
Abstract
The human malaria parasite Plasmodium vivax is responsible for 25–40% of the ∼515 million annual cases of malaria worldwide. Although seldom fatal, the parasite elicits severe and incapacitating clinical symptoms and often causes relapses months after a primary infection has cleared. Despite its importance as a major human pathogen, P. vivax is little studied because it cannot be propagated continuously in the laboratory except in non-human primates. We sequenced the genome of P. vivax to shed light on its distinctive biological features, and as a means to drive development of new drugs and vaccines. Here we describe the synteny and isochore structure of P. vivax chromosomes, and show that the parasite resembles other malaria parasites in gene content and metabolic potential, but possesses novel gene families and potential alternative invasion pathways not recognized previously. Completion of the P. vivax genome provides the scientific community with a valuable resource that can be used to advance investigation into this neglected species.
Similar content being viewed by others

Main
Plasmodium vivax is the major cause of malaria outside Africa, mainly afflicting Asia and the Americas1. Principally a disease affecting poor people living on the margins of developing economies, vivax malaria traps many societies in a relentless cycle of poverty. Intermittent transmission makes protective immunity rare, and the disease strikes all ages. Repeated acute febrile episodes of debilitating intensity can occur for months. In children this can lead to life-long learning impairment, whereas incapacitation of adults has tremendous direct economic consequences through lost productivity and depletion of meagre financial reserves. Drug resistance in P. vivax is spreading, hindering management of clinical cases, and reports of severe pathology, including respiratory distress and coma, are challenging the description of P. vivax malaria as ‘benign’ (ref. 1).
Several biological characteristics underlie the distinct pathogenic and epidemiological nature of vivax malaria. In contrast to P. falciparum, P. vivax is only capable of infecting reticulocytes, causing severe anaemia by dyserythropoiesis and destruction of infected and uninfected erythrocytes despite much lower parasitaemias. P. vivax cannot infect Duffy-blood-group-negative reticulocytes (a trait shared with the closely related monkey malaria parasite P. knowlesi), and is thus absent from West Africa where Duffy negativity predominates2. Differences in Anopheles mosquito dynamics allow P. vivax transmission in temperate climates not tolerated by P. falciparum. In such regions P. vivax infects hepatocytes but may persist as dormant hypnozoites for months or years before initiating blood-stage infections (relapses) during another transmission season.
Because P. vivax kills infrequently and is not amenable to continuous in vitro culture, it has been relatively little studied in comparison to P. falciparum. The P. vivax genome sequence we report here, and comparative analyses with sequenced malaria parasites P. falciparum3, the rodent parasite P. yoelii yoelii4,5, and the primate parasite P. knowlesi6 (an excellent model for in vivo studies of human malaria), provide important insights into the biology of this neglected parasite.
Genome sequencing and characteristics
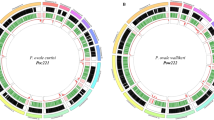
The ∼26.8-megabase (Mb) nuclear genome sequence of P. vivax (Salvador I) was sequenced by whole-genome shotgun methods to tenfold coverage, with targeted gap closure and finishing, and manual curation of automated annotation. Details of these and other methods are given in Supplementary Information. Large contigs totalling ∼22.6 Mb were assigned to the 14 P. vivax chromosomes; ∼4.3 Mb of small subtelomeric contigs remain unassigned due to their repetitive nature (Supplementary Table 1). P. vivax chromosomes are unique among human Plasmodium species in exhibiting a form of isochore structure7, with subtelomeric regions of low G+C content and chromosome internal regions of significantly higher G+C content. We finished the subtelomeric ends of several P. vivax chromosomes, allowing us to define their isochore boundaries (Fig. 1).

To view a larger version of this image download the pdf (2.27MB).
Putative orthologues were computed between P. falciparum (Pf), P. vivax (Pv), P. knowlesi (Pk) and P. y. yoelii (Py) proteomes and used to define blocks of synteny (shaded regions) between Py–Pk, Pv–Pk and Pv–Pf chromosomes. Genes on contigs that could not be assigned to chromosomes are not shown (see Supplementary Information). The composite rodent malaria parasite (cRMP) chromosomes generated in ref 18 are shown. Plots below the Pv chromosomes display the following: MS, the position of polymorphic microsatellites; (G+C)-skew, the base composition ((G-C)/(G+C)) within each strand in a 5-kb window; G+C, the percentage of bases in a 2-kb window that are G and C (%). Tracks within the Pv–Pk bracket show two evolutionary parameters: dS (number of synonymous substitutions per synonymous site; top) and ω (ratio of dN (non-synonymous substitutions per non-synonymous site) to dS; bottom). Horizontal red lines on the G+C (%), dS and ω tracks indicate the average value for that chromosome. a–d, Distribution of selective constraints (ω) for the gene ontology classifications biological process (a), molecular function (b) and cellular component (c). Selective constraint is also shown for several motifs (d): proteins containing predicted transmembrane domains (TM) and/or signal peptides (SP); GPI-anchored proteins; and proteins predicted to be exported (exportome). Each grey box represents the interquartile range, which contains the sample’s 25% to 75% range (quartiles Q1 to Q3, respectively), and the median is indicated (black horizontal bar within the interquartile range). Horizontal tick marks outside of the interquartile range show the range of all elements within Q1 - 1.5 × interquartile range and Q3 + 1.5 × interquartile range (∼99.3% interval of a normal distribution).
