lead to genetic drift and fixation (Supplementary Table 1). To lessen the problem of fixation due to genetic drift within populations, we conducted sweep analyses with pooled sequence data from three groups of populations selected for similar purposes: all domestic lines, the two commercial broiler lines (CB-1 and CB-2) and three layer populations (WL-A, WL-B and RIR). We calculated the pooled heterozygosity, Hp, in sliding 40-kb windows along the autosomes from sequence reads corresponding to the most and least frequently observed alleles at all SNP positions (Methods).
The distributions of observed Hp values and the Z transformations of Hp, ZHp, are plotted in Fig. 2a for the three comparisons. Owing to the complex and partly unknown demography of these populations, it is difficult to define a strict threshold that distinguishes true sweeps from regions of homozygosity caused by drift. We propose that the best way to evaluate the many putative sweeps is to cross-reference these results with others such as verification of the sweep in additional chicken populations and data on co-localization with major quantitative trait loci (QTLs) and differentially expressed genes. We focus our description of putative sweeps on those reaching a ZHp score of -6 or less, as these are in the extreme lower end of the distribution (Fig. 2a).

a, Distributions of pooled heterozygosity values, Hp, and corresponding Z transformations, ZHp, for all 40-kb windows (n = 46,906). Bins of Hp and ZHp are presented along the x axes. μ, mean; σ, standard deviation. b, Selective sweep at the yellow skin/BCDO2 locus4. Genes in the vicinity of the sweep are shown below the graph. * marks the sweep interval defined using sequence data from all pools of domestic chickens. ** marks the 23-kb interval previously associated with yellow skin4. c, The negative end of the ZHp distribution presented along chromosomes 1–28. The horizontal dashed lines indicate the threshold at ZHp = -6. Genes residing within 20 kb of a window with ZHp ≤ -6 are indicated by their gene names. The location of TSHR is also indicated in the plots for commercial broilers and layers, even though ZHp did not pass the threshold of -6. Ensembl genes are denoted ENS:XXXXX, where XXXXX are the last five digits of the Ensembl chicken gene annotation (ENSGALG:11 digits). For windows with no gene overlap, the presence of PhastCons evolutionary conserved regions (ECRs) and expressed sequence tags (ESTs) is indicated with a single asterisk. ** indicates a gene candidate close to a window with no genes overlapping ±20 kb.
Only ∼0.1% of the windows (n = 58) had a ZHp score of less than or equal to -6 in the all-domestic comparison, and the corresponding fractions were ∼0.05% for the broilers and ∼0.01% for the layers (Fig. 2c and Supplementary Table 3). We used the yellow skin allele of BCDO2 locus as a proof of principle showing that this approach could reveal an established sweep4; all domestic birds in this study were assumed to be homozygous for the yellow skin allele. We observed an Hp value of 0.09 (ZHp = -8.2) over BCDO2 (Fig. 2b), and the region perfectly overlapped the previously defined sweep4. The region did not show complete fixation, as a few domestic birds carried a wild-type haplotype.
The number of putative sweeps reaching the ZHp-score threshold of -6 was highest for the all-domestic comparison: 58 windows representing 21 loci passed this threshold. Further genetic and functional studies of these loci are well justified because many of them, as well as some that did not reach the significance threshold, may have contributed significantly to chicken domestication. Three putative sweeps had ZHp scores that were more extreme (that is, lower) than that of yellow skin/BCDO2. One was located on chromosome 1 in a non-coding region upstream of SEMA3A, which encodes semaphorin 3A, an axon guidance molecule with an essential role in brain development9. The second occurred in a non-coding region 160 kb upstream of the gene for V-set and transmembrane-domain-containing protein 2A (VSTM2A), which is a predicted target-SNARE gene on chromosome 2, and the third occurred at the locus encoding thyroid stimulating hormone receptor (TSHR) on chromosome 5. We decided to further evaluate TSHR because it had the lowest ZHp score (-9.2) and because of the well-established biological significance of TSHR signalling for metabolic regulation and reproduction10,11,12,13. The sweep region showed almost complete fixation over a 40-kb region (Fig. 3). The TSHR region also approached significance for the individual broiler and layer comparisons, with ZHp scores of 4.7 and 4.9, respectively. Both groups showed complete homozygosity at the TSHR locus but failed to reach the threshold of -6 because the sweep regions did not cover a full 40-kb window. In the all-domestic comparison, the consistent sweep signal from all eight populations resulted in an extremely low ZHp score even though the region of complete homozygosity across all populations did not cover an entire 40-kb window.

