
Abstract
The cancer genome is moulded by the dual processes of somatic mutation and selection. Homozygous deletions in cancer genomes occur over recessive cancer genes, where they can confer selective growth advantage, and over fragile sites, where they are thought to reflect an increased local rate of DNA breakage. However, most homozygous deletions in cancer genomes are unexplained. Here we identified 2,428 somatic homozygous deletions in 746 cancer cell lines. These overlie 11% of protein-coding genes that, therefore, are not mandatory for survival of human cells. We derived structural signatures that distinguish between homozygous deletions over recessive cancer genes and fragile sites. Application to clusters of unexplained homozygous deletions suggests that many are in regions of inherent fragility, whereas a small subset overlies recessive cancer genes. The results illustrate how structural signatures can be used to distinguish between the influences of mutation and selection in cancer genomes. The extensive copy number, genotyping, sequence and expression data available for this large series of publicly available cancer cell lines renders them informative reagents for future studies of cancer biology and drug discovery.
Similar content being viewed by others


Main
The genome of a cancer cell usually carries several somatic changes1. A subset, termed ‘driver’ mutations, confer clonal growth advantage, are positively selected and are causally implicated in cancer development. The remainder are ‘passengers’.
A central challenge presented by catalogues of structural abnormalities from cancer genomes is distinguishing drivers from passengers. One approach is to assume that passenger mutations are randomly distributed, while drivers are restricted to a subset of genes. Thus mutational catalogues from several cancer samples will show non-random accumulation of drivers in certain genes. However, the underlying assumption that passengers are randomly distributed need not always be correct. A cluster of somatic mutations may also be attributable to an increased local mutation rate. Passenger mutations arising in these circumstances may be confused with drivers.
This problem may apply to any type of structural alteration. For example, clusters of base substitution somatic mutations occur in some lymphoid neoplasms owing to a hypermutation phenotype involved in the generation of immune diversity2,3. Similarly, small insertions and deletions occur at polynucleotide tracts in cancers with defective DNA mismatch repair. Some, within the coding exons of genes, have been proposed as drivers4,5,6. However, the presence of probable passenger insertions and deletions at similar frequencies in intronic and intergenic polynucleotide tracts renders this interpretation uncertain.
Distinguishing driver from passenger mutations has particularly compromised the search for somatically mutated recessive cancer genes (tumour suppressor genes). Large homozygous deletions (HDs) of recessive cancer genes result in their inactivation and are driver mutations. The identification of several recessive cancer genes including CDKN2A7, RB1 (ref. 8), SMAD4 (ref. 9), SMARCB1 (ref. 10), MAP2K4 (ref. 11) and PTEN12 has primarily depended on detection of HDs in cancer genomes. However, similar HDs in cancer genomes occur over common fragile sites—regions that show increased rates of chromosome breakage in normal cells in response to agents causing replicative stress13.
One approach to distinguish between HD clusters due to recessive cancer genes and fragile sites is to investigate the presence of alternative inactivating changes such as protein-truncating point mutations. These are expected in recessive cancer genes but not in fragile sites. However, the prevalence of truncating point mutations in recessive cancer genes is variable, limiting the utility of this approach. Here we have characterized copy number changes at high resolution in a large series of cancer cell lines and extracted structural features that facilitate discrimination between HDs over recessive cancer genes and fragile sites.
Genomic features of cancer cell lines
We analysed 746 publicly available cancer cell lines derived from many different cancer classes (Supplementary Table 1). The series includes most lines used in studies of cancer biology or cancer drug sensitivity, notably the NCI-60 set that has been exposed to tens of thousands of potential anti-cancer compounds.
DNA was hybridized to Affymetrix SNP6.0 arrays to extract information on genotype status and copy number changes. A total of 121,340 breakpoints were detected, each mapped to an average resolution of <5 kilobases (kb). These breakpoints mark the boundaries of segments of copy number increase (including amplifications), reduction (including hemizygous and homozygous deletions) and losses of heterozygosity (LOH) (Supplementary Figs 1–3 and Supplementary Table 2). We observed 1,198 individual regions of >10-fold amplification (Supplementary Table 3). Of these, 138 were over MYC, MYCN, ERBB2, MYCL1, EGFR, MDM2, AKT2, ALK, MITF and REL, cancer genes that are known to contribute to oncogenesis through amplification (http://www.sanger.ac.uk/genetics/CGP/Census/). The 746 cancer cell lines have also been sequenced for mutations in the coding exons of 46 known cancer genes and analysed for gene expression. In total, 1,753 putative oncogenic mutations were identified including 1,302 point mutations and 378 HDs in addition to the amplifications described earlier (Supplementary Tables 3–5; http://www.sanger.ac.uk/genetics/CGP/CellLines/, http://www.sanger.ac.uk/cgi-bin/genetics/CGP/cghviewer/CghHome.cgi).
The landscape of HDs in cancer genomes
The detection of HDs in primary cancers is compromised by the presence of non-neoplastic tissue. Cancer cell lines are free of this confounder, providing advantageous reagents with which to study the origins of HDs, even though further genomic alterations may have occurred during culture14.
A total of 6,058 HDs were observed. DNA from normal tissue from the same individual is not available for most lines, therefore it is not possible to evaluate directly whether either or both of the deletion events constituting a HD were somatically acquired or were present in the germ line. To address this, we analysed 466 DNAs from normal tissues (Supplementary Table 6) and derived filters discriminating between germline and somatic deletions.