In many aspects, the genomes of mammalian Plasmodium species (P. falciparum, P. knowlesi, P. vivax, P. y. yoelii) are uniform, ranging from 23 to 27 Mb across 14 chromosomes, and comprising ∼5,500 genes, most of which (∼51%) contain at least one intron (Table 1). However, differences in nucleotide bias can be extreme (for example, P. vivax and P. falciparum average a G+C content of ∼42.3% and ∼19.4%, respectively), and a large gene family found in P. y. yoelii raised its gene count to ∼5,880 (ref. 4). A remarkable 77% of genes are orthologous between the four species (Supplementary Fig. 1); almost one-half of these encode conserved hypothetical proteins of unknown function. In P. falciparum, the high incidence of tandem repeats and low complexity regions (LCRs) in proteins, especially antigens, has led researchers to propose that LCRs are involved in immune evasion mechanisms, such as antigen diversification8 and reducing the host’s antibody response to critical epitopes by acting as a ‘smokescreen’9. We found that LCRs tend to constitute a smaller proportion of P. vivax proteins on average (39%) than P. falciparum proteins (60%; Supplementary Fig. 2), and that LCR expansion partly accounts for the slightly larger size of P. falciparum proteins (Supplementary Table 2), but how this relates to differences in immune evasion mechanisms between P. vivax and P. falciparum is unclear.

Notwithstanding the recent functional characterization of the apicomplexan AP2 family of transcriptional regulators in Plasmodium10, the parasite seems to lack most of the standard eukaryotic transcriptional machinery, such as transcription-associated proteins (TAPs)11, but is rich in regulatory sequences12, fostering the idea that gene expression regulation in Plasmodium is complex and unusual. Our initial studies found no significant differences in the TAP repertoire between P. falciparum, P. vivax and P. knowlesi, indicating that transcriptional mechanisms are similar in all three species (Supplementary Table 3). Genes encoding messenger RNA stability proteins containing a CCCH-zinc finger were abundant in all three species, affirming the importance of post-transcriptional regulation in the control of gene expression across Plasmodium. A genome scan of P. vivax for known core promoter elements such as TATA and CAAT boxes identified some candidates, but many of them lacked positional specificity. Similarly, a search for novel promoter elements in regions upstream of ∼1,800 mapped transcription start sites (5′ UTRs), and for RNA binding elements in ∼1,300 3′ UTRs, also failed to produce convincing candidates (data not shown). To determine whether binding sites are conserved between P. falciparum and other primate Plasmodium species, we searched for over-represented nucleotide ‘words’ in regions upstream of clusters of potentially co-regulated genes conserved in P. vivax, P. falciparum, P. knowlesi and P. y. yoelii (Supplementary Information). Seven putative novel regulatory binding sites conserved across at least two species were identified (Supplementary Table 4). These binding sites were associated with core eukaryotic processes such as dephosphorylation and with parasite-specific functions such as cell invasion. Independent support for two of our predicted sites comes from a recent report of the sporozoite-associated motif 5′-TGCATGCA-3′ and the merozoite invasion-related 5′-GTGTGCACAC-3′ motif13. In our analysis these two sites, together with the dephosphorylation-associated motif 5′-GCACGCGTGC-3′, were conserved across the four Plasmodium species.
Examination of natural parasite population structure is key to understanding transmission dynamics, the spread of drug resistance, and to design and test malaria control efforts. Many population studies have exploited the abundant polymorphic microsatellites in the P. falciparum genome, primarily simple sequence repeats such as [TA] dinucleotide and polyA/polyT14. We screened the P. vivax genome for microsatellites, identifying ∼160 that are polymorphic between eight P. vivax laboratory lines (Fig. 1 and Supplementary Table 5). P. vivax microsatellites average 27.5% G+C, with an average repeat unit length of 3.1 nucleotides and an average copy number of 19.1. We found fewer microsatellites in P. vivax than in P. falciparum (as noted previously15), probably due to the more conventional nucleotide composition of the former. Even so, these genome-wide polymorphic markers are already facilitating studies of P. vivax population structure and genetic diversity16,17.