a, Pooled heterozygosities, Hp, in the all-domestic data set, plotted for 40-kb windows on chromosome 5. The most highly fixed window (grey box) overlaps the TSHR gene. b, Degree of haplotype sharing in pairwise comparisons among populations. SNP frequencies in individual lines were used to calculate identity scores in 40-kb windows. Greyscale/purple boxes (left) indicate the comparison performed on that row according to the shade/colour assigned to each chicken line (left versus right column). Heat-map colours (right) indicate identity scores. LR, layers; BR, broilers; RJ, red jungle fowl pool; Ref, reference genome haplotype; Dom, domestic chicken; OS, obese strain; High/Low, high/low growth line. c, A TSHR missense mutation Gly558Arg is highly frequent in domestic chicken. Dashes indicate positions identical to the red jungle fowl sequence. Polar and positively charged amino acids are green and blue, respectively. d, Schematic structure of chicken TSHR based on the model provided by the Glycoprotein-hormone Receptors Information System (http://gris.ulb.ac.be/). In the magnification, the amino acids are coloured according to their properties: blue, basic; red, acidic; green, polar; grey, hydrophobic. The amino-acid substitution is indicated by the red arrowhead (amino acid 558 in UniProt Q3V5M1).
We decided to screen eight SNPs from the TSHR region using 271 birds representing 36 populations with geographic origins ranging from Iceland to China (Supplementary Table 4). Every domestic chicken tested, representing commercial as well as local populations, carried at least one copy of the sweep haplotype; seven birds were heterozygous for the haplotype and 264 were homozygous. This is remarkable given the extensive genetic diversity present in the domestic chicken for most parts of the genome. Thus, TSHR may be a domestication locus in chicken, that is, a locus where essentially all individuals of a domesticated species carry a mutant allele. We observed the sweep haplotype at an intermediate frequency in red jungle fowl representing zoo populations (Supplementary Table 4), and believe that the most likely explanation for this is that many zoo populations have a history of some hybridization with domestic chicken.
We next searched for candidate mutations that may have been the target for the TSHR sweep and identified a non-conservative amino-acid substitution, namely glycine to arginine at residue 558 (Fig. 3c). This residue is located at the border between transmembrane region 4 and the following extracellular loop (Fig. 3d). Glycine at this position is conserved among all known vertebrate TSHR sequences and it is almost completely conserved among other members of this family of glycoprotein-hormone receptors (see the Glycoprotein-hormone Receptors Information System; http://gris.ulb.ac.be/). A bioinformatic analysis using DASher14 indicated that the glycine-to-arginine substitution pushes this residue outwards from the membrane and may therefore influence ligand interaction. Thus, this missense mutation is a candidate causal mutation for the TSHR sweep. Currently, we cannot formally exclude the possibility that the identified missense substitution is a slightly deleterious mutation that has hitch-hiked on a linked causative mutation. However, careful examination, including Sanger-based resequencing of the 40-kb sweep region to close any sequence gaps, did not reveal any strong suspects (GenBank accession numbers GU323554–GU323556).
A possibility is that the domestic TSHR allele confers an advantage as regards increased metabolic activity and growth. However, this is unlikely, because the TSHR locus does not coincide with any of the 13 growth loci detected in our previous QTL study of an intercross between White Leghorn and red jungle fowl15, where the two TSHR alleles are segregating. It is now well established that TSHR signalling between the pars tuberalis, of the pituitary gland, and ependymal cells in the hypothalamus regulates photoperiod control of reproduction in birds and mammals11,12,13. The TSHR sweep may therefore be related to a classical feature of domestic animals, namely the absence of the strict regulation of seasonal reproduction found in natural populations16. We plan to follow up the discovery of the TSHR sweep with functional receptor studies as well as careful phenotypic characterization of our White Leghorn/red jungle fowl intercrosses.
The screen for selective sweeps in populations of commercial broilers revealed many loci that make sense in relation to selection for muscle growth (Fig. 2c). A region on chromosome 1 with strong support contains both the gene for insulin-like growth factor 1 (IGF1), which is a candidate gene for growth, as well as the gene for pro-melanin-concentrating hormone (PMCH), which has important roles in appetite and metabolic regulation17. IGF1 also shows a strong indication of a selective sweep in the layer comparison (ZHp < -5.6; Supplementary Table 3) but not in the all-domestic comparison, as different haplotypes are close to fixation in broilers and layers. A functionally related candidate gene affecting growth traits and with a central role for insulin signalling, insulin receptor (INSR), also lies within a sweep region.