We classified 3,630 HDs as germ line. These formed 205 clusters of two or more HDs and 109 singletons. In total, 23 megabases (Mb) of the reference human genome fell within HDs of germline origin, containing part or all of 246 (1%) protein-coding genes.
In total, 2,428 HDs were classified as somatic (Supplementary Table 7). In aggregate, these delete 519 Mb (17.3%) of the reference genome and contain part or all of the coding sequences of 2,369 (11%) protein-coding genes that are presumably not mandatory for the survival of human cells in tissue culture. Somatic HDs were ∼10-fold larger than those of germline origin (average size 510 kb compared to 49 kb).
The distribution of somatic HDs across the genome is illustrated in Fig. 1. On the basis of overlapping genomic footprints, 1,689 out of 2,428 (70%) fell into 206 clusters, and 739 (30%) were singletons overlapping no other deletion (Supplementary Fig. 4). We were able to attribute the likely underlying cause to 38 of the 206 (18%) clusters.

Deletion patterns in the 746 cancer cell lines across the autosomal chromosomes. The homozygous deletion (red) and small hemizygous deletion (blue) counts are calculated for 100-kb windows across the genome. All homozygous deletion clusters containing six or more homozygous deletions are annotated. Each tick-mark on the vertical axis denotes ten deletions, and the horizontal axis represents each chromosome from p-telomere to q-telomere.
Fourteen (including 389 HDs) were located over the known recessive cancer genes CDKN2A, PTEN, RB, SMAD4, NF1, MAP2K4, MSH2, TP53, NF2, MLH1, SMARCB1, PIK3R1, BMPR1A and CDH1 (Supplementary Tables 2, 4 and 7). CDKN2A (p16) exhibits more HDs than other recessive cancer genes (Fig. 1). This may be because it is adjacent to a large gene desert and therefore HDs are associated with less negative selection. However, it is also possible that inactivation of CDKN2B (which is adjacent to CDKN2A) and p14ARF (which shares an overlapping coding sequence) confers further selective advantage15. This combination of inactivated genes is more likely to occur through large deletions overlying CDKN2A than by point mutations in its coding sequence.
Nineteen of the 38 HD clusters (including 419 HDs) were over the 39 fragile sites whose genomic location has been fine-mapped (Supplementary Table 7). There was considerable variation in the activity of fragile sites in cancer genomes. For example, many HDs were observed over FRA2F, FRA3B, FRA4F, FRA5H and FRA16D, but few over FRA2B, FRA4B and FRA16C (Fig. 1).
Five of the 38 HD clusters (38 HDs) mapped over regions known to be somatically rearranged in lymphoid cells to generate immunological diversity. The deepest of these is over the T-cell-receptor-α gene on chromosome 14. Most HDs in these clusters (33 out of 38) are in cancers of haematological or lymphoid system origins.
There were 168 (82%) clusters (including 843 HDs) and 712 singletons that were ‘unexplained’. Some include large numbers of HDs (Fig. 1). The possibility that this distribution of unexplained HDs across the genome has been randomly generated was excluded by simulations (P < 0.0001).
Structural signatures of HD clusters
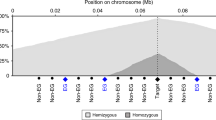
We compared the patterns of homozygous and hemizygous deletion over known recessive cancer genes and fragile sites (Fig. 1). For these analyses, hemizygous deletions were separated into two classes: small hemizygous deletions (less than 1 Mb) that require two DNA breakage events close to each other and therefore reflect local fragility, and large hemizygous deletions (greater than 1 Mb) that are due to several mechanisms including whole chromosome loss (Fig. 2).

At any locus each parental chromosome can have one of three deletion states: wild type (W); large deletion (>1 Mb, L); or small deletion (<1 Mb, S). Therefore, in principle, six combinations can exist in a diploid genome: WW, both chromosomes are wild type; WL, one chromosome has a large hemizygous deletion; WS, one chromosome has a small hemizygous deletion; LL, homozygous deletion in which both chromosomes have large overlapping deletions; LS, homozygous deletion in which one chromosome has a large deletion overlapping a small deletion in the other; SS, homozygous deletion in which both chromosomes have small overlapping deletions.
Over known recessive cancer genes there were 298 HDs and 70 small hemizygous deletions (ratio of 4.3). In contrast, over known fragile sites there were 193 HDs and 320 small hemizygous deletions (ratio of 0.6). The difference probably reflects the influences of selection and mutation rate at the two classes of locus. Over recessive cancer genes the rate of generation of small deletions is low and most do not confer selective growth advantage because the second allele is still wild type. Therefore, small hemizygous deletions over recessive cancer genes are rarely seen in cancer genomes. However, when converted into HDs by loss of the other allele they confer clonal selective advantage and thus manifest frequently.
In contrast, over fragile sites the rate of DNA breakage is high. Consequently, small hemizygous deletions are observed in cancers, despite the fact that they do not result in homozygous inactivation of an underlying gene. By chance, however, some are converted to HDs by a small or a large deletion on the other parental chromosome and the observed frequency of HDs reflects the rates of these two processes. The deepest unexplained HD clusters observed, on chromosomes 1 (72 Mb), 3 (118 Mb), 4 (181 Mb), 8 (5 Mb), 9 (12 Mb), 16 (6 Mb) and 20 (15 Mb), are all co-located with deep clusters of small hemizygous deletions (Fig. 1) and show a ratio of homozygous to hemizygous deletions (0.7) similar to fragile sites.