Chromosome synteny and genome evolution
Previous studies have indicated significant conservation of gene synteny between Plasmodium parasites4 in direct proportion to their genetic distance. We generated a synteny map of P. vivax, P. knowlesi, P. falciparum, and the rodent malaria parasites P. y. yoelii, P. berghei and P. chabaudi (considered as a single lineage18 owing to their virtually complete synteny; Fig. 1). The P. vivax and P. knowlesi chromosomes are highly syntenic except for microsyntenic breaks at species-specific genes (in particular the P. knowlesi kir and SICAvar genes; see ref. 6); a previous study identified such breaks as foci for the evolution of host–parasite interaction genes18. The karyotypes of P. vivax and P. knowlesi correspond to the most parsimonious reconstruction of the ancestral form of the six species; the karyotypes of P. falciparum and the rodent malaria parasites can be reconstructed from this form through nine and six chromosomal rearrangements, respectively (Supplementary Fig. 3). No ‘hotspots’ of synteny breakage were identified, indicating that intersyntenic breakpoints were not ‘reused’ during the divergence of the species, and no obvious motifs except for (A+T)-rich regions and LCRs were identified in regions of the P. vivax genome predicted to have recombined to give single P. falciparum chromosomes. Of the 3,336 orthologues between all six species, 3,305 (99%) were found to be positionally conserved (Supplementary Table 6).
We used 3,322 high-quality P. vivax/P. knowlesi orthologues to obtain maximum likelihood estimates of the rate of substitution at synonymous (dS) and non-synonymous (dN) sites, as well as ω (dN/dS; Supplementary Table 7 and Fig. 1). P. vivax chromosomes differ significantly in their average values for both dS and dN, but the two variables are strongly correlated within and between chromosomes (Supplementary Fig. 4). The chromosomes also differ significantly in average %GC4 (the G+C content in third codon positions of fourfold degenerate amino acids). This variable is positively correlated with average dS and inversely correlated with chromosome length, such that synonymous sites in genes on the smallest chromosomes (∼1 Mb) evolve ∼1.5 times faster than genes on the two largest (∼3 Mb) chromosomes (Supplementary Fig. 5). These observations strongly indicate the existence of heterogeneous mutation rates across the genome. It is unclear if this is due to cytosine-to-thymine deamination, which is more probable in (G+C)-rich regions, as it is not known whether DNA methylation occurs in P. vivax. The degree of selective constraint (ω) also varies across classes of genes. Genes encoding glycosylphosphatidylinositol (GPI)-anchored proteins, cell adhesion proteins, exportome proteins (proteins predicted to be exported) and proteins with transmembrane or signal peptide motifs, all of which are at least partly extracellular, were found to evolve significantly faster than genes involved in, for example, carbohydrate metabolism, enzyme regulation and cell structure (Fig. 1a–d and Supplementary Table 8). The host immune system, by targeting extracellular peptides, seems to have strongly influenced evolutionary rate variation between gene classes in Plasmodium.
A highly conserved Plasmodium metabolome
We found that key metabolic pathways, housekeeping functions and the repertoire of predicted membrane transporters are highly conserved between the P. vivax and P. falciparum3 proteomes (Supplementary Table 9), suggesting that the two species have much the same metabolic potential. Conservation of metabolic processes also extends to the apicoplast, an apicomplexan plastid secondarily acquired from an ancient cyanobacterium. The apicoplast has lost photosynthetic function, but is essential to the parasite’s metabolism, hosting nuclear-encoded proteins that are targeted to the apicoplast lumen by a conserved bipartite amino-terminal presequence. The complete genome sequence of P. vivax offers an opportunity to update and improve the apicoplast proteome that was predicted in silico3. Apicoplast-targeted proteins conserved in P. vivax participate in major metabolic processes previously recognized in P. falciparum19, such as complete type II fatty acid synthesis, isopentenyl diphosphate and iron sulphur cluster assembly pathways, and a fragmented haem synthesis pathway distributed between the apicoplast and mitochondria. Conservation of these pathways in P. vivax is important because synthetic pathways for type II fatty acid synthesis and isopentenyl diphosphates are targets for antimalarial chemotherapeutics20. The revised Plasmodium apicoplast proteome (Supplementary Table 10) also clarifies the localization of two important processes. We show thiamine pyrophosphate biosynthesis, previously thought to take place in the apicoplast19, to be cytosolic. Conversely, we confirm a glyoxalase pathway in the apicoplast, with glyoxalase I and glyoxalase II enzymes being targeted there21; both enzymes are potential drug targets. Thus, comparison of overall apicoplast metabolic capabilities shows very few differences between P. vivax and P. falciparum.
P. vivax can form hypnozoites, a latent hepatic stage responsible for patent parasitaemia relapses months or even years after an initial mosquito-induced infection22. Hypnozoites survive most drugs that kill blood-stage parasites; complete elimination of P. vivax infections (radical cure) requires primaquine, the only licensed drug that can kill hypnozoite stages. However, resistance to the drug is spreading23, and its use is contra-indicated in pregnant women or patients with glucose-6-phosphate dehydrogenase deficiency, which is common in malaria-endemic regions. After an initial examination of P. vivax-specific proteins failed to identify leads (Supplementary Table 11), we hypothesized that the genetic switch for hypnozoite formation may involve P. vivax homologues of dormancy genes. Analysis of the predicted P. vivax proteome revealed some candidates (Supplementary Table 12). However such an association remains speculative, and investigation of hypnozoite formation and activation will require continued development of in vitro systems for culturing P. vivax liver stages24.