The sweep at the TBC1D1 (TBC1 (tre-2/USP6, BUB2, cdc16) domain family, member 1) locus is particularly interesting, because it is located at the major QTL explaining differences in growth between broilers and layers in three independent studies18,19,20. Strong further support for the TBC1D1 sweep comes from the fact that the sweep haplotype is also fixed in the high and low growth lines (Fig. 1). This implies that the sweep took place during the early development of broiler chickens, as the high and low growth lines were established in 1957 and have been kept as closed populations since then. TBC1D1 has been associated with susceptibility to obesity in humans21, and a loss-of-function mutation in this gene causes leanness in mice22. It has recently been shown that insulin-stimulated phosphorylation of TBC1D1 is required for the translocation of the main glucose transporter, GLUT4 (also known as SLC2A4), to the cell surface of mouse C2C12 myoblasts23. This study implies that billions of broiler chickens around the world carry a mutant TBC1D1 haplotype and could be used for in-depth functional studies.
It has been proposed that loss of function may be an important factor in rapid evolution, such as occurs during domestication24. Deleterious mutations may also accumulate as a result of relaxed selection or hitch-hiking during selective sweeps25. We used the resequencing data to look for two specific loss-of-function mutations: stop-codon mutations and deletions (Supplementary Information and Supplementary Tables 5 and 6). We identified almost 1,300 deletions that were fixed or close to fixation in at least one population. Only 16 of these were longer than 6.4 kb and none was longer than 67 kb (Supplementary Fig. 3). We found little evidence that selection for loss-of-function mutations has had a prominent role in chicken domestication. However, we detected seven deletions in coding sequences (Supplementary Table 7), one of which has previously been described as functional and another of which shows strong evidence of function. The deletion in the growth hormone receptor (GHR) gene has previously been reported to be a causative mutation for sex-linked dwarfism26 and has been used in some commercial broiler lines to reduce growth and feed consumption in parental lines.
We also found a novel deletion that removes all but the first exon of the gene SH3RF2 (SH3 domain containing ring finger 2) (Fig. 4a). The deletion is fixed in the high growth line and occurs at a low frequency in the low growth line and in CB-1. SH3RF2 lies within a QTL region for body weight detected in a cross between the high and low growth lines27. We genotyped 400 birds from generation F8 of this intercross, which provides a much better mapping resolution than generation F2 (ref. 28). The analysis revealed a highly significant association between the presence of the deletion and increased growth (P < 0.001; Fig. 4b). Body weight at 70 days for Del/Del birds (600 g) was 20% greater than for WT/WT birds (500 g). We could take advantage of the fact that the deletion was also present at a low frequency in the low growth line. If this is the causative mutation, it should be associated with the same QTL effect in generation F2 whether it was inherited from an F0 chicken from the high growth line or the low growth line. This provides high resolution in QTL mapping because the lines were crossed after generation 41 of the selection experiment, meaning that they are separated by 82 generations of meioses, disrupting the associations between the QTL and linked markers. The estimated effects of the deletion haplotype originating from the high growth line were indistinguishable from those of it originating from the low growth line (Fig. 4c). This shows that the deletion or a mutation in the very near vicinity must be the causative mutation for the QTL. Expression analysis using hypothalamus messenger RNA revealed SH3RF2 expression in the low growth line but not in the high growth line, which is expected because the latter is fixed for the deletion (Fig. 4d). The result is of interest as it is well established that chickens from the high growth line have a genetic defect in hypothalamic appetite regulation29.

a, Gene annotation and human protein homology with depth of sequence coverage in the high growth line (top), the low growth line (middle) and in the reference bird (bottom). The 18,961-bp deletion (red arrow) in the high growth line deletes all but the first exon of SH3RF2. b, Growth curve for WT/WT, WT/Del and Del/Del F8 birds from a cross between the high and low growth lines. WT, wild-type allele; Del, deletion allele. Error bars, s.e.m. c, Estimated allele substitution effect on body weight in day-70 F2 animals, comparing the respective weight gains observed when the deletion is inherited from high- and low-growth-line parental birds. Error bars, s.e.m. d, SH3RF2 mRNA expression in male (n = 3 in each line) and female (n = 7 in each line) high- and low-growth-line chickens.