To explore these signatures further, we examined the nature of the two deletions that constitute each HD (Fig. 3). Over recessive cancer genes, most HDs were composed of a large deletion on one parental chromosome and a small deletion on the other. Only 5.7% were constituted of two small deletions. In contrast, over fragile sites 33.7% were constituted of two small deletions, a pattern consistent with an increased rate of local breakage. Over the deepest of the unexplained HD clusters, the fraction composed of two small deletions was 32.6%, similar to fragile sites.

Deletion clusters over known fragile sites, over known recessive cancer genes and that are unexplained are shown. The counts for small hemizygous deletions (blue), homozygous deletions arising from two small deletions (orange) and homozygous deletions with at least one large deletion (red) are given for the 100-kb window at the deepest point of each homozygous deletion cluster.
We also searched for truncating point mutations on the retained allele opposite small hemizygous deletions by sequencing the 746 cancer cell lines through 19 known recessive cancer genes, two genes that lie within fragile sites, and two genes that lie within clusters of unexplained HDs. In 25% (15 out of 59) of samples with a small hemizygous deletion over a recessive cancer gene, a truncating point mutation was found on the other allele. In contrast, in two genes that lie within known fragile sites, FHIT (FRA3B) and WWOX (FRA16D), we found no truncating point mutations in the retained allele opposite 149 small hemizygous deletions (P < 0.001). Two deep unexplained HD clusters fall completely within the genomic footprint of single genes, NEGR1 (chromosome (chr) 1, 72 Mb) and MACROD2 (chr 20, 15 Mb). Only one truncating point mutation was found in the retained allele opposite 87 small hemizygous deletions in these two genes (1%) a similar pattern to fragile sites (Supplementary Tables 8 and 9).
Thus, the deepest unexplained HD clusters bear structural features reminiscent of fragile sites. Some may represent fragile sites that have previously been localized cytogenetically but lack higher resolution mapping information. Others, however, are distant from any known fragile site and may reflect regions with a cancer-specific propensity for DNA double-strand breakage. To evaluate this possibility further we examined cytogenetically the genomic regions containing unexplained HD clusters on chromosomes 3, 4, 9, 16 and 20 for chromosome breakage in lymphoblasts under aphidicolin-induced replicative stress (Table 1). None showed evidence of fragility in this assay. Therefore, their fragility may be limited to the internal milieu of cancer cells.
Ranking homozygous deletion clusters
At any autosomal position in the genome each sample can be classified into one of six states (Fig. 2). The observed frequency of each state in the set of cell lines at a particular genomic position will depend on the local small and large deletion rates and the effects of positive and negative selection. To assess individual HD clusters, we constructed a statistical model that allows the small deletion rate ( ) and the selection pressure relative to the average for fragile sites (λ) to be estimated from the observed counts (Table 2 and Supplementary Table 7). These are indicators of local fragility and positive selection over recessive cancer genes respectively.
) and the selection pressure relative to the average for fragile sites (λ) to be estimated from the observed counts (Table 2 and Supplementary Table 7). These are indicators of local fragility and positive selection over recessive cancer genes respectively.
Deletion clusters over fragile sites generally showed a high rate of small deletion and low selection. Conversely, clusters over known recessive cancer genes had the opposite profile, a low deletion rate with high selection. The exception to this rule was TP53. This seems to be due to the coincidence of an exceptionally high prevalence of inactivating point mutations (63%), leading to selection of occasional small hemizygous deletions that occur on the other allele, coupled to a disproportionately small number of HDs over this gene.
A few unexplained HD clusters exhibited a signature indicative of a recessive cancer gene (Table 2). The three deepest HD clusters with this pattern were over TGFBR2, CDKN2C and CTNNA1 (Table 2 and Figs 1 and 3), all of which have been proposed previously as recessive cancer genes but for which the evidence is incomplete.
TGFBR2 encodes a cell-surface receptor for transforming growth factor-β (TGF-β). The TGF-β-regulated pathway is frequently inactivated in cancer and TGFBR2 often exhibits truncating somatic mutations in gastrointestinal cancers. However, these are usually deletions or insertions at polynucleotide tracts in tumours with defective DNA mismatch repair16, therefore their status as driver or passenger mutations has been unclear.
CDKN2C encodes p18, a cell-cycle control protein. We recently provided functional evidence for CDKN2C as a recessive cancer gene in glioma17. However, truncating point mutations in CDKN2C were not found previously18 and we only detected a single homozygous protein truncating variant among 786 cancer cell lines (Supplementary Table 9).
CTNNA1 encodes α-catenin, which interacts with CDH1, the product of a known recessive cancer gene involved in maintaining cell adhesion19. Two truncating point mutations in CTNNA1 have previously been reported20,21 together with biological evidence supporting its role as a recessive cancer gene22. We sequenced the coding exons of CTNNA1 through 746 cancer cell lines and 354 primary cancers. Only two proven somatic mutations were found, in a primary renal cell cancer and a breast cancer cell line, but both are predicted to cause premature protein truncation (Supplementary Table 9) and three further homozygous truncating variants were found among the cell lines. Thus, for all three genes the structural signatures of their HD clusters provide persuasive support for the otherwise inconclusive evidence that they function as recessive cancer genes.