Gene families shape Plasmodium biology
Plasmodium lineages display differential gene family expansion that has shaped the specific biology of each species. Phenotypes illustrating this include parasite invasion of red blood cells and antigenic variation. Invasion of erythrocytes by extracellular Plasmodium merozoites, crucial to the development of malaria in an infected individual, depends on specific interactions between merozoite ligands and erythrocyte surface receptors (Fig. 2). Plasmodium species-specific mechanisms act mostly during the preliminary phases of invasion (for example, merozoite attachment and orientation). In P. vivax, but not P. falciparum, invasion is restricted to Duffy-positive reticulocytes2. P. vivax Duffy-binding protein (DBP25) and reticulocyte-binding proteins (RBPs26) are the archetypes of two distinct Plasmodium families of cell-binding proteins involved in erythrocyte selection (referred to as the Duffy-binding-like (DBL) and reticulocyte-binding-like (RBL) families, respectively). Homologues of rbp1 and rbp2, two genes originally identified in P. vivax, include the P. falciparum rh/nbp genes (reviewed in ref. 27) and the Py235 family in P. y. yoelii (reviewed in ref. 28). Unexpectedly, we identified additional rbp genes in the P. vivax genome (Supplementary Table 13), including multiple rbp2 genes, which could provide P. vivax with a diversity of invasion mechanisms comparable to that of P. falciparum. This finding dispels a view that P. vivax has a relatively uncomplicated erythrocyte invasion mechanism. Instead, P. vivax probably has alternative invasion pathways, as differential expression of rbp homologues in P. falciparum29 and P. y. yoelii30 is closely linked to switching of invasion pathways (Fig. 2). All rbp2 loci occur in the subtelomeric regions of P. vivax chromosomes: non-syntenic, dynamic regions of the genome in which species-specific genes are generated (Supplementary Fig. 6).

RBL and DBL invasion families predicted from several Plasmodium proteomes are shown above a Plasmodium merozoite colliding and re-orientating on the red blood cell surface. Species-specific RBL families interact with an array of species-specific DBL proteins that use both alternative (crossed arrows) and fixed (straight arrows) pathways with known or predicted receptors on the surface of erythrocytes. Blocking these receptor–ligand interactions offers a potential mechanism to prevent clinical malaria. DARC, Duffy antigen receptor for chemokine-dependent (+) and -independent (-) pathways; GPA/B/C, P. falciparum glycophorin A/B/C receptors; RH SA+/-, rhesus sialic-acid-dependent (+) and -independent (-) pathways; ‘X’, ‘Y’, predicted receptors. The asterisk indicates that the presence of this pathway is controversial.
The final phase of invasion, merozoite entry into an intraerythrocytic vacuole, uses an intracytoplasmic molecular motor (components of which are highly conserved between Plasmodium species) coupled to simultaneous shedding of crucial merozoite surface proteins (MSPs). There are at least ten distinct MSPs (Supplementary Table 14), and P. vivax genome analysis reveals two particularly interesting MSP families, MSP3 and MSP7. Eleven members of the msp3 gene family occur in tandem on a ∼60-kb region of P. vivax chromosome 10 (Supplementary Fig. 7), and show weak similarity to four msp3 gene family members on P. falciparum chromosome 10 and to two P. knowlesi msp3 genes located on different chromosomes. Thus, there has been a significant expansion of the msp3 gene family in P. vivax, perhaps as a means to enhance immune evasion, as P. falciparum and P. vivax msp3 gene family members have been shown to be antigenic and to partially immunize non-human primates against blood-stage parasites31. In P. falciparum, MSP6 (a member of the MSP3 family that lacks heptad repeats) non-covalently binds with MSP1, but there is no counterpart to MSP6 in P. vivax. MSP7, another P. falciparum antigen that binds to MSP1 on the surface of merozoites, has also been expanded in P. vivax, with eleven copies on chromosome 12, compared to six and three members in P. falciparum and P. y. yoelii, respectively; it is not known if any P. vivax MSP7 proteins bind to MSP1.
The surface coats of merozoites and extracellular forms of Plasmodium parasites are composed largely of GPI-anchored proteins, many of which are important targets of protective immune responses and thus constitute promising vaccine candidates. When we predicted the GPI-anchored proteome of P. vivax and compared it to validated P. falciparum GPI-anchored proteins32, 29 of the 30 GPI-anchored proteins identified in P. falciparum had counterparts in P. vivax (Supplementary Fig. 8), an extraordinary level of conservation. MSP2 (the second most abundant merozoite surface protein in P. falciparum) is absent in the P. vivax genome, and P. vivax contains one additional GPI-anchored protein that seems to be a member of the ‘six cysteine’ apicomplexan-specific gene family33. Both the P. vivax and P. knowlesi genomes encode an apparently paralogous gene next to msp1, which is the largest and most abundant protein on the P. falciparum merozoite surface. P. vivax MAP1 is not closely related to MSP1 (11% identity, 22% similarity), although their sizes, a predicted GPI-attachment site, and structural features such as a carboxy-terminal double EGF module are similar.