The results strongly suggest that the deletion, eliminating most of the coding sequence of SH3RF2, is the causative mutation for a QTL contributing to the remarkable difference in growth between the high and low growth selection lines. The nature of the mutation, that is, loss of function of a gene that is well conserved among birds and mammals, suggests that the deletion is unlikely to be just a neutral linked marker. SH3RF2 encodes SH3 domain containing ring finger 2 and is expressed in brain and muscle. The high and low growth lines now provide the first animal model with which the functional significance of this gene may be explored.
The present study casts light on the genetic basis of domestication, but also has implications both for the use of chicken as a model organism for biomedical research and for the application of genomics to practical chicken breeding (Supplementary Information). Chicken was selected for this study because the fairly small genome size (∼1 Gb) made it easier to achieve sufficient sequence coverage to obtain a reasonable power to detect sweeps, deletions and high-quality SNPs. However, the successful outcome of this approach suggests that it should be applied to other domesticated species as well as to natural populations, where it may reveal the genetic basis for rapid evolutionary adaptations.
Methods Summary
SOLiD sequencing
We prepared libraries from pools of genomic DNA (except for the UCD 001 reference individual) and sequenced them using SOLiD (version 2 was used for all runs except two WL-A runs, for which we used version 1) according to standard manufacturer protocols.
SNP detection and analysis
We called SNPs using the Corona Lite pipeline from Life Technologies. To call a SNP, at least three reads with different start sites supporting the non-reference allele had to be present. We called SNPs on each individual line and also by bioinformatically combining the data from multiple pools to improve sensitivity to rare alleles. All reported SNPs are unique sites from the union of all runs.
Selective-sweep analysis
We used allele counts at SNP positions to identify signatures of selection in sliding 40-kb windows, for pools of sequence data. For each pool and SNP, we determined the numbers of reads corresponding to the most and least abundant allele (nMAJ and nMIN). For each window in each breed pool, we calculated a pooled heterozygosity score: Hp = 2ΣnMAJΣnMIN/(ΣnMAJ + ΣnMIN)2, where ΣnMAJ and ΣnMIN are the sums of nMAJ and, respectively, nMIN for all SNPs in the window. Individual Hp values were then Z-transformed as follows: ZHp = (Hp - μHp)/σHp.
Detection of deletions and stop codons
We first scanned the genome for putative deletions in each line. A putative deletion was defined as 100 bp or greater with no read coverage. We then tested all of these regions against coverage by the reference bird to remove those that were uncovered because of sequencing or alignment bias.
Online Methods
Animals
We included genomic DNA samples from the following chicken populations. Four layer lines were sampled: eleven males from White Leghorn line 13 (WL-A) developed at the Swedish University of Agricultural Sciences30 and previously used for QTL mapping experiments15; eight males from a commercial White Leghorn line (WL-B); ten males from the obese strain (OS) established in 1955 from a White Leghorn line as a model for autoimmune thyroiditis31; and eight males from a commercial Rhode Island Red population (RIR; Hubbard ISA) collected as part of the AvianDiv project32. Four broiler lines were sampled: ten males from a commercial broiler line (Ross 308, denoted CB-1 here), ten females from a second commercial line (CB-2), maintained by Hubbard ISA and collected by the AvianDiv project32; and seven males and four females each from the high and low growth selection lines, both established from White Plymouth Rock chickens in 195729 and used in previous QTL mapping experiments27,33. Eight males representing two different zoo populations of red jungle fowl (RJ) were pooled. Finally, the single RJ female from the partly inbred UCD 001 line used to generate the chicken genome sequence8 was also included.
SOLiD sequencing
Libraries were prepared from pools of birds (except the UCD 001 reference individual) and sequenced using SOLiD, version 2 (except for two WL-A runs, for which we used version 1), according to standard manufacturer protocols. Reads were aligned in colour space to the reference assembly (version 2.1, Washington University) using the MAPREADS program allowing for up to three mismatches (including ‘valid adjacent’ changes as a single mismatch) and no indels. Only reads aligning uniquely in the genome were retained.
SNP detection and analysis
SNPs were called using the Corona Lite pipeline from Life Technologies. To call a SNP, at least three reads with different start sites supporting the non-reference allele had to be present. We called SNPs on each individual line and also by bioinformatically combining the data from multiple pools to improve sensitivity to rare alleles. All reported SNPs are unique sites from the union of all runs. Following determination of all unique variant sites, we in silico genotyped all variant sites in all lines using custom PERL scripts, to compare all uniquely aligned reads in colour space to both the reference and variant alleles to count reference and variant alleles observed in each line.