Discussion
We have shown that simple structural signatures can provide clues to the dominant forces underlying clusters of HDs. The results provide supporting evidence for TGFBR2, CTNNA1 and CDKN2C as recessive cancer genes. However, many unexplained HDs in cancer genomes have signatures indicative of fragility. In support of this conclusion, some unexplained HD clusters overlie gene-free regions (for example, chr 16, 6 Mb) and others overlie genes with large genomic footprints (MACROD2, 2 Mb) a known feature of fragile sites. Indeed, recent analyses of fibroblasts treated with aphidicolin provide independent support for the fragility of some of the unexplained HD clusters reported here23.
Several genes located under unexplained HD clusters have been previously proposed as recessive cancer genes. For example, PTPRD partially overlaps the deepest unexplained HD cluster at chr 9 (12 Mb). An in vitro study has provided evidence of tumour-suppressor activity associated with PTPRD24. However, our results indicate that this region is fragile. Moreover, screening for truncating point mutations in PTPRD in hundreds of samples has shown only three heterozygous truncating and a few somatic missense variants24,25,26,27, and PTPRD knockout mice have the same cancer incidence as wild-type animals (N. Uetani and M. L. Tremblay, personal communication).
It is possible, however, that by coincidence some genes located in fragile regions may also confer clonal growth advantage and contribute to cancer development when inactivated by HDs. This has been proposed for FHIT (FRA3B) and WWOX (FRA16D) for which knockout mice show a modestly increased tumour burden at long latency28. We did not find evidence of more selection over FHIT and WWOX than over other fragile sites. However, it remains possible that the low levels of positive selection conferred by HDs over these genes are not detectable using our model. Moreover, our analysis applies exclusively to the selective advantage conferred by biallelic inactivation of classical recessive cancer genes. The conclusions do not pertain to putative haploinsufficient cancer genes or dominant cancer genes that might be activated through deletions. Indeed, DNA double-strand breaks at fragile sites may, by themselves, contribute to oncogenesis by triggering genomic instability, without any selective advantage conferred by inactivation of genes they have disrupted29.
The structural features that determine fragility and the underlying defects in cancer cells that result in the accumulation of deletions over fragile sites are not well understood30. ATR is involved in detection and repair of DNA damage occurring during replication of these regions, and absence of ATR31 or other components of DNA double-strand break repair32,33 leads to expression of fragile sites. Because some cancer genes are known to be implicated in DNA repair, we investigated whether fragility in the 746 cancer cell lines was associated with cancer-causing mutations in any of the 46 known cancer genes sequenced (Supplementary Table 4). We found no clear associations, although the gene set does not include ATR or many of its downstream targets.
The forces of mutation and selection that shape cancer genomes are complex. Mutational signatures remain clues that require cautious and informed interpretation in the light of all available information. Nevertheless, with complete cancer genome sequencing on a large scale, a realistic prospect over the next few years, increasingly informed and sophisticated structural signatures will provide the primary, pragmatic approach to the identification of driver mutations, after which they can be subjected to more detailed evaluation by functional experiments.
Methods Summary
Analysis was performed on 746 cancer cell lines (Supplementary Table 1 and http://www.sanger.ac.uk/genetics/CGP). Genotyping/copy number analysis was performed using Affymetrix Genome-Wide Human SNP Array 6.0 and expression analysis using the HT-HU133A expression array. Copy number and genotype analysis were performed using PICNIC34 (http://www.sanger.ac.uk/genetics/CGP/Software/PICNIC/), which uses hidden Markov models to allow estimation of absolute copy number of each parental copy at each point in the genome. The locations of fragile sites used in this study were obtained from the literature (Supplementary Table 10). Known cancer genes were screened by PCR amplification/sequencing and by multiplex PCR (Supplementary Table 11). Genome fragility was assessed in metaphases derived from two lymphoblastoid lines grown in the presence on 0.6 μM aphidicolin.
Online Methods
Samples
Analysis was performed on 746 cancer cell lines, most of which were sourced from public repositories, and 466 DNA samples from normal tissue (Supplementary Table 1 and http://www.sanger.ac.uk/genetics/CGP).
Mapping of fragile sites and known recessive cancer genes
In total, 127 fragile sites were identified from either the NCBI (http://www.ncbi.nlm.nih.gov/) or the literature (Supplementary Table 10), and 95 of these were classified as common fragile sites. All the fragile sites are mapped to cytogenetic bands, the sequence coordinates of which were obtained using Ensembl (http://www.ensembl.org/Homo_sapiens/Info/Index, release 52 (December 2008)) based on the NCBI 36 assembly of the human genome (November 2005).
Refined mapping positions were obtained from the literature for 38 of the common fragile sites mapping to the autosomal chromosomes. Positions of these sites were mapped onto the NCBI 36 assembly of the human genome using the mapping information (clones/genes) present in the relevant paper. Where possible, the extent of fragile sites was taken as those probes, distal and proximal to the fragile site that showed no indication of breakage in metaphase chromosomes when grown under conditions of replicative stress (see Supplementary Table 10 for a full list of the publications used). The criteria used here in defining the fragile sites may lead to a larger apparent footprint for some fragile sites for which refined mapping has localized the hotspot of fragility to a small genomic region (notably FRA3B and FRA16D). However, such mapping data are not present for all fragile sites. We have therefore used the wider definition to maintain consistency. However, restricting the definitions of well-mapped fragile sites to hotspots of fragility was not found to alter the results of the analysis. HD clusters were assigned to known fragile sites if they overlapped partially or completely with the interval of the fragile site.