A second notable parasite phenotype is antigenic variation: the ability to vary surface proteins during the course of an infection to evade the host’s immune response. In P. falciparum, antigenic variation is mediated by species-specific gene families such as var, members of which are expressed clonally and regulated epigenetically34. In P. vivax, the largest multigene family vir, part of the pir (Plasmodium interspersed repeats) superfamily found in several Plasmodium species5, has been implicated in antigenic variation; 35 gene copies were previously identified35. We identified 346 vir genes in the P. vivax genome located within (A+T)-rich subtelomeric regions of chromosomes (Fig. 1). Structurally, vir genes vary greatly, ranging from 156 to 2,316 bp in length and containing 1–5 exons. VIR proteins were previously classified into six subfamilies (A–F) on the basis of sequence similarity35, and representatives of these subfamilies were identified in patient isolates36. Clustering the VIRs in the Salvador I genome yielded six new subfamilies (G–L) and we confirmed gene expression for several of these in natural infections (Supplementary Table 15). Motif analysis of the total VIR repertoire (Fig. 3) showed that approximately one-half (171) contain a transmembrane domain, and one-half (160) contain a motif similar to the PEXEL/VSP sequence linked to export of parasite proteins37,38. Introns from 25 vir genes contain a conserved motif proximal to the donor splice site, suggesting possible functionality of the sequence in the control of vir gene expression, as has been shown for P. falciparum var introns39. Motif-shuffling among the sequences is apparent, particularly among large VIR proteins that have undergone an expansion of some motifs at the amino terminus. Similarly to P. falciparum var genes, in situ hybridization analysis has shown that P. vivax chromosome ends localize to the nuclear periphery40, where ectopic recombination favours the generation of variants and gene expansion. Although the repeat structure of P. vivax subtelomeric regions is not as extensive as that seen in P. falciparum6, P. vivax probably uses chromosomal exchange as a mechanism for generating antigenic diversity. VIR proteins represent an extremely diverse family, members of which currently appear more divergent than members of other partially characterized PIR families such as the P. chabaudi CIR (135 members) and the P. berghei BIR (245 members) families (Supplementary Fig. 9). Shared structural characteristics have been shown between VIR subfamily D proteins and the P. falciparum Pfmc-2tm family located at Maurer’s clefts, and VIR subfamily A proteins and the P. falciparum SURFIN family found on the surface of infected erythrocytes41. We speculate that the extreme diversity and sub-structuring of VIR proteins indicate members’ different subcellular localizations and functions, including immune evasion.

The structure of an archetypal vir gene is shown at the top, followed by VIR motifs in each of the subfamilies A–E and G–L (subfamily F is not shown), arranged from the N terminus (left) to the C terminus (right). Consensus motif sequences numbered in decreasing order of statistical significance are shown colour coded below the figure. Motif 2, transmembrane (TM) domain; motif 3, PEXEL/VSP-like motif; all remaining motifs are predicted to be exposed globular domains. The overall organization and order of the motifs is maintained, with the central core motifs 9, 1, 3, 6 and 10 followed by C terminus motifs 7, 2, 4, 8 and 5 embedded in a variant-sized portion of the molecule. Motifs are listed in the Supplementary Information.
We identified eight novel gene families (Pv-fam-a to Pv-fam-e and Pv-fam-g to Pv-fam-i; Supplementary Table 16) in the P. vivax genome, most of which are located in subtelomeric regions (Fig. 1). Of particular interest are (1) the PvTRAG (Pv-fam-a) gene family (36 genes), one member of which was previously identified (it encodes a protein localized to the caveola–vesicle complex of infected erythrocytes, and has been shown to elicit a humoral immune response during the course of natural infections42); and (2) the Pv-fam-e family (Supplementary Fig. 10), 36 copies of which are found in two loci on either side of the predicted centromere on chromosome 5, with one 10-gene locus present in a 47% G+C region, and a second 26-gene locus present in a 36% G+C region. Whereas P. vivax proteins have a fairly balanced codon composition, using all 61 sense codons almost equally (effective number of codons, Nc = 54.2), their orthologues in P. falciparum are more biased (Nc = 37.5), with G- and C-ending codons nearly absent from fourfold degenerate amino acids (Supplementary Table 17). However, P. vivax gene families, which are predominantly located in (A+T)-rich regions, have a codon composition of Nc = 47. This pattern suggests a strong influence of local mutation pattern on the nucleotide composition of genes and indicates a potential for differential gene expression.