Genetic differentiation was measured between lines with pairwise fixation index (FST), using an estimator introduced in ref. 34. This statistic was calculated across all SNPs identified in any line within the assembled chromosome sequences (that is, SNPs assigned to the ‘_random’ and ‘Un’ sequences were excluded). Then a phylogenetic tree was constructed from these estimates using the neighbour-joining method implemented in PHYLIP35.
SNP validation
A VeraCode GoldenGate assay (Illumina) was designed targeting 384 SNPs that were called in the White Leghorn/red jungle fowl comparison. All SNPs were located in putative selective sweep regions identified in an initial screen for chromosomal regions showing a high degree of homozygosity. A total of 271 domestic chickens representing 36 populations and 51 red jungle fowl from a number of zoo populations were genotyped using the standard protocol provided by Illumina. The GoldenGate assays were read using a BeadXpress Reader (Illumina) and data were analysed on the GENOMESTUDIO V2009.1 software (Illumina). To estimate the false-negative rate of our SNP-calling pipeline, the sequence from the yellow skin allele4 (GenBank EU334162) determined by classical Sanger-sequencing was used. By comparing the sequence against the reference genome sequence, a total of 175 SNP positions were detected. Those SNP positions were cross-referenced with the SNP positions called in the all-domestic comparison.
Estimation of total SNP errors
All calls made at known SNP sites on the Z chromosome (ascertained in lines other than the reference bird) were reviewed and the number of valid base calls of each type made at that position in the reference bird was counted. For all sites with two or more calls, we assumed that any minority calls were errors (rather than using the reference base, to avoid counting errors at miscalled reference bases). As expected given that the Z chromosome in the reference female is hemizygous, we predominantly saw positions with two or three reads. We took the single-read, per-position error rate to be that seen at three reads of coverage (0.00644). We note that this includes both sequence detection errors and artefacts introduced by alignment errors and is approximately ten times worse than the manufacturer’s specification of rates of accuracy after dibase correction (99.94%); independent assessment of single-base accuracy from invalid colour changes suggests that raw sequencing accuracy for these runs was very close to the manufacturer’s specification. Because errors should be random, it was assumed that one-third of them would represent each incorrect base, and we thus computed for each depth of coverage in the genome the probability of seeing three or more of any given erroneous base. These probabilities were all multiplied by the total number of bases seen at each depth and summed over all depths.
Selective-sweep analysis
Allele counts at identified SNP positions were used to identify signatures of selection in sliding 40-kb windows with a step size of 20 kb for three pools of sequence data (breed pools): all eight domestic lines, the two commercial broiler lines (CB-1 and CB-2) and three layer populations (WL-A, WL-B and RIR). At each detected SNP position, we counted the numbers of reads corresponding to the most and least frequently observed allele (nMAJ and nMIN, respectively) in each breed pool. This was done even if a particular SNP had not been originally detected by reads from the particular breed pool where the selective-sweep scan was performed. To prevent windows containing very few SNPs from adding spurious fixation signals, we omitted seven windows where only 1–10 SNPs had been detected and analysed 46,906 windows along chicken chromosomes 1–28 represented in the version 2.1 draft assembly. For each window, we calculated the pooled heterozygosity, Hp, values using the formula Hp = 2ΣnMAJΣnMIN/(ΣnMAJ + ΣnMIN)2, where ΣnMAJ and ΣnMIN are breed-pool-specific sums of nMAJ and, respectively, nMIN counted at all SNPs in the window. The distributions of Hp values resembled normal distributions (Fig. 2a), and we therefore transformed the Hp values into Z scores: ZHp = (Hp - μHp)/σHp. The Z transformation allowed us to place the three breed pools into the same framework because ZHp values indicate the number of standard deviations by which Hp deviates from the mean. We applied a threshold of ZHp ≤ -6 for putative selective sweeps because windows below this threshold represent the extreme lower end of the distribution (Fig. 2a).
Calculation of identity scores
We calculated identity scores, IS, to visualize haplotype sharing in pairwise comparisons at the TSHR locus (Fig. 3b). For each identified SNP, we determined the fraction of reads that corresponded to the reference genome allele, F, in each chicken line. The IS values of individual SNPs were then calculated as IS = 1 - (|Fline1 - Fline2|), with SNPs assessed only if at least one read was obtained in each line. The IS value for a window was the mean of all SNP IS values observed in the window for a specific comparison.