The list of known recessive cancer genes was taken from the Cancer Gene Census (http://www.sanger.ac.uk/genetics/CGP/), and represents those genes known to be somatically mutated in cancer (n = 38). Mapping positions given were obtained from Ensembl (http://www.ensembl.org/Homo_sapiens/Info/Index, release 52 (December 2008)) based on the NCBI 36 assembly of the human genome (November 2005).
PCR and sequencing
PCR primers were designed to amplify the exons and flanking intronic sequences of the 46 cancer genes together with two genes from known fragile sites (FHIT in FRA3B and WWOX in FRA16D) and two genes from unexplained deletion clusters (NEGR1 and MACROD2). PCR products were 500 bp in length, with several overlapping amplimers for larger exons. PCR amplification of genomic DNA templates and bidirectional direct sequencing were done as described previously35.
Sequence traces were analysed using in-house software, AutoCSA35, followed by manual inspection of putative variants. All putative disease-causing mutations were confirmed by bidirectional sequencing of a second independently amplified PCR product. Matched normal cell lines were available for 40 of the cell lines. The somatic status of variants in these 40 cell lines was determined by sequencing DNA from the corresponding normal.
Classification of sequencing results for the 46 known cancer genes
Putative oncogenic mutations are classified as either likely oncogenic mutations (LOM) or tentative oncogenic variants (TOV). LOM are sequence changes that have previously been shown to be somatic mutations in human cancer and/or those consistent with the position and type of mutations for a given gene. This class includes homozygous and compound heterozygous inactivating mutations and splice site mutations affecting positions -1, -2, +1, +2 or +5 (where the wild-type nucleotide is a G) in tumour-suppressor genes. TOV are inactivating mutations present as heterozygous variants in tumour-suppressor genes (other than missense mutations in TP53 and FBXW7 that are included in the LOM class).
Multiplex PCR detection of HDs
Exon deletions in CDKN2A (p14, Ensembl transcript ID (build 52) ENST00000380150; p16, ENST00000304494), PTEN (ENST00000371953), RB1 (ENST00000267163), STK11 (ENST00000326873), MAP2K4 (ENST00000353533) and SMAD4 (previously known as MADH4; ENST00000342988) were identified by multiplex PCR. The primer sequences together with PCR conditions are given in Supplementary Table 11. Control PCR amplimers were designed for β-actin and random intergenic genomic sequences. PCR products were resolved on 2% agarose gels. All multiplex PCR experiments were done in duplicate.
Copy number and genotype analysis
The SNP6 Genome Wide Affymetrix arrays were segmented with a Bayesian hidden Markov model (PICNIC34, http://www.sanger.ac.uk/cgp/software/PICNIC). This enabled simultaneous identification of actual allelic copy numbers (including LOH) and genotypes.
Copy number polymorphism identification (filters)
DNA from normal tissue from the same individual is not available for most of the cancer cell lines, therefore it is not possible to evaluate directly whether either single hemizygous deletions or overlapping hemizygous deletions that give rise to a HD were somatically acquired or were present in the germ line. To address this, we used several different analyses to identify probable copy number polymorphisms (CNPs).
A series of 466 normal DNAs (Supplementary Table 1) was analysed and regions of copy number alteration of less than 1 Mb were classed as CNPs. CNPs were also taken from the ref. 36 CNP analysis of the 270 HapMap samples available from Affymetrix. These CNPs were remapped on to build 36 of the genome. Segments within the series of cancer cell lines that matched within ±5 features of one of these ‘known’ CNPs were flagged as CNPs. Segments of less than 1 Mb that occurred more than once with identical boundaries within the cancer cell line data set were also flagged as CNPs.
Some other segments within the data set were excluded from the analysis, although not classed as CNPs. These included regions that matched (±5 features) segments of less than 30 Mb that were scored as having copy neutral LOH within the normal samples. Also, segments of less than 1 Mb that shared (±1 feature) either the start or end of a previously identified CNP.
Array validation
To assess the effectiveness of the CNP filters, we examined 141 putative somatic HDs found in 40 cancer cell lines for which a lymphoblastoid cell line from the same individual is available. Only 10 (7%) were found in the lymphoblastoid cell lines, indicating that the filters are successful in limiting the extent of misclassification. These remaining CNPs are rare and dispersed through the genome, and hence do not significantly influence the analysis of HD clusters in cancer genomes.
The sensitivity and specificity of the Affymetrix SNP6 array for identification of HDs was determined against those HDs identified by multiplex PCR. In this analysis, most of the coding exons of six recessive cancer genes (CDKN2A, MAP2K4, PTEN, RB1, SMAD4 and STK11) together with the CDKN2C were screened through the series of 746 cancer cell lines (81 exons in total, of which 79 worked). Two multiplex PCR assays were also performed for each of four of the common fragile sites (FRA3B, FRA16D, FRA6E and FRA6F). The results from these genomic loci were compared against the HDs identified using the SNP6 array. Of the 398 HDs identified by multiplex PCR, 327 were also detected using the SNP6 array, giving a sensitivity of 82.2%. Of the 71 deletions not picked up by the SNP6 array, 24 (34%) were single exon deletions. Some deletions within the SNP6 data overlapping these loci were not identified in the multiplex analysis (n = 13), giving an estimate for specificity of 96.2%.