Plasmodium drug interaction genes
The sexual stages of P. vivax are produced before the onset of clinical symptoms, permitting mosquito transmission early in an infection. Such early parasite transmission may delay development of resistance to many of the antimalarial drugs used to treat vivax malaria, despite the extensive long-term use of these drugs in regions endemic for both P. vivax and P. falciparum43. Nevertheless, P. vivax can develop resistance to most of the current antimalarial drugs. To understand the interactions between antimalarial drugs and the parasite proteins implicated in drug binding and resistance, we examined crystal structures and developed homology models for several P. vivax proteins in the predicted proteome, and compared the predicted binding sites and reported mutations with those of their P. falciparum orthologues (Table 2).

Currently, the most efficacious novel antimalarial drugs are derivatives of artemisinin (qinghaosu) and atovaquone, used predominantly in combination therapies. Arteminsinin derivatives, the most potent drugs recommended for treatment, may target a sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA)-type protein, ATPase6 (ref. 44). We constructed homology models of P. vivax and P. falciparum ATPase6 and identified two residues in the putative active sites for artemisinin that differ between the two species (P. vivax A263 and S1008, equivalent to L263 and N1039 in P. falciparum). A change in residue 263 from leucine to alanine results in a threefold increase in susceptibility to artemisinin by Xenopus laevis expressing PfATPase6 (ref. 44), although the IC50 (concentration required to inhibit parasite growth by 50%) for some P. vivax field isolates appears higher than the IC50 for P. falciparum45. However, it should be noted that clinical resistance of any human Plasmodium species to artemisinin derivatives has yet to be documented. Atovaquone, used in combination with the antifolate proguanil, selectively inhibits mitochondrial electron transport at the cytochrome bc(1) complex; mutations in the cytochrome b (cytb) gene can interfere with this inhibition, causing resistance. We constructed a homology model of P. vivax CYTB and compared it to the P. falciparum CYTB homology model46, revealing almost identical structures, including the predicted atovaquone active sites. Although there are no reports of atovaquone treatment failures for P. vivax infections, our studies indicate that should resistance arise, the same sites in P. vivax CYTB may be implicated.
Towards a policy shift for P. vivax malaria
Despite the insights into parasite biology provided by the P. vivax genome, many important questions remain that can only be addressed by functional studies. For example, we were unable to find differences in the predicted P. vivax proteome that might explain the rheological behaviour of P. vivax-infected erythrocytes, which remain flexible and can repeatedly pass through the spleen, unlike P. falciparum-infected reticulocyte, the rigidity of which facilitates cytoadherence and avoidance of splenic clearance47. Studies of the hypnozoite transcriptome, although technically challenging, would radically increase our inadequate knowledge of the biology of this dormant form. Studies are currently underway to develop new in vitro culture systems48, which could provide badly needed biological material for such functional studies.
The malaria research and control communities were challenged recently to establish once again the eradication of malaria as a policy goal49. Given the significant contribution of P. vivax to the global malaria situation1,43, it is imperative that these efforts include elimination of P. vivax as well as P. falciparum. Elimination of P. vivax presents special challenges, in particular the parasite’s production of dormant hypnozoites that enables relapses long after the initial parasitaemia has cleared. Indeed, an important aspect of P. vivax eradication will be the development of new drugs to replace primaquine for radical cure. Although the development of new drugs targeting P. vivax liver stages is a formidable task, recent developments offer hope that this goal can be accomplished50.
Methods Summary
Genome sequencing, assembly, mapping and annotation
Saimiri boliviensis boliviensis monkeys were infected with the Salvador I strain of P. vivax isolated from a patient from El Salvador. Extracted parasite DNA was used to make genomic DNA libraries for shotgun sequencing. Reads were assembled into scaffolds, inter-scaffold gaps closed, and scaffolds assigned to P. vivax chromosomes through hybridization of scaffold-specific probes to pulsed-field gel separated chromosomes. Gene prediction algorithms were used to predict gene models, and each model was manually checked for structural inconsistencies. Gene function was assigned using an automated annotation pipeline with subsequent manual curation.
Genome analysis
Methods for the in silco analysis of the genome sequence are described in the Supplementary Information.
Studies requiring laboratory experimentation
For polymorphic microsatellite identification, primers flanking 333 microsatellites identified from the genome sequence and designed for field studies where access to capillary electrophoresis equipment may not be possible were used to amplify the loci from eight worldwide P. vivax laboratory strains adapted to growth in monkeys (Brazil I, Miami II, Pakchong, Panama I, Nica, Thai II, Vietnam IV and Indonesia XIX). Amplicons were separated by electrophoresis on agarose gels and scored for size differences. For vir gene expression studies, cDNA was generated from total RNA extracted from the Salvador I isolate and from three patient isolates from Brazil. Primers were designed to eight vir gene subfamilies and used to amplify the loci.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
Sequence and annotation data for the genome are deposited in GenBank under the project accession number AAKM00000000 and are also available at the Plasmodium genome sequence database PlasmoDB (http://plasmodb.org). A minimal tiling path of clones covering each chromosome is available through the malaria repository MR4 (http://www.mr4.org), and a long-oligo array through the Pathogen Functional Genomics Resource Center (http://pfgrc.jcvi.org).