QTL analysis
The phenotypic effect of the SH3RF2 deletion was estimated in two ways. First, the genotype–phenotype map for the three genotypes (Del/Del, Del/WT and WT/WT) was estimated without accounting for the line origin of the alleles and accounting for the observed allele frequencies using the R package NOIA36,37, version 0.92. Second, the effect of the deletion was estimated independently within the low growth and high growth lines by computing the IBD (relationship) matrix following ref. 38, under the assumption of three subsets of alleles in generation F2 (Del originating in F0 birds from the high growth or low growth lines and wild type (WT) always originating in F0 birds from the low growth line).
Detection of deletions and stop codons
We first scanned the genome for putative deletions in each line. A putative deletion was defined as 100 bp or greater with no read coverage. We then tested all of these regions against coverage by the reference bird to remove those that were uncovered because of sequencing or alignment bias. We computed the binomial p value of an N:0 split of reference and line reads given the chromosomal total reference:line ratio. To correct for testing ∼1,000,000 putative deletions in all lines, we required a P value of <10-8 to consider a region deleted. However, if a putative deleted region reached this significance threshold in one line, a P value of <10-5 was considered sufficient to call the presence of an overlapping deletion in other lines. Then we merged by single linkage all overlapping deletion events in multiple lines, on the assumption that such events were likely to be identical. Finally, we merged runs of consecutive deletions that occurred in the same line(s) if the sum of deleted bases was at least 90% of the total span length; manual inspection revealed that such events were usually incorrectly split because of sequences that attracted spurious read placement, or possibly because the line was not 100% fixed for the event. Two such events overlapped genes and were successfully validated as single deletions, supporting this ‘bridging’. Because of the absence of W sequences in male birds and the large amount of unplaced W sequences, the W chromosome and all random and unplaced chromosomes were excluded from this analysis. To prevent regions of unusually high coverage in the reference bird from causing false-positives, we ignored reads in any line overlapping any region that had coverage in any line exceeding that expected given all of our sequencing (Poisson P ≤ 0.05, Bonferroni corrected for sites observed across all lines).
To detect stop codons, the Ensembl gene set was filtered to include only models that made a complete protein product of ≥100 amino acids. We then computed the impact of the alternate allele of every detected SNP on its in-frame codon for each transcript overlapping it. If either the reference or the alternate codon was a stop, it was retained for manual review. During manual review, we discarded any putative stop variant if the transcript failed to show homology to any known protein or if the variant codon lay outside a region of conservation in the alignment and was not supported by chicken EST evidence.
Validation of loss-of-function mutations
Putative deletions or introduced stop codons were amplified using approximately 20 ng of genomic DNA from individuals taken from the appropriate pool in 10-μl reactions containing 10 mM Tris-HCL (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, 0.2 mM dNTP, 1 μM each of forward and reverse primer and 0.5 U AmpliTaq Gold (Applied Biosystems). Validation of products was performed by direct sequencing using BigDye Terminator v3.1 chemistry (Applied Biosystems) and DNA sequences were aligned and edited using CODONCODE ALIGNER (version 3.0.2, CodonCode). The true break points of validated deletions that affected coding sequencing were used to design multiplex PCR assays. These reactions were amplified as above, but with 1 μM of each multiplex primer (m1, m2 and m3) and visualized on 2% agarose gels. Multiplex assays were also applied to a diversity panel (Supplementary Table 9) to assess the prevalence of the mutation in a global setting. All primers were designed in PRIMER339 (version 0.4.0) and are given in Supplementary Table 9, and new sequences have been deposited in GenBank (accession numbers GU323512–GU323553).
SH3RF2 expression study
We dissected hypothalamus from four-day-old birds from both the high growth and low growth lines (three male and seven female chicks from each line, for a total of 20 samples). RNA was isolated with TRIzol (Invitrogen) and purified with RNeasy Mini Kit (Qiagen). The quality of the total RNA was checked with the Agilent 2100 Bioanalyser (Agilent Technologies) and the RNA samples were frozen at -70 °C until used. Complementary DNA synthesis from total RNA and two-step quantitative PCR were performed as described previously40. Briefly, quantitative PCR with reverse transcription was performed using the iQ SYBR Green Supermix (Bio-Rad Laboratories) in combination with the MyiQ Single-Colour Real-Time PCR Detection System (Bio-Rad Laboratories), according to the manufacturer’s protocol. Primers were designed with PRIMER EXPRESS software (version 1.5, Applied Biosystems) and are listed in Supplementary Table 9. The collected data were analysed using the 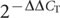 method41 in comparison with the average of the four-day-old high-growth-line male expression level, and all results were normalized to the chicken β-actin gene.
method41 in comparison with the average of the four-day-old high-growth-line male expression level, and all results were normalized to the chicken β-actin gene.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The SOLiD reads have been submitted to the NCBI Sequence Read Archive under the study accession number SRP001870. SNPs reported in this work can be accessed from dbSNP using the query http://www.ncbi.nlm.nih.gov/projects/SNP/snp_viewBatch.cgi?sbid=〈batch〉, in which 〈batch〉 is each of 1049678, 105005–7, 1050009, 1050013–4, 1050016, 1050018–9, 1050029–50. Data generated by Sanger-based resequencing of the TSHR region, putative deletions and introduced stop codons have been submitted to GenBank under the accession numbers GU323512–GU323556.