Classification of genomic amplifications
A genomic amplification was defined as a region containing one or more contiguous genomic segments defined by PICNIC34 that had a total copy number of ten or more. If an extended region of such segments was split by a genomic segment below total copy number of ten then this would be classed as two or more independent amplicons and counted separately.
Statistical significance of HD clustering
The significance of genomic clustering of HDs was assessed as follows. All deletions occurring within the footprint of clusters involving known fragile sites and recessive cancer genes were removed. The positions of all the deletions were randomized (ensuring that they did not overlap the removed known clusters). For each cluster of overlapping deletions (including occurrences of a single deletion), the genomic position of the 100-kb window that overlapped the maximum number of deletions was obtained, and the number of overlapping deletions recorded. This associated each cluster with an overlap statistic. This process was repeated approximately 1,000 times to obtain the null distribution of the overlap statistics. The significance of each overlap statistic for all the original clusters was then derived from this distribution. To identify the clusters with significant overlap, the Benjamini–Hochberg algorithm was used37 with a false discovery rate (FDR) of 0.1%.
Patterns of hemizygous and homozygous deletions
To analyse patterns of deletions (both hemizygous and homozygous deletions arising from two overlapping deletions) we classified deletions into two classes. These were small deletions less than 1 Mb in size and large deletions greater than 1 Mb that included loss of chromosome arms and whole chromosome loss. The cut-off was set at 1 Mb as it best differentiated known fragile sites from known recessive cancer genes (Fig. 2). Hemizygous deletions were defined as the loss of a genomic region from one parental chromosome giving rise to a region of loss of heterozygosity, and included the full range of such possible events from whole chromosome loss to small hemizygous deletions of less than 1 Mb. Consistent with this definition, copy number neutral loss of heterozygosity was also classed as a hemizygous deletion. However, such events did not contribute substantially to the overall results and most hemizygous deletions were associated with a reduction in copy number.
Modelling selection and mutation
Any cluster of homozygous or hemizygous deletions results from processes of selection and mutation. The mutation process will generate hemizygous deletions at a locus. In some samples both chromosomal copies at this locus will be deleted resulting in homozygous deletion. If the region in question does not contain a recessive cancer gene, for example, clusters arising owing to fragility, the ratio of homozygous to hemizygous deletion will be indicative of the expected value independent of selection. Conversely, the presence of a recessive cancer gene will mean that HDs will be observed in cancer more frequently than expected, resulting in an increase in this ratio. The relative increase in this ratio can be used to represent the strength of the selection at this locus. Although it is possible that haploinsufficiency may result in a degree of selection on hemizygous deletions at recessive cancer genes, we have made the simplifying assumption that selection at recessive cancer genes is on HDs. Thus, selection due to haploinsufficiency has not been modelled.
To investigate these effects in more detail we have introduced a model that resolves the deletion counts into measurements of mutation rate and relative selection pressure. In this context, a mutation rate represents the probability that a randomly chosen chromosome from the set of cancer cell lines will contain a deletion at a locus at completion of oncogenesis, and the selection pressure measures the relative increase in these rates compared to the expected. These counts were observed to be variable across the genomic footprint of individual clusters, so the 100-kb window within each cluster overlapping the maximum number of deletions was selected to represent the probable target of selection/fragility. The homozygous and hemizygous deletion totals were then obtained by counting the number of such deletions overlapping this window. To calculate selection pressure we took the average ratio of homozygous to hemizygous deletions over all fragile sites as a benchmark. The increase of the ratio of homozygous to hemizygous deletions of any cluster compared to this benchmark represents the relative selection at this cluster. However, this is complicated by two confounding factors. First, deletion rates differ depending on both the size of the deletion and the sample in question. To adjust for the size variation we considered two classes of deletion, depending on whether their size is smaller or larger than 1 Mb. This results in small and large deletion rates. These have mean values ( ), when averaged across samples. We capture the variation in these rates arising from sample differences with a dispersion parameter (ε). Second, although HDs are clearly positively selected by recessive cancer genes, we observed that very large HDs generally tend to be selected against, presumably due to inactivation of neighbouring genes that reduces cell viability. To account for this we incorporated an HD selection pressure (λ) and a cell-viability parameter (γ). We make the simplifying assumption that both of these measurements of selection are constant across all samples. Using this model the HD selection pressure (λ) and the small averaged deletion rate (
), when averaged across samples. We capture the variation in these rates arising from sample differences with a dispersion parameter (ε). Second, although HDs are clearly positively selected by recessive cancer genes, we observed that very large HDs generally tend to be selected against, presumably due to inactivation of neighbouring genes that reduces cell viability. To account for this we incorporated an HD selection pressure (λ) and a cell-viability parameter (γ). We make the simplifying assumption that both of these measurements of selection are constant across all samples. Using this model the HD selection pressure (λ) and the small averaged deletion rate ( ) can be estimated for any HD cluster as shown in Table 2 and Supplementary Table 7.