References
Price, R. N. et al. Vivax malaria: Neglected and not benign. Am. J. Trop. Med. Hyg. 77 (suppl. 6). 79–87 (2007)
Miller, L. H., Mason, S. J., Clyde, D. F. & McGinniss, M. H. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295, 302–304 (1976)
Gardner, M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum . Nature 419, 498–511 (2002)
Carlton, J. M. et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii . Nature 419, 512–519 (2002)
Hall, N. et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 307, 82–86 (2005)
Pain, A. et al. The genome of the simian and human malaria parasite Plasmodium knowlesi . Nature 10.1038/nature07306 (this issue)
McCutchan, T. F., Dame, J. B., Miller, L. H. & Barnwell, J. Evolutionary relatedness of Plasmodium species as determined by the structure of DNA. Science 225, 808–811 (1984)
Hughes, A. L. The evolution of amino acid repeat arrays in Plasmodium and other organisms. J. Mol. Evol. 59, 528–535 (2004)
Anders, R. F. Multiple cross-reactivities amongst antigens of Plasmodium falciparum impair the development of protective immunity against malaria. Parasite Immunol. 8, 529–539 (1986)
De Silva, E. K. et al. Specific DNA-binding by Apicomplexan AP2 transcription factors. Proc. Natl Acad. Sci. USA 105, 8393–8398 (2008)
Coulson, R. M., Hall, N. & Ouzounis, C. A. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum . Genome Res. 14, 1548–1554 (2004)
van Noort, V. & Huynen, M. A. Combinatorial gene regulation in Plasmodium falciparum . Trends Genet. 22, 73–78 (2006)
Young, J. A. et al. In silico discovery of transcription regulatory elements in Plasmodium falciparum . BMC Genomics 9, 70 (2008)
Ferdig, M. T. & Su, X. Z. Microsatellite markers and genetic mapping in Plasmodium falciparum . Parasitol. Today 16, 307–312 (2000)
Feng, X. et al. Single-nucleotide polymorphisms and genome diversity in Plasmodium vivax . Proc. Natl Acad. Sci. USA 100, 8502–8507 (2003)
Imwong, M. et al. Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J. Infect. Dis. 195, 927–933 (2007)
Joy, D. A. et al. Local adaptation and vector-mediated population structure in Plasmodium vivax malaria. Mol. Biol. Evol. 25, 1245–1252 (2008)
Kooij, T. W. et al. A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog. 1, e44 (2005)
Ralph, S. A. et al. Tropical infectious diseases: Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nature Rev. Microbiol. 2, 203–216 (2004)
Sato, S. & Wilson, R. J. The plastid of Plasmodium spp.: a target for inhibitors. Curr. Top. Microbiol. Immunol. 295, 251–273 (2005)
Akoachere, M. et al. Characterization of the glyoxalases of the malarial parasite Plasmodium falciparum and comparison with their human counterparts. Biol. Chem. 386, 41–52 (2005)
Krotoski, W. A. The hypnozoite and malarial relapse. Prog. Clin. Parasitol. 1, 1–19 (1989)
Baird, J. K. & Hoffman, S. L. Primaquine therapy for malaria. Clin. Infect. Dis. 39, 1336–1345 (2004)
Sattabongkot, J. et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax . Am. J. Trop. Med. Hyg. 74, 708–715 (2006)
Fang, X. D., Kaslow, D. C., Adams, J. H. & Miller, L. H. Cloning of the Plasmodium vivax Duffy receptor. Mol. Biochem. Parasitol. 44, 125–132 (1991)
Galinski, M. R., Medina, C. C., Ingravallo, P. & Barnwell, J. W. A. Reticulocyte-binding protein complex of Plasmodium vivax merozoites. Cell 69, 1213–1226 (1992)
Cowman, A. F. & Crabb, B. S. Invasion of red blood cells by malaria parasites. Cell 124, 755–766 (2006)
Gruner, A. C. et al. The Py235 proteins: glimpses into the versatility of a malaria multigene family. Microbes Infect. 6, 864–873 (2004)
Duraisingh, M. T. et al. Phenotypic variation of Plasmodium falciparum merozoite proteins directs receptor targeting for invasion of human erythrocytes. EMBO J. 22, 1047–1057 (2003)
Preiser, P. R., Jarra, W., Capiod, T. & Snounou, G. A rhoptry-protein-associated mechanism of clonal phenotypic variation in rodent malaria. Nature 398, 618–622 (1999)
Roussilhon, C. et al. Long-term clinical protection from falciparum malaria is strongly associated with IgG3 antibodies to merozoite surface protein 3. PLoS Med. 4, e320 (2007)
Gilson, P. R. et al. Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum . Mol. Cell. Proteomics 5, 1286–1299 (2006)
Sanders, P. R. et al. Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum . J. Biol. Chem. 280, 40169–40176 (2005)
Dzikowski, R., Templeton, T. J. & Deitsch, K. Variant antigen gene expression in malaria. Cell. Microbiol. 8, 1371–1381 (2006)
del Portillo, H. A. et al. A superfamily of variant genes encoded in the subtelomeric region of Plasmodium vivax . Nature 410, 839–842 (2001)