References
Darwin, C. On the Origins of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life (Murray, 1859)
Darwin, C. The Variation of Animals and Plants under Domestication (Murray, 1868)
Andersson, L. Genetic dissection of phenotypic diversity in farm animals. Nature Rev. Genet. 2, 130–138 (2001)
Eriksson, J. et al. Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet. 4, e1000010 (2008)
Crawford, R. D. Poultry Breeding and Genetics (Elsevier Science, 1990)
International Chicken Polymorphism Map Consortium A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature 432, 717–722 (2004)
Havenstein, G. B., Ferket, P. R. & Qureshi, M. A. Growth, livability, and feed conversion of 1957 versus 2001 broilers when fed representative 1957 and 2001 broiler diets. Poult. Sci. 82, 1500–1508 (2003)
International Chicken Genome Sequencing Consortium Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004)
Tran, T. S., Kolodkin, A. L. & Bharadwaj, R. Semaphorin regulation of cellular morphology. Annu. Rev. Cell Dev. Biol. 23, 263–292 (2007)
Dobney, K. & Larson, G. Genetics and animal domestication: new windows on an elusive process. J. Zool. 269, 261–271 (2006)
Yoshimura, T. et al. Light-induced hormone conversion of T4 to T3 regulates photoperiodic response of gonads in birds. Nature 426, 178–181 (2003)
Nakao, N. et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature 452, 317–322 (2008)
Hanon, E. A. et al. Ancestral TSH mechanism signals summer in a photoperiodic mammal. Curr. Biol. 18, 1147–1152 (2008)
Messina, D. N. & Sonnhammer, E. L. L. DASher: a stand-alone protein sequence client for DAS, the Distributed Annotation System. Bioinformatics 25, 1333–1334 (2009)
Kerje, S. et al. The twofold difference in adult size between the red junglefowl and white leghorn chickens is largely explained by a limited number of QTLs. Anim. Genet. 34, 264–274 (2003)
Price, E. O. Animal Domestication and Behavior (CABI, 2002)
Shimada, M. et al. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396, 670–674 (1998)
Sewalem, A. et al. Mapping of quantitative trait loci for body weight at three, six, and nine weeks of age in a broiler layer cross. Poult. Sci. 81, 1775–1781 (2002)
Zhou, H. et al. Genome-wide linkage analysis to identify chromosomal regions affecting phenotypic traits in the chicken. I. Growth and average daily gain. Poult. Sci. 85, 1700–1711 (2006)
Ambo, M. et al. Quantitative trait loci for performance traits in a broiler × layer cross. Anim. Genet. 40, 200–208 (2009)
Stone, S. et al. TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum. Mol. Genet. 15, 2709–2720 (2006)
Chadt, A. et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nature Genet. 40, 1354–1359 (2008)
Peck, G. R. et al. Insulin-stimulated phosphorylation of the Rab GTPase-activating protein TBC1D1 regulates GLUT4 translocation. J. Biol. Chem. 284, 30016–30023 (2009)
Olson, M. V. When less is more: gene loss as an engine of evolutionary change. Am. J. Hum. Genet. 64, 18–23 (1999)
Lu, J. et al. The accumulation of deleterious mutations in rice genomes: a hypothesis on the cost of domestication. Trends Genet. 22, 126–131 (2006)
Agarwal, S. K., Cogburn, L. A. & Burnside, J. Dysfunctional growth hormone receptor in a strain of sex-linked dwarf chicken: evidence for a mutation in the intracellular domain. J. Endocrinol. 142, 427–434 (1994)
Jacobsson, L. et al. Many QTLs with minor additive effects are associated with a large difference in growth between two selection lines in chickens. Genet. Res. 86, 115–125 (2005)
Darvasi, A. Experimental strategies for the genetic dissection of complex traits in animal models. Nature Genet. 18, 19–24 (1998)
Dunnington, E. A. & Siegel, P. B. Long-term divergent selection for eight-week body weight in white Plymouth rock chickens. Poult. Sci. 75, 1168–1179 (1996)
Liljedahl, L.-E., Kolstad, N., Sørensen, P. & Maijala, K. Scandinavian selection and crossbreeding experiment with laying hens. I. Background and general outline. Acta Agric. Scand. 29, 273–286 (1979)
Cole, R. K. Hereditary hypothyroidism in the domestic fowl. Genetics 53, 1021–1033 (1966)
Hillel, J. et al. Biodiversity of 52 chicken populations assessed by microsatellite typing of DNA pools. Genet. Sel. Evol. 35, 533–557 (2003)