) can be estimated for any HD cluster as shown in Table 2 and Supplementary Table 7.
We now describe the model in more detail. We make the simplifying assumption that two chromosomes are present and each chromosome can contain one of three states: no deletion (w), a small deletion (s) or a large deletion (l). We use W, S and L to represent the probabilistic events, respectively. Therefore each sample can be classified into one of six categories. The number of samples in each category can then be calculated (Fig. 2).
We assume that  describes the probabilities that any single chromosome is wild type, has a large deletion, or a small deletion, respectively
describes the probabilities that any single chromosome is wild type, has a large deletion, or a small deletion, respectively  . These probabilities are unlikely to be constant across samples because different samples probably have different rates of deletion. We capture this variance by assuming these probabilities arise from a Dirichlet distribution:
. These probabilities are unlikely to be constant across samples because different samples probably have different rates of deletion. We capture this variance by assuming these probabilities arise from a Dirichlet distribution:

Here,  ,
, and
and  are the sample averaged deletion rates
are the sample averaged deletion rates  and ε is a dispersion parameter capturing the sample variance of these deletion rates.
and ε is a dispersion parameter capturing the sample variance of these deletion rates.
Then we construct the multinomial conditional likelihood:

Where we have:






The deletion rate parameters  ,
, and
and  are assumed to vary by cluster, reflecting the inherently distinct deletion rates across clusters. The dispersion parameter captures sample variation in these parameters and is assumed fixed across the genome.
are assumed to vary by cluster, reflecting the inherently distinct deletion rates across clusters. The dispersion parameter captures sample variation in these parameters and is assumed fixed across the genome.
To estimate the dispersion parameter, deletion counts from clusters over the (assumed) selective neutral fragile sites were calculated and the empirical Bayes estimate of ε was calculated. This was assumed to be constant in all subsequent analyses.
To extend this model to incorporate selection we proceed as follows. If C denotes the event that a particular deletion type results in an observed cancer sample, we assume that: ,
,  ,
,  ,
,  ,
,  and
and  .
.
The selection pressure parameter λ models the increased likelihood of observing HDs at recessive cancer gene loci. The viability parameter γ models selection against large deletions in the genome, in which the increased size of HDs may disrupt a plethora of gene functions and negatively affect cell viability. Both these parameters are unity under a neutral model of evolution. Then a Bayes law inversion gives us, for each cluster:

The parameters were given uninformative priors and the maximum a priori estimates and confidences were obtained from 100,000 Metropolis–Hastings Markov chain Monte Carlo iterations.
In particular, this produces the small deletion rate and HD selection pressure for each cluster, as displayed in Table 2 and Supplementary Table 7.
Analysis of truncating point mutations
The observed counts of truncating point mutations of the known tumour suppressor genes were compared to the mutation counts of the WWOX and FHIT genes within fragile sites (allowing for differences in hemizygous status) using an exact Cochran–Mantel–Haenszel test.
Estimation of fragility in the presence of aphidicolin
Mapping of new fragile sites was performed on two Epstein-Barr virus (EBV)-transformed lymphoblast cell lines (GM10851 and GM15510), obtained from Coriell Cell Repositories. Cells were grown in RPMI medium 1640 supplemented with 16% fetal calf serum, 100 U ml-1 penicillin, 100 μg ml-1 streptomycin. The induction of fragile sites and metaphase preparation essentially followed ref. 38. In brief, 24 h before collection the DNA polymerase inhibitor aphidicolin (Sigma) was added to cell cultures in the presence of 0.5% ethanol at final concentrations of 0.2, 0.4 and 0.6 μM. Chromosome spreads were prepared according to the conventional cytogenetic techniques.
Human bacterial artificial chromosomes (BACs) corresponding to unexplained HD clusters and control fragile sites were chosen according to their positions in the hg17 genome sequence assembly using the UCSC Genome Browser. All BAC clones were provided by the clone archive team of the Wellcome Trust Sanger Institute. BAC DNA was extracted using the PhasePrep BAC DNA kit (Sigma) following the protocols provided by the manufacturer.
Probe labelling, and FISH were carried out as described previously39,40. In brief, 10–20 ng DNA from each BAC clone was first subject to a round of amplification using a whole-genome amplification kit WGA2, (Sigma), then labelled with Cy3-, Cy5-dUTP (GE Healthcare), Spectrum Green-dUTP (Abbott Molecular) or Chromatide Texas Red-12-dUTP (Invitrogen) using a modified whole-genome reamplification kit without dNTP WGA3 (Sigma). Approximately 100 ng of each labelled probe was used in multi-colour FISH experiments. After post-hybridization washes, slides were mounted with SlowFade Gold mounting solution containing DAPI (4′,6-diamidino-2-phenylindole, Invitrogen). Metaphases were examined with a Zeiss Axioplan Imaging microscope. Images were captured and processed with the Smart Capture software (Digital Scientific).