Fernandez-Becerra, C. et al. Variant proteins of Plasmodium vivax are not clonally expressed in natural infections. Mol. Micro. 58, 648–658 (2005)
Marti, M., Good, R. T., Rug, M., Knuepfer, E. & Cowman, A. F. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 306, 1930–1933 (2004)
Hiller, N. L. et al. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 306, 1934–1937 (2004)
Frank, M. et al. Strict pairing of var promoters and introns is required for var gene silencing in the malaria parasite Plasmodium falciparum . J. Biol. Chem. 281, 9942–9952 (2006)
Scherf, A., Figueiredo, L. & Freitas-Junior, L. H. Genomes and Molecular Cell Biology of Malaria Parasites (Horizon Press, 2004)
Merino, E. F. et al. Multi-character population study of the vir subtelomeric multigene superfamily of Plasmodium vivax, a major human malaria parasite. Mol. Biochem. Parasitol. 149, 10–16 (2006)
Jalah, R. et al. Identification, expression, localization and serological characterization of a tryptophan-rich antigen from the human malaria parasite Plasmodium vivax. . Mol. Biochem. Parasitol. 142, 158–169 (2005)
Mendis, K., Sina, B. J., Marchesini, P. & Carter, R. The neglected burden of Plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 64, 97–106 (2001)
Uhlemann, A. C. et al. A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nature Struct. Mol. Biol. 12, 628–629 (2005)
Russell, B. et al. Determinants of in vitro drug susceptibility testing of Plasmodium vivax . Antimicrob. Agents Chemother. 52, 1040–1045 (2008)
Korsinczky, M. et al. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob. Agents Chemother. 44, 2100–2108 (2000)
Suwanarusk, R. et al. The deformability of red blood cells parasitized by Plasmodium falciparum and P. vivax . J. Infect. Dis. 189, 190–194 (2004)
Udomsangpetch, R., Kaneko, O., Chotivanich, K. & Sattabongkot, J. Cultivation of Plasmodium vivax . Trends Parasitol. 24, 85–88 (2008)
Roberts, L. & Enserink, M. Malaria. Did they really say. eradication? Science 318, 1544–1545 (2007)
Carraz, M. et al. A plant-derived morphinan as a novel lead compound active against malaria liver stages. PLoS Med. 3, e513 (2006)
Acknowledgements
We thank the P. vivax research community for their support, and in particular M. Gottlieb and V. McGovern for facilitating financial support. Funding came from the following sources: P. vivax sequencing, assembly and closure, US Department of Defense and National Institute of Allergy and Infectious Diseases; genome mapping, Burroughs Wellcome Fund; and selective constraint analysis, National Institute of General Medical Sciences. We wish to thank TIGR’s SeqCore, Closure and IFX core facilities, E. Lee, J. Sundaram, J. Orvis, B. Haas and T. Creasy for engineering support, R. K. Smith Jr for annotation support, E. Lyons and H. Zhang for technical assistance, H. Potts for statistical analysis, T. McCutchan for rDNA sequence annotation, and S. Perkins for the Plasmodium phylogeny.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
This file contains data shown in Figure 1, Expanded Table 2, Supplementary Methods, Supplementary Results, Supplementary Figures 1-10, Supplementary Tables 1-4, 7-9, 11-17 (see separate files for Tables 5, 6 and 10) and Supplementary References (PDF 4734 kb)
Supplementary Table 5
This files contains Supplementary Table 5 (for Legend, see page 41 of nature07327-s1.pdf) (XLS 67 kb)
Supplementary Table 6
This files contains Supplementary Table 6 (for Legend, see page 42 of nature07327-s1.pdf) (XLS 6290 kb)
Supplementary Table 10
This files contains Supplementary Table 10 (for Legend, see page 51 of nature07327-s1.pdf) (XLS 253 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Carlton, J., Adams, J., Silva, J. et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763 (2008). https://doi.org/10.1038/nature07327
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature07327
This article is cited by
-
Profiling the antibody response of humans protected by immunization with Plasmodium vivax radiation-attenuated sporozoites
Scientific Reports (2024)
-
Systems biology of malaria explored with nonhuman primates
Malaria Journal (2022)
-
Evolutionary history of Plasmodium vivax and Plasmodium simium in the Americas
Malaria Journal (2022)
-
Immunogenicity and antigenicity of a conserved fragment of the rhoptry-associated membrane antigen of Plasmodium vivax
Parasites & Vectors (2022)
-
The presence of circulating antibody secreting cells and long-lived memory B cell responses to reticulocyte binding protein 1a in Plasmodium vivax patients
Malaria Journal (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