Carlborg, Ö. et al. Epistasis and the release of genetic variation during long-term selection. Nature Genet. 38, 418–420 (2006)
Karlsson, E. K. et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nature Genet. 39, 1321–1328 (2007)
Felsenstein, J. PHYLIP – Phylogeny inference package (version 3.2). Cladistics 5, 164–166 (1989)
Álvarez-Castro, J. M. & Carlborg, Ö. A unified model for functional and statistical epistasis and its application in quantitative trait loci analysis. Genetics 176, 1151–1167 (2007)
Le Rouzic, A. & Álvarez-Castro, J. M. Estimation of genetic effects and genotype-phenotype maps. Evol. Bioinform. Online 4, 225–235 (2008)
Rönnegård, L., Besnier, F. & Carlborg, Ö. An improved method for quantitative trait loci detection and identification of within-line segregation in F2 intercross designs. Genetics 178, 2315–2326 (2008)
Rozen, S. & Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386 (2000)
Ka, S. et al. Proviral integrations and expression of endogenous Avian leucosis virus during long term selection for high and low body weight in two chicken lines. Retrovirology 6, 68 (2009)
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the method. Methods 25, 402–408 (2001)
Acknowledgements
Thanks are due to J. Altimiras, J. Dodgson, F. Imsland and S. Kerje for providing genomic DNA samples. We would also like to thank E. Sonnhammar for help with the bioinformatic analysis of the TSHR sequence, the Uppsala Genome Center for SOLiD sequencing and the Uppsala Multidisciplinary Center for Advanced Computational Science for help with data storage and analysis. This work was supported by grants from the Swedish Foundation for Strategic Research, the Knut and Alice Wallenberg Foundation, the Swedish National Infrastructure for Computing and The Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning. K.L.-T. and Ö.C. are supported by European Young Investigator awards.
Author Contributions C.-J.R. was responsible for the selective-sweep analysis; M.C.Z. was responsible for detection of deletions and stop-codon mutations, and contributed to the selective-sweep analysis and SNP detection; J.E. was responsible for SNP verification and experimental validation of selective sweeps, J.R.S.M. was responsible for experimental validation of deletions and stop-codon mutations; E.S. and T.S. contributed to bioinformatic analysis; M.T.W. was responsible for the phylogenetic analysis; L.J. and M.I. contributed to SNP detection; S.K. and F.H. contributed to the experimental analysis of SH3RF2; F.B. and Ö.C. performed the QTL analysis of SH3RF2; B.B., M.T.B., P.J. and P.S. provided biological samples of critical importance; K.L.-T. contributed to the experimental design and analysis; and L.A. designed and coordinated the study. L.A. wrote the paper together with all co-authors, who contributed sections and edited drafts to yield the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A provisional patent application (no. 61/247,782) has been filed concerning the commercial use of some of the results presented in this paper.
Supplementary information
Supplementary Information
This file contains Supplementary Text and References, Supplementary Tables 1-9, and Supplementary Figures 1-4 with legends. (PDF 1847 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Rubin, CJ., Zody, M., Eriksson, J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010). https://doi.org/10.1038/nature08832
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature08832
This article is cited by
-
Genomic analysis of Nigerian indigenous chickens reveals their genetic diversity and adaptation to heat-stress
Scientific Reports (2024)
-
Genomic insights into Yak (Bos grunniens) adaptations for nutrient assimilation in high-altitudes
Scientific Reports (2024)
-
Archaeological and molecular evidence for ancient chickens in Central Asia
Nature Communications (2024)
-
Genomic insight into the influence of selection, crossbreeding, and geography on population structure in poultry
Genetics Selection Evolution (2023)
-
Integrating genomics and transcriptomics to identify candidate genes for high egg production in Wulong geese (Anser cygnoides orientalis)
BMC Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