References
Stratton, M. R., Campbell, P. J. & Futreal, P. A. The cancer genome. Nature 458, 719–724 (2009)
Pasqualucci, L. et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 412, 341–346 (2001)
Liu, M. et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841–845 (2008)
Rampino, N. et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275, 967–969 (1997)
Bader, S. et al. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene 18, 8044–8047 (1999)
Melo, S. A. et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nature Genet. 41, 365–370 (2009)
Kamb, A. et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nature Genet. 8, 22–26 (1994)
Fung, Y.-K. T. et al. Structural evidence for the authenticity of the human retinoblastoma gene. Science 236, 1657–1661 (1987)
Thiagalingam, S. et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nature Genet. 13, 343–346 (1996)
Versteege, I. et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206 (1998)
Teng, D. H. F. et al. Human mitogen-activated protein kinase kinase 4 as a candidate tumor suppressor. Cancer Res. 57, 4177–4182 (1997)
Li, J. et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997)
Lukusa, T. & Fryns, J. P. Human chromosome fragility. Biochim. Biophys. Acta 1779, 3–16 (2008)
Solomon, D. A. et al. Sample type bias in the analysis of cancer genomes. Cancer Res. 69, 5630–5633 (2009)
Krimpenfort, P. et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a . Nature 448, 943–946 (2007)
Markowitz, S. et al. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 268, 1336–1338 (1995)
Wiedemeyer, R. et al. Feedback circuit among INK4 tumor suppressors constrains human glioblastoma development. Cancer Cell 13, 355–364 (2008)
Solomon, D. A., Kim, J.-S., Jean, W. & Waldman, T. Conspirators in a capital crime: co-deletion of p18INK4c and p16INK4a/p14ARF/p15INK4b in glioblastoma multiforme. Cancer Res. 68, 8657–8660 (2008)
Ozawa, M., Baribault, H. & Kemler, R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 8, 1711–1717 (1989)
Vermeulen, S. J. et al. The αE-catenin gene (CTNNA1) acts as an invasion-suppressor gene in human colon cancer cells. Oncogene 18, 905–915 (1999)
Bullions, L. C., Notterman, D. A., Chung, L. S. & Levine, A. J. Expression of wild-type α-catenin protein in cells with a mutant α-catenin gene restores both growth regulation and tumor suppressor activities. Mol. Cell. Biol. 17, 4501–4508 (1997)
Liu, T. X. et al. Chromosome 5q deletion and epigenetic suppression of the gene encoding α-catenin (CTNNA1) in myeloid cell transformation. Nature Med. 13, 78–83 (2006)
Arlt, M. F. et al. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 84, 339–350 (2009)
Solomon, D. A. et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 68, 10300–10306 (2008)
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008)
Weir, B. A. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898 (2007)
Sjöblom, T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006)
Iliopoulos, D. et al. Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett. 232, 27–36 (2006)
Gorgoulis, V. G. et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 (2005)
Ried, K. et al. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet. 9, 1651–1663 (2000)
Casper, A. M., Nghiem, P., Arlt, M. F. & Glover, T. W. ATR regulates fragile site stability. Cell 111, 779–789 (2002)
Freudenreich, C. H. Chromosome fragility: molecular mechanisms and cellular consequences. Front. Biosci. 12, 4911–4924 (2007)
Schwartz, M. et al. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 19, 2715–2726 (2005)
Greenman, C. D. et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics 11, 164–175 (2010)
Dicks, E. et al. AutoCSA, an algorithm for high throughput DNA sequence variant detection in cancer genomes. Bioinformatics 23, 1689–1691 (2007)
McCarroll, S. A. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genet. 40, 1166–1174 (2008)
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995)
Sawińska, M. et al. Novel aphidicolin-inducible common fragile site FRA9G maps to 9p22.2, within the C9orf39 gene. Genes Chromosom. Cancer 46, 991–999 (2007)
Freeman, J. L. et al. Definition of the zebrafish genome using flow cytometry and cytogenetic mapping. BMC Genomics 8, 195 (2007)
Perry, G. H. et al. Copy number variation and evolution in humans and chimpanzees. Genome Res. 18, 1698–1710 (2008)
Acknowledgements
We would like to acknowledge the support of the Wellcome Trust under grant reference 077012/Z/05/Z.
Author Contributions Experiments were designed by G.R.B., P.A.F. and M.R.S. Experiments were performed and overseen by G.R.B., H.D., S.E., J.M.A., G.B., L.C., C.L., S.W., C.F., B.F., G.L.D., P.D. and F.Y. Data were analysed by G.R.B., C.D.G., H.D., A.P.B., L.C., D.B., J.H. and M.R.S. B.T.T. supplied samples. The manuscript was written by M.R.S., G.R.B., C.D.G., P.J.C. and P.A.F.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-4 with Legends, Supplementary Tables 1-11 and Supplementary References. (PDF 8597 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Bignell, G., Greenman, C., Davies, H. et al. Signatures of mutation and selection in the cancer genome. Nature 463, 893–898 (2010). https://doi.org/10.1038/nature08768
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature08768
This article is cited by
-
Pan-cancer genomic analysis shows hemizygous PTEN loss tumors are associated with immune evasion and poor outcome
Scientific Reports (2023)
-
Replication stress causes delayed mitotic entry and chromosome 12 fragility at the ANKS1B large neuronal gene in human induced pluripotent stem cells
Chromosome Research (2023)
-
Co-dependent regulation of p-BRAF and potassium channel KCNMA1 levels drives glioma progression
Cellular and Molecular Life Sciences (2023)
-
Novel insights into the pathogenesis of follicular lymphoma by molecular profiling of localized and systemic disease forms
Leukemia (2023)
-
NRG1 fusions in breast cancer
Breast Cancer Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
